

Abstract
Spinocerebellar Ataxia Type 3 (SCA3) is one of nine polyglutamine (polyQ) diseases that are all characterized by progressive neuronal dysfunction and the presence of neuronal inclusions containing aggregated polyQ protein, suggesting that protein misfolding is a key part of this disease. Ataxin-3, the causative protein of SCA3, contains a globular, structured N-terminal domain (the Josephin domain) and a flexible polyQ-containing C-terminal tail, the repeat-length of which modulates pathogenicity. It has been suggested that the fibrillogenesis pathway of ataxin-3 begins with a non-polyQ-dependent step mediated by Josephin domain interactions, followed by a polyQ-dependent step. To test the involvement of the Josephin domain in ataxin-3 fibrillogenesis, we have created both pathogenic and nonpathogenic length ataxin-3 variants with a stabilized Josephin domain, and have both stabilized and destabilized the isolated Josephin domain. We show that changing the thermodynamic stability of the Josephin domain modulates ataxin-3 fibrillogenesis. These data support the hypothesis that the first stage of ataxin-3 fibrillogenesis is caused by interactions involving the non-polyQ containing Josephin domain and that the thermodynamic stability of this domain is linked to the aggregation propensity of ataxin-3.
Keywords: ataxin-3, polyglutamine, proten misfolding, fibrillogenesis
Introduction
The polyglutamine (polyQ) diseases are a group of inherited neurodegenerative diseases, whereby the expansion of a polyQ tract beyond a specific threshold leads to disease. These diseases are characterized by the presence of neuronal nuclear inclusions that contain fibrillar protein aggregates.1 Nine polyQ-containing proteins are known to be involved in these diseases, and aside from their expanded polyQ tract, the proteins share no sequence or known structural homology.2 Increasingly, it has become clear that the domains and sequences flanking the polyQ tract are a significant factor in the aggregation mechanism of these proteins. For example, recent studies have demonstrated that a number of the polyQ proteins contain domains with an intrinsic aggregation propensity.3,4 The Josephin domain of ataxin-3,5,6 ataxin-1/HBP1 (AXH) domain of ataxin-1,7 and first 17 amino acids of huntingtin8 all have a propensity to aggregate under physiological conditions. In addition, these domains also play a key role during aggregation of the full-length protein in vivo and in vitro. In a cellular environment, the amount of aggregates formed by ataxin-1 is decreased when the AXH domain is substituted with a nonfibrillogenic homologue.7,9 Similarly, when the Josephin domain of ataxin-3 is substituted with the Hb1 domain, the formation of large aggregates is inhibited.10 These studies indicate the importance of the flanking domains in the aggregation of polyQ proteins.
An early study suggested that the expansion of the polyQ tract led to protein destabilization and subsequent aggregation;11 however, a number of recent studies using both disease and model proteins have investigated the effects of expanding the polyQ tract on overall protein stability, with mixed results. The model system CRABP1-huntingtin exon-1 only showed a decrease in global stability with an expanded polyQ tract,12 whereas no change in stability was observed in a SpA-polyQ model system with various length polyQ tracts.13 In ataxin-3, the addition of a polyQ tract destabilized the protein, yet expansion into the pathogenic range had no further effect.14,15 A comprehensive study into the relationship between flanking domain stability and polyQ aggregation has not however been reported.
It has been suggested that the polyQ protein ataxin-3 aggregates via a two-stage aggregation mechanism.16,17 The first stage is characterized by interactions possibly involving the flanking Josephin domain, and the second step involves the expanded polyQ tract. This two-stage mechanism, involving non-polyQ domains first, appears to be similar for ataxin-17 and huntingtin.8 Thus, we hypothesize that intermolecular interactions between Josephin domains are the key first step responsible for initiating the ataxin-3 aggregation process, and that the stability of this domain therefore modulates polyQ aggregation kinetics. If this hypothesis is correct, changing the Josephin domain stability will correspondingly affect ataxin-3 aggregation kinetics. We have rationally designed both stabilizing and destabilizing mutations within the Josephin domain of pathogenic and non-pathogenic length ataxin-3 using the PoPMuSiC algorithm,18,19 and show that in support of our hypothesis, the corresponding in vitro aggregation kinetics of these mutants are modulated.
Results and Discussion
The aim of this study is to analyze the effects of changing the thermodynamic stability of Josephin on the aggregation kinetics of ataxin-3. To achieve this, we utilized an in silico approach to predict mutations that would effect the stability of Josephin and then investigated the stability and aggregation pathways of the mutants.
Prediction of mutations with changed stability using PoPMuSiC
Using the PoPMuSiC program, we introduced successively all possible single-site mutations in the Josephin domain (PDB code 1YZB20) and computed the resulting change in folding free energy (ΔΔGcomputed). To reduce the error on the computed ΔΔG values due to dataset limitations, we used two different datasets, that is, DB1 and DB2 (see Experimental procedures), to evaluate two ΔΔG values: ΔΔGDB1 and ΔΔGDB2. The ΔΔGcomputed's presented in this article refer to the average of these two values.
We excluded mutations involving prolines from our analysis, as they are likely to provoke backbone structural rearrangements, which are not modeled by the PoPMuSiC program. We also excluded mutations close to the catalytic triad (Cys14, His119, and Asn134) to avoid alteration of the biological activity of the protein. We selected the stabilizing mutations that present a ΔΔGcomputed value lower than −1.0 kcal mol−1 and the destabilizing mutations with a ΔΔGcomputed value larger than +2.0 kcal mol−1. Using this selection criteria, we derived a list of five predicted stabilizing mutations (Q24G, Q24Y, S81A, R103G, and Q129I) and 232 predicted destabilizing mutations.
We chose two of the most stabilizing mutations (Table I), and four destabilizing mutations that were predicted to have varying stabilities. Of the destabilizing mutations, only the L169H mutation resulted in soluble expression and was used in this study. The lack of soluble expression of the destabilized recombinant proteins suggests protein stability is strongly linked to solubility and/or aggregation.
Table I.
PoPMuSiC Predicted Changes in Josephin Stability
| Predicted changes in Josephin stability | ||
|---|---|---|
| Mutation type | Mutation | ΔΔGcomputed (kcal mol−1) |
| Stabilising | R103G | −1.37 |
| Stabilising | S81A | −1.1 |
| Destabilising | L169H | 3.0 |
In the Josephin domain, ataxin-3(Q15) and ataxin-3(Q64) variants, three potentially stabilizing mutants were utilized; the single mutants R103G and S81A and the double mutant R103G S81A, in addition to the destabilizing mutation L169H (Fig. 1). Soluble expression was not achieved for L169H in either ataxin-3(Q15) or ataxin-3(Q64). Purification of Josephin L169H resulted in a yield 10-fold lower than that typically achieved for wild-type Josephin; however, this was sufficient for use throughout this study. All of the proteins used in this study were correctly folded, with their secondary structure and intrinsic fluorescence unchanged from wild type (data not shown). They were also biologically active, as determined by measurement of ubiquitin-protease activity.14
Figure 1.
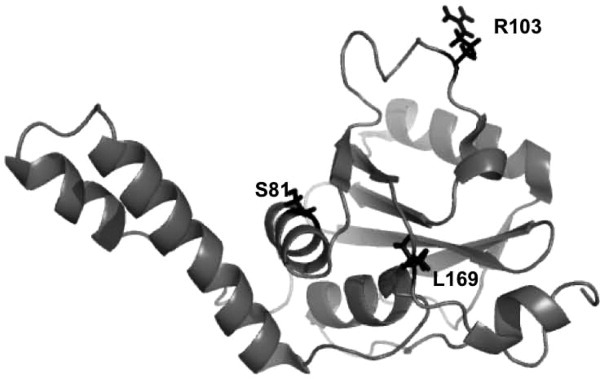
Predicted stability mutations in the Josephin domain. The two stabilizing mutations R103G and S81A and the destabilizing mutation L169H are highlighted on the Josephin structure.
Stability Mutations in Josephin correspond as predicted in silico
Thermal denaturation was used to determine whether the mutations resulted in a change to the thermodynamic stability of Josephin (Fig. 2). Although thermal denaturation of Josephin is not reversible, due to aggregation, the midpoints of denaturation give a relative measure of the protein's stability. For wild-type Josephin, the midpoint of the transition was determined to be 51.3 ± 0.6°C [Fig. 2(A)]. The two stabilizing mutants Josephin R103G and Josephin S81A both showed an increase in stability with a shift in the transition to midpoints to 55.9 ± 0.2°C and 54.1 ± 0.4°C respectively, whereas the destabilizing mutant Josephin L169H showed a decrease in stability with a transition midpoint of 47.5°C (Table II). These changes in stability are in agreement with the PoPMuSiC predictions. Interestingly, the double mutant Josephin R103G S81A showed the same thermal stability as Josephin S81A, with a midpoint of 55.4 ± 0.2°C. This trend was also observed for ataxin-3(Q15) [Fig. 2(B)] and ataxin-3(Q64) [Fig. 2(C), Table II].
Figure 2.
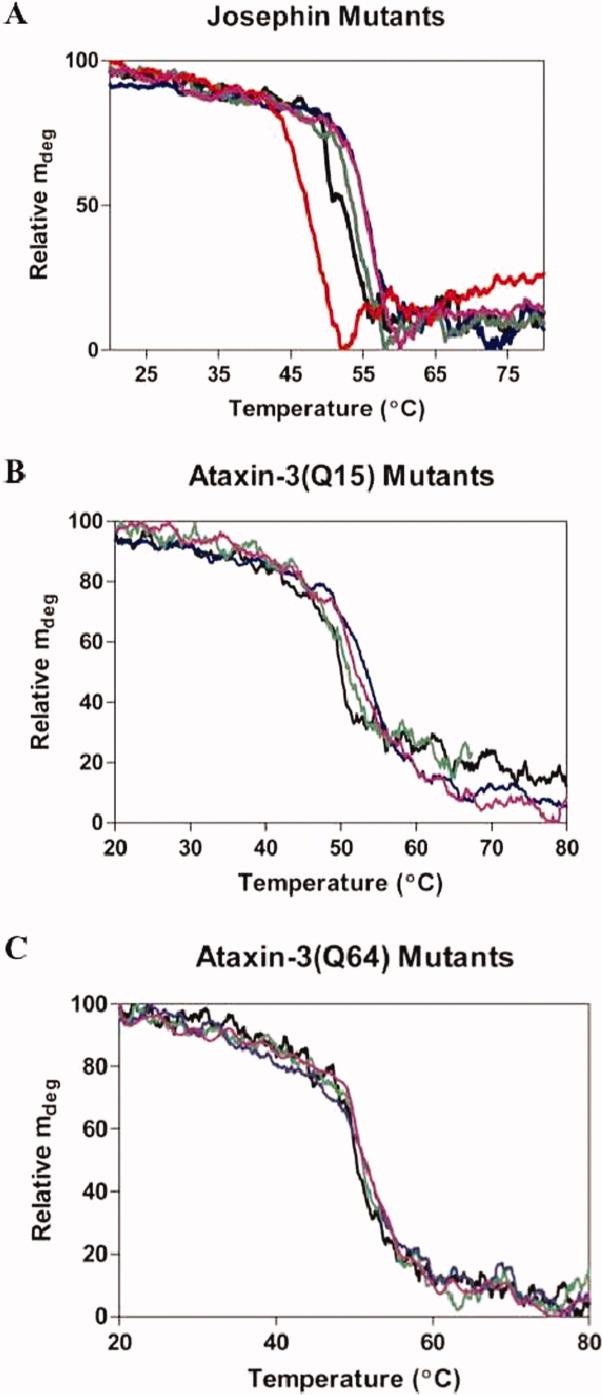
Thermal denaturation of Josephin stability mutants. Thermal denaturation was followed at 280 nm using near-UV CD. The temperature was increased by 1°C min−1 in a stoppered cuvette with a sample concentration of 1 mg mL−1. Representative traces are shown for (A) Josephin, (B) ataxin-3(Q15), and (C) ataxin-3(Q64) with wild-type (black), R103G (blue), S81A (green), R103G S81A (purple), and L169H (red).
Table II.
Thermal Denaturation and Aggregation Midpoints
| Thermal denaturation midpoint (°C) | ThioT midpoint (h) | SDS-stability midpoint (h) | |
|---|---|---|---|
| Josephin | 51.3 ± 0.6 | 79.4 ± 6.7 | |
| Josephin R103G | 55.9 ± 0.2 | 147.5 ± 7.3 | |
| Josephin S81A | 54.1 ± 0.4 | 115.2 ± 6.9 | |
| Josephin R103G S81A | 55.4 ± 0.2 | >150 h | |
| Josephin L169H | 47.5 ± 0.1 | 4.0 ± 0.9 | |
| Ataxin-3(Q15) | 49.2 ± 0.1 | 24.0 ± 1.3 | |
| Ataxin-3(Q15) R103G | 52.9 ± 0.8 | 48.9 ± 1.8 | |
| Ataxin-3(Q15) S81A | 50.6 ± 0.9 | 32.2 ± 1.9 | |
| Ataxin-3(Q15) R103G S81A | 53.4 ± 0.4 | 50.0 ± 2.8 | |
| Ataxin-3(Q64) | 51.0 ± 0.3 | 11.7 ± 1.2 | 35.0 ± 2.7 |
| Ataxin-3(Q64 R103G | 52.1 ± 0.6 | 25.3 ± 2.1 | 42.9 ± 2.6 |
| Ataxin-3(Q64) S81A | 50.8 ± 0.2 | 26.3 ± 4.9 | 41.0 ± 7.1 |
| Ataxin-3(Q64) R103G S81A | 52.4 ± 0.3 | 40.6 ± 2.2 | 48.7 ± 1.3 |
Stage 1 aggregation kinetics are modulated by the stability of Josephin
The first stage of ataxin-3 aggregation results in the formation of sodium dodecyl sulfate (SDS)-soluble, curvilinear protofibrils, which are detectable using ThioflavinT (ThT) fluorescence.17 As demonstrated previously, the aggregation kinetics of the ataxin-3 variants differed, with ataxin-3(Q64) showing the fastest aggregation rate, followed by ataxin-3(Q15) and lastly the isolated Josephin domain.6,16 All of the ataxin-3 variants including the stabilized mutants showed the sigmoidal aggregation curve, consisting of a lag phase followed by rapid elongation, indicative of nucleated polymerization kinetics (Fig. 3).
Figure 3.
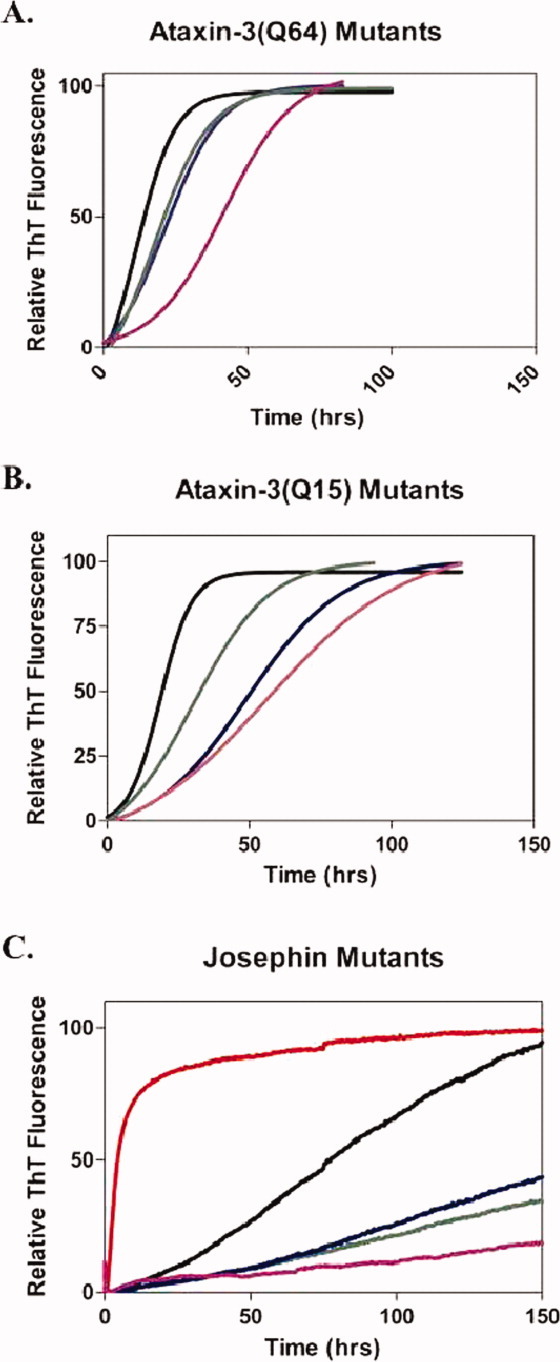
Ataxin-3 Stage 1-aggregation kinetics are changed by the Josephin mutants. Stage 1 aggregation was monitored using thioT fluorescence at 30 μM and pH 7.4 with a fluorescence plate reader. Wild-type (black), R103G (blue), S81A (green), R103G S81A (purple), and L169H (red) are shown in (A) Ataxin-3(Q64), (B) ataxin-3(Q15), and (C) Josephin.
For Josephin, ataxin-3(Q15) and ataxin-3(Q64) introduction of both stabilizing mutations cause a decreased rate of aggregation in comparison to their wild-type counterparts (Fig. 3, Table II). The aggregation rates of the R103G variants are slower than that of the S81A variants and this correlates with the slightly greater degree of stabilization of the R103G mutants (Fig. 2). The aggregation rate of the double mutant R103G S81A was suppressed even further compared to the single mutants. This was unexpected given that the R103G S81A double mutant had the same thermal stability as the R103G mutant. This highlights the complex nature of the aggregation mechanism in polyQ proteins, and demonstrates that thermodynamic stability is one of numerous factors influencing the Josephin-dependent aggregation stage of ataxin-3.
The destabilized mutant Josephin L169H showed a dramatic change in aggregation kinetics, with the almost complete elimination of the lag phase and an aggregation midpoint of 4.0 ± 0.9 h [Fig. 3(C)]. Considering that the change in the midpoint of thermal denaturation is similar in magnitude for both the stabilized Josephin R103G and destabilized Josephin L169H, for this set of mutants, it appears that destabilizing the Josephin domain has a greater impact on aggregation.
Stage 2 SDS-insoluble aggregation is correspondingly slowed by the stabilizing mutations
The SDS-insoluble aggregates formed by pathogenic length ataxin-3 during the second stage of the aggregation process can be monitored by a filter trap assay.17 In this study, the stabilized ataxin-3(Q64) mutants showed slower formation of SDS-insoluble fibrils. During Stage 1 of aggregation, there was a twofold to fourfold decrease in the aggregation rates of the stabilizing mutants compared to wild type ataxin-3(Q64) [Fig. 3(C)]. In Stage 2 of the aggregation pathway, the aggregation midpoints of the stabilized ataxin-3(Q64) mutants only increased by 20–40% compared to wild-type ataxin-3(Q64).
Morphology of end-point fibrils of the ataxin-3(Q64) stabilizing mutants is unchanged
The morphology of end point ataxin-3(Q64) fibrils was observed using transmission electron microscopy (TEM). The aggregates were analyzed after 90 h of incubation. The diameters of the aggregates ranged from 45 to 90 nm, with average lengths of 2 to 4 μm (Fig. 5). These images confirm that all of the stabilized ataxin-3(Q64) mutants form large, filamentous stage 2 aggregates, as previously described.17
Figure 5.
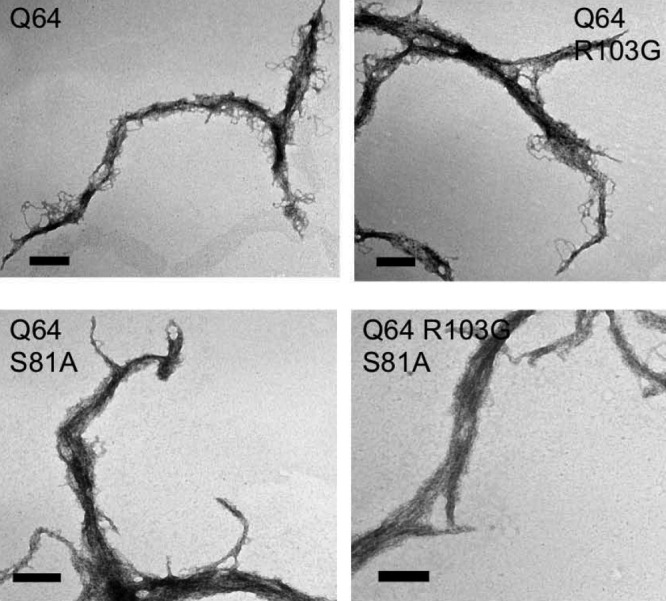
Morphology of endpoint aggregates in unchanged. Transmission electron microscopy of (A) ataxin-3(Q64), (B) ataxin-3(Q64) R103G, (C) ataxin-3(Q64) S81A, and (D) ataxin-3(Q64) R103G S81A. After 90 h of aggregation, samples were negatively stained using 1% uranyl acetate. Scale bar represents 200 nm.
Figure 4.

SDS-insoluble aggregation is slowed by a stabilized Josephin domain. Formation of SDS-insoluble aggregates was analyzed over time, taking aliquots from an aggregation assay at 30 μM and pH 7.4. (A) A representative filter trap membrane shows the formation of SDS-insoluble aggregates formed from ataxin-3(Q64) and the stabilizing ataxin-3(Q64) mutants. (B) Densitometry is shown for ataxin-3(Q64) (black), ataxin-3(Q64) R103G (blue), ataxin-3(Q64) S81A (green), and ataxin-3(Q64) R103G S81A (purple). Analysis of the membrane by densitometry was completed on three independent experiments and fit to a sigmoidal curve.
Implications for the aggregation mechanism of ataxin-3
Our study provides direct experimental evidence that conformational change within the Josephin domain is involved in the first step of the ataxin-3 aggregation pathway, as predicted by the multi-stage aggregation model.16,17 Specifically, increasing the thermodynamic stability of the Josephin domain slowed down the rate of stage-1 ataxin-3 fibrillogenesis and vice versa (Fig. 3). Thus, in the aggregation pathway of the Josephin domain, changing the stability of the protein significantly affects the kinetics of aggregation. Although only stabilizing mutations were studied for ataxin-3(Q15) and ataxin-3(Q64), the stabilizing mutations slowed the aggregation rate in both cases. This suggests that in the context of full-length ataxin-3, the Josephin domain is involved in the initial interactions of Stage 1 aggregation.
Our observations complement recent studies in which it was shown that interaction of Jospehin with another protein, either a chaperone6 or a binding partner, ubiquitin,21 reduces or prevents aggregation of ataxin-3. In these cases, it is likely that the effects observed are due in part to stabilization of the Josephin domain through protein–protein interactions, similar to that achieved in this work through mutagenesis. The stabilization leads to reduced rate of conformational change and accordingly a decreased rate of aggregation. Therefore, conformational change within the polyQ flanking domain, Josephin, is directly involved in aggregation of the full length protein, which suggests that targeting this folded domain with compounds that stabilize it may provide a potential therapeutic strategy.
Materials and Methods
Materials
Phenylmethylsulfonyl fluoride, β-mercaptoethanol and thioflavin T (ThioT) were all obtained from Sigma.
PoPMuSiC algorithm
The PoPMuSiC program aims at predicting the folding free energy changes (ΔΔG = ΔGmutant − ΔGwild-type) resulting from single-site mutations in a protein.22,23 All the possible point mutations are successively introduced in silico in the protein and their ΔΔG's are computed by means of statistical potentials derived from datasets of known protein structures. Two datasets are used here, referred to as DB1 and DB2: the first contains 141 well-resolved X-ray structures (resolution ≤ 2.5 Å) of protein chains with less than 25% pairwise sequence identity,24 and the other 735 high-resolution X-ray structures (resolution ≤ 2.0 Å) of protein chains with no more than 20% pairwise sequence identity.25 Two types of potentials are considered. ΔGtorsion is a torsion potential that describes local interactions along the sequence and is obtained from the frequencies of association between residues or residue pairs and backbone torsion angle domains. ΔGdistance is a distance potential computed from the propensities of residue pairs to be separated by a spatial distance between average side chain centroids Cμ. These potentials take implicitly the solvent into account as they are derived from solution structures. The folding free energy changes ΔΔG's are evaluated by linear combinations of torsion and distance potentials:
where α, β and γ are weighting factors depending on the solvent accessibility of the mutated residue.22
The PoPMuSiC program requires as input the structure of the target protein in PDB format and uses a simplified representation of protein structures consisting of the backbone atoms and the pseudo-atom Cμ The backbone structure is assumed to be unchanged upon mutation.
Mutagenesis
Using the QuickChange mutagenesis method, the following mutations were introduced into Josephin, ataxin-3(Q15) and ataxin-3(Q64): L169H, S81A, and R103G.
Expression and Purification of Ataxin-3 variants
All ataxin-3 variants were expressed and purified as previously described,26 and the proteins were stored at −80°C. Following purification the deubiquitinating activity of the proteins was measured15 and before use all were analyzed using gel filtration to ensure that no multimeric species were present.
Thermal denaturation
Thermal unfolding was monitored on a thermostatted Jasco-810 spectropolarimeter and performed by increasing the temperature by 1°C per min from 20 to 80°C. Changes in the near-UV CD signal were monitored at 280 nm, using a stoppered quartz cuvette with a path length of 1 cm. Each protein was at a concentration of 1mg mL−1 in PBS (137 mM NaCl, 10.1 mM NaH2PO4, 2.68 mM KCl, 1.76 mM KH2PO4, 10% (v/v) glycerol, ph 7.4).
Fibrillogenesis Time Course Assays
Ataxin-3 variants (30 μM) were incubated in TBSG (80 mM Tris, 100 mM NaCl, 10% (v/v) glycerol, pH 7.4) containing 5 mM of ethylenediaminetetraacetic acid (EDTA), 15 mM of β-mercaptoethanol, and 2 mM of phenylmethylsulfonyl fluoride. Samples were incubated without shaking at 37°C in air-tight containers to prevent evaporation.
ThioT Fluorescence
ThioT fluorescence measurements were recorded on a BMG Fluorostar plate reader, using the buffering conditions described previously with the addition of 30 μM thioT. Excitation and emission wavelengths of 430 and 480 nm with a cut off filter of 455 nm were used, and both excitation and emission were read from the underneath of a black clear bottom plate. All reactions were completed at 37°C with no shaking.
Membrane Filter Trap Assay
Aliquots containing 7.4 μg of protein were taken from the fibrillogenesis reaction, diluted 1:1 with a 4% (w/v) SDS/100 mM DTT solution and then boiled for 5 min at 100°C. About 200 μL of 2% (w/v) SDS was then added to each of the samples, and 2.5 μg of protein were filtered through a 0.2-μm cellulose acetate membrane (Schleicher and Schuell) using a Bio-Rad Bio-Dot SF microfiltration unit. The membrane was then washed twice by filtering 200 μL of 0.1% (w/v) SDS, and blotted with a hexahistidine (His6) antibody (Serotec). Densitometry was completed using the program ImageQuant.
Transmission Electron Microscopy
TEM images were obtained using a Hitachi TEM with an accelerating voltage of 80 kV. Samples were adsorbed onto a carbon-coated grid, and stained with 1% (w/v) uranyl acetate.
Acknowledgments
The authors thank the Protein Production Unit at Monash University for their help in protein purification.
References
- 1.Diaz-Hernandez M, Moreno-Herrero F, Gomez-Ramos P, Moran MA, Ferrer I, Baro AM, Avila J, Hernandez F, Lucas JJ. Biochemical, ultrastructural, and reversibility studies on huntingtin filaments isolated from mouse and human brain. J Neurosci. 2004;24:9361–9371. doi: 10.1523/JNEUROSCI.2365-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Robertson AL, Bate MA, Androulakis SG, Bottomley SP, Buckle AM. PolyQ: a database describing the sequence and domain context of polyglutamine repeats in proteins. Nucleic Acids Res. 2011;39:D272–D276. doi: 10.1093/nar/gkq1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Robertson AL, Bottomley SP. Towards the treatment of polyglutamine diseases: the modulatory role of protein context. Curr Med Chem. 2010;17:3058–3068. doi: 10.2174/092986710791959800. [DOI] [PubMed] [Google Scholar]
- 4.Saunders HM, Bottomley SP. Multi-domain misfolding: understanding the aggregation pathway of polyglutamine proteins. Protein Eng Des Sel. 2009;22:447–451. doi: 10.1093/protein/gzp033. [DOI] [PubMed] [Google Scholar]
- 5.Masino L, Nicastro G, Menon RP, Dal Piaz F, Calder L, Pastore A. Characterization of the structure and the amyloidogenic properties of the Josephin domain of the polyglutamine-containing protein ataxin-3. J Mol Biol. 2004;344:1021–1035. doi: 10.1016/j.jmb.2004.09.065. [DOI] [PubMed] [Google Scholar]
- 6.Robertson AL, Headey SJ, Saunders HM, Ecroyd H, Scanlon MJ, Carver JA, Bottomley SP. Small heat-shock proteins interact with a flanking domain to suppress polyglutamine aggregation. Proc Natl Acad Sci USA. 2010;107:10424–10429. doi: 10.1073/pnas.0914773107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Chiara C, Menon RP, Dal Piaz F, Calder L, Pastore A. Polyglutamine is not all: the functional role of the AXH domain in the ataxin-1 protein. J Mol Biol. 2005;354:883–893. doi: 10.1016/j.jmb.2005.09.083. [DOI] [PubMed] [Google Scholar]
- 8.Thakur AK, Jayaraman M, Mishra R, Thakur M, Chellgren VM, IJ LB, Anjum DH, Kodali R, Creamer TP, Conway JF, Gronenborn AM, Wetzel R. Polyglutamine disruption of the huntingtin exon 1 N terminus triggers a complex aggregation mechanism. Nat Struct Mol Biol. 2009;16:380–389. doi: 10.1038/nsmb.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Chiara C, Menon RP, Adinolfi S, de Boer J, Ktistaki E, Kelly G, Calder L, Kioussis D, Pastore A. The AXH domain adopts alternative folds the solution structure of HBP1 AXH. Structure. 2005;13:743–753. doi: 10.1016/j.str.2005.02.016. [DOI] [PubMed] [Google Scholar]
- 10.Menon RP, Pastore A. Expansion of amino acid homo-sequences in proteins: insights into the role of amino acid homo-polymers and of the protein context in aggregation. Cell Mol Life Sci. 2006;63:1677–1685. doi: 10.1007/s00018-006-6097-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bevivino AE, Loll PJ. An expanded glutamine repeat destabilizes native ataxin-3 structure and mediates formation of parallel beta -fibrils. Proc Natl Acad Sci USA. 2001;98:11955–11960. doi: 10.1073/pnas.211305198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ignatova Z, Gierasch LM. Extended polyglutamine tracts cause aggregation and structural perturbation of an adjacent beta barrel protein. J Biol Chem. 2006;281:12959–12967. doi: 10.1074/jbc.M511523200. [DOI] [PubMed] [Google Scholar]
- 13.Robertson AL, Horne J, Ellisdon AM, Thomas B, Scanlon MJ, Bottomley SP. The structural impact of a polyglutamine tract is location-dependent. Biophys J. 2008;95:5922–5930. doi: 10.1529/biophysj.108.138487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chow MK, Ellisdon AM, Cabrita LD, Bottomley SP. Polyglutamine expansion in ataxin-3 does not affect protein stability: implications for misfolding and disease. J Biol Chem. 2004;279:47643–47651. doi: 10.1074/jbc.M405799200. [DOI] [PubMed] [Google Scholar]
- 15.Chow MK, Mackay JP, Whisstock JC, Scanlon MJ, Bottomley SP. Structural and functional analysis of the Josephin domain of the polyglutamine protein ataxin-3. Biochem Biophys Res Commun. 2004;322:387–394. doi: 10.1016/j.bbrc.2004.07.131. [DOI] [PubMed] [Google Scholar]
- 16.Ellisdon AM, Pearce MC, Bottomley SP. Mechanisms of ataxin-3 misfolding and fibril formation: kinetic analysis of a disease-associated polyglutamine protein. J Mol Biol. 2007;368:595–605. doi: 10.1016/j.jmb.2007.02.058. [DOI] [PubMed] [Google Scholar]
- 17.Ellisdon AM, Thomas B, Bottomley SP. The two-stage pathway of ataxin-3 fibrillogenesis involves a polyglutamine-independent step. J Biol Chem. 2006;281:16888–16896. doi: 10.1074/jbc.M601470200. [DOI] [PubMed] [Google Scholar]
- 18.Cabrita LD, Gilis D, Robertson AL, Dehouck Y, Rooman M, Bottomley SP. Enhancing the stability and solubility of TEV protease using in silico design. Protein Sci. 2007;16:2360–2367. doi: 10.1110/ps.072822507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gilis D, McLennan HR, Dehouck Y, Cabrita LD, Rooman M, Bottomley SP. In vitro and in silico design of alpha1-antitrypsin mutants with different conformational stabilities. J Mol Biol. 2003;325:581–589. doi: 10.1016/s0022-2836(02)01221-4. [DOI] [PubMed] [Google Scholar]
- 20.Nicastro G, Menon RP, Masino L, Knowles PP, McDonald NQ, Pastore A. The solution structure of the Josephin domain of ataxin-3: structural determinants for molecular recognition. Proc Natl Acad Sci USA. 2005;102:10493–10498. doi: 10.1073/pnas.0501732102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Masino L, Nicastro G, Calder L, Vendruscolo M, Pastore A. Functional interactions as a survival strategy against abnormal aggregation. FASEB J. 2011;25:45–54. doi: 10.1096/fj.10-161208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gilis D, Rooman M. PoPMuSiC, an algorithm for predicting protein mutant stability changes: application to prion proteins. Protein Eng. 2000;13:849–856. doi: 10.1093/protein/13.12.849. [DOI] [PubMed] [Google Scholar]
- 23.Kwasigroch JM, Gilis D, Dehouck Y, Rooman M. PoPMuSiC, rationally designing point mutations in protein structures. Bioinformatics. 2002;18:1701–1702. doi: 10.1093/bioinformatics/18.12.1701. [DOI] [PubMed] [Google Scholar]
- 24.Wintjens RT, Rooman MJ, Wodak SJ. Automatic classification and analysis of alpha alpha-turn motifs in proteins. J Mol Biol. 1996;255:235–253. doi: 10.1006/jmbi.1996.0020. [DOI] [PubMed] [Google Scholar]
- 25.Hobohm U, Scharf M, Schneider R, Sander C. Selection of representative protein data sets. Protein Sci. 1992;1:409–417. doi: 10.1002/pro.5560010313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chow MK, Ellisdon AM, Cabrita LD, Bottomley SP. Purification of polyglutamine proteins. Methods Enzymol. 2006;413:1–19. doi: 10.1016/S0076-6879(06)13001-3. [DOI] [PubMed] [Google Scholar]