

Abstract
The lysine-specific crosslinker 3,3′-dithiobis(sulfosuccinimidylpropionate) (DTSSP) is commonly used in the structural characterization of proteins by chemical crosslinking and mass spectrometry and we here describe an efficient two-step LC-MALDI-TOF/TOF procedure to detect crosslinked peptides. First MS data are acquired, and the properties of isotope-labeled DTSSP are used in data analysis to identify candidate crosslinks. MSMS data are then acquired for a restricted number of precursor ions per spot for final crosslink identification. We show that the thiol-catalyzed exchange between crosslinked peptides, which is due to the disulfide bond in DTSSP and known to possibly obscure data, can be precisely quantified using isotope-labeled DTSSP. Crosslinked peptides are recognized as 8 Da doublet peaks and a new isotopic peak with twice the intensity appears in the middle of the doublet as a consequence of the thiol-exchange. False-positive crosslinks, formed exclusively by thiol-exchange, yield a 1:2:1 isotope pattern, whereas true crosslinks, formed by two lysine residues within crosslinkable distance in the native protein structure, yield a 1:0:1 isotope pattern. Peaks with a 1:X:1 isotope pattern, where 0 < X < 2, can be trusted as true crosslinks, with a defined proportion of the signal [2X/(2 + X)] being noise from the thiol-exchange. The thiol-exchange was correlated with the protein cysteine content and was minimized by shortening the trypsin incubation time, and for two molecular chaperone proteins with known structure all crosslinks fitted well to the structure data. The thiol-exchange can thus be controlled and isotope-labeled DTSSP safely used to detect true crosslinks between lysine residues in proteins.
Keywords: chemical crosslinking, DTSSP, disulfide, mass spectrometry, MALDI-TOF/TOF, protein structure, protein–protein interactions, small heat shock protein, isotope pattern
Introduction
Chemical crosslinking in combination with mass spectrometry has been emerging as a promising strategy for the structural characterization of protein complexes and protein–protein interactions over the last decade.1–3 Proteins or protein complexes can be crosslinked with different crosslinking reagents that are reactive toward specific amino acids. The crosslinked proteins can be digested resulting in a mixture of unmodified peptides and peptides modified by the crosslinking reagent, which can be identified with mass spectrometry. The identification of two crosslinked amino acid residues yields a distance constraint useful in structural characterization, especially if the two residues reside on two different peptides (interpeptide crosslinks). The major challenges in the field include the detection of interpeptide crosslinks, which are low abundant compared with unmodified peptides and dead-end and intrapeptide crosslinks, and the unambiguous, confident assignment of peaks from mass spectrometry data to crosslinks.
Although the high-throughput interpretation of MS and MSMS data from crosslinking experiments has recently been improved by the development of several software programs that were specifically designed to handle crosslinking data1,4 and the increasing mass accuracy of today's mass spectrometers,5–7 the detection of low abundant interpeptide crosslinks is still not trivial. To address the problem of recognizing crosslinks in very complex peptide mixtures, a lot of progress has been made on the design of crosslinking reagents. Specialized reagents can be used that are isotope-labeled8 or give a specific signature or ion-tag during MSMS fragmentation.6,9–12 Other strategies include isotope-labeling with modification reagents or 18O of the N- or C-termini of peptides (of which there are two for interpeptide crosslinks).13,14 Crosslinking reagents can be cleavable, usually through reduction of a disulfide bond in the linker region. The disappearance of crosslinks in the mass spectra upon reduction and the appearance of the reduced forms of the species aids in the identification of crosslinks. Disulfide bond containing crosslinks also yield a specific 66 Da doublet signature in MALDI-TOF/TOF (matrix-assisted laser desorption/ionization, time-of-flight) spectra that can be used for their unambiguous identification.15 In addition, the cleavability of these crosslinking reagents offers the possibility to assess crosslinking efficiency by SDS-PAGE (sodium dodecyl sulfate polyacrylamide gel electrophoresis) or gel filtration in the absence and presence of a reducing agent.
One commonly used thiol-cleavable crosslinker, that is commercially available in both an unlabeled and an isotope-labeled form, is 3,3′-dithiobis(sulfosuccinimidylpropionate) (DTSSP). DTSSP contains two sulfo-N-hydroxysuccinimide esters that react with the primary amines in the side chain of lysines and the protein N-terminus. There are several studies reporting the use of this crosslinker for protein structural characterization.15–20 Despite the advantages of the disulfide bond in DTSSP, it can also complicate the results, as the disulfide bonds of the reagent may undergo thiol-exchange leading to disulfide bond scrambling, especially promoted at the alkaline pH used during digestion with trypsin, which is most active at around pH 8. Digestion with pepsin at an acidic pH is commonly used in the determination of native disulfide bonds by mass spectrometry,21 but the specificity of this protease is much lower, which would be adding to the already high complexity of crosslinked peptide mixtures, making data analysis even more difficult. The possibility of disulfide bond scrambling of DTSSP crosslinks has been recognized as potentially compromising the validity of identified crosslinks,2,3 but to our knowledge quantitative data on the phenomenon is currently lacking.
We have previously used crosslinking and mass spectrometry to study protein interactions within and between heat shock proteins.20,22 In this work, we have used the isotope-labeled crosslinking reagent DTSSP in a novel two-step offline LC-MALDI-TOF/TOF procedure to efficiently detect numerous crosslinks, and we have systematically evaluated the extent of disulfide scrambling by thiol-exchange using various proteins that are being used for protein–protein interaction studies. The disulfide scrambling strongly correlated with the protein cysteine content and the isotope pattern of crosslinks could be used as a quantitative measure of the scrambling, allowing controlled and safe detection of true crosslinks. We show that the distance constraints obtained from DTSSP crosslinking can be as reliable as information from nondisulfide containing crosslinking reagents.
Results and Discussion
Workflow to detect DTSSP crosslinks by two-step LC-MALDI-TOF/TOF
Data were collected for different proteins that were crosslinked with isotope-labeled DTSSP, digested into peptides, separated by offline LC (liquid chromatography), and analyzed by a two-step procedure for MALDI-TOF/TOF mass spectrometry using the program FINDX specifically developed for this workflow (Fig. 1). The disulfide bond in DTSSP allows the parallel processing and analysis of three samples per experiment: the crosslinked sample, a non-crosslinked reference sample, and a third sample in which the disulfide bonds in the DTSSP crosslinks were reduced by dithiothreitol (DTT).16,20 First MS data were acquired for the three samples and analyzed with FINDX in MS mode, to identify peaks matching peptide masses with DTSSP modifications (main modified species expected: dead-end (type 0), intrapeptide (type 1), and interpeptide (type 2) crosslinks). Matching peaks fulfilling two requirements, not being present in the reference and the DTT-treated sample, and showing an 8 Da doublet from isotope-labeled DTSSP, were regarded as candidate crosslinks by FINDX and selected as precursors for MSMS fragmentation.
Figure 1.
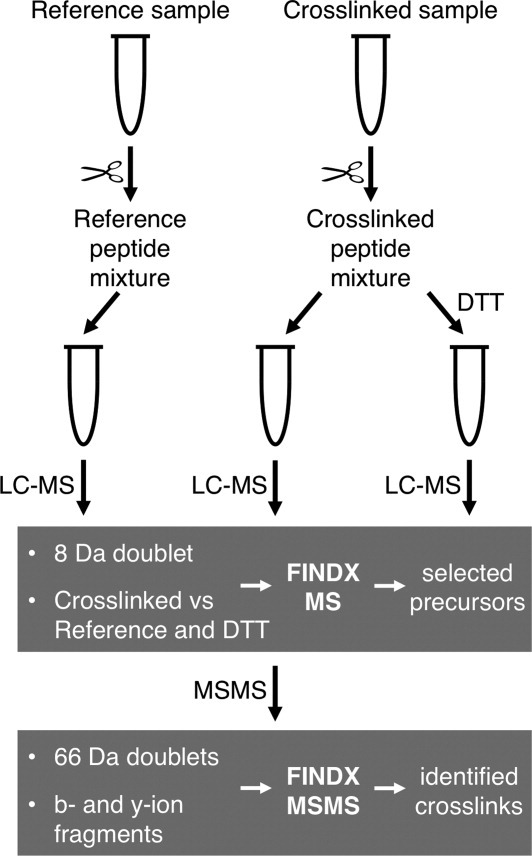
Crosslinking mass spectrometry workflow using DTSSP and two-step offline LC-MALDI-TOF/TOF. Protein is crosslinked with DTSSP and subsequently digested with trypsin, in parallel with the digestion of a reference non-crosslinked sample. The digested crosslinked sample is divided into two halves, one of which is treated with DTT to reduce the DTSSP disulfide bond. The three resulting peptide mixtures are separated by reversed phase LC, and acquired MS data are analyzed by FINDX in MS mode (first step). Based on two criteria, peak appearance as an 8 Da doublet and peak absence in the reference and DTT samples, peaks are selected for a candidate crosslink inclusion list of precursor ions, which is submitted for the acquisition of MSMS data. MSMS data are subsequently analyzed by FINDX in MSMS mode (second step). Based on two criteria, presence of 66 Da doublets15 and b- and y-ion fragments, crosslinks are finally identified.
Because the quality of MSMS data from MALDI-TOF/TOF is highly dependent on the number of precursors per spot on the MALDI target plate, the possibility in this workflow to select a restricted number of precursors is promoting high quality MSMS data. In the second step, MSMS data were acquired for the selected precursors and subsequently analyzed with FINDX in MSMS mode, confirming their identities by matching b- and y-ions (possibly including the DTSSP modification and the other peptide) and one or two 66 Da doublets resulting from the asymmetric fragmentation around the disulfide bond in DTSSP in MALDI-TOF/TOF.15 The crosslinks were thus identified and validated by information from MS data (not present in reference and DTT samples, and having 8 Da doublet), and MSMS data [b-/y-ion fragments and 66 Da doublet(s)].
The isotope pattern gives a quantitative measure of the proportion of native crosslinks and false-positive crosslinks originating from thiol-exchange in DTSSP
As with native disulfide bonds in proteins, the disulfide bonds in DTSSP crosslinked peptides can rearrange themselves by continuous dissociation and formation, depending on the experimental conditions. When using DTSSP crosslinking and mass spectrometry to study low-resolution protein structure, it is essential to know for each interpeptide crosslink whether the two amine groups were indeed crosslinked in the native protein structure or the detected crosslink has formed after trypsin digestion due to such rearrangement of disulfide bonds by thiol-exchange, which we from here on refer to as scrambling. In the latter case, an interpeptide crosslink can arise from any two peptides that have been modified with DTSSP, and it gives no or false information about the protein structure, so we refer to it as a false-positive crosslink. Using isotope-labeled DTSSP, the isotope pattern of peaks representing crosslinks can be used to recognize and validate native crosslinks and discard false-positives, by a quantitative assessment of the actual contribution from false-positive crosslinks that originate from scrambling by thiol-exchange, as outlined in Figure 2.
Figure 2.

Thiol-exchange causing disulfide scrambling of DTSSP crosslinks quantified by isotope pattern analysis. Lysine residues (LYS) within close proximity in the native protein structure are crosslinked with the disulfide bond-containing crosslinker DTSSP. Thiol-exchange with protein cysteine residues (CYS) can cause disulfide bond scrambling of DTSSP crosslinks upon digestion, leading to the formation of false-positive crosslinks (red). The isotope pattern of candidate crosslink peaks can be used to quantify the proportion between true crosslinks (black) that reflect native protein structure, and false-positives (red), as explained in more detail in Figure 3.
The commercially available isotope-labeled DTSSP reagent is a mixture of 50% light and 50% heavy reagent with the heavy reagent being 8.05016 Da heavier than the light reagent. The two times four hydrogen atoms on either side of the disulfide bond have been substituted with eight deuterium atoms in total, resulting in the 8 Da doublet peak for DTSSP-modified peptides in MS spectra [Fig. 3(A)]. When crosslinking aiming to probe protein structure, light or heavy molecules of DTSSP react with and crosslink two lysines within close spatial proximity in the native protein structure, and after digestion the two particular peptides that are crosslinked appear in the MS spectrum as an 8 Da doublet with similar intensities of the two peaks [Fig. 3(B), native crosslink].
Figure 3.

Isotope pattern of crosslinked peptides allows distinction between true-positive and false-positive crosslinks. A: The isotope-labeled crosslinking reagent DTSSP (Creative Molecules). B: Example of an isotope pattern with a 1:0:1 ratio for the +0, +4, and +8 Da triplet. The crosslink reflects a crosslinking event in the native protein. C: Example of an isotope pattern with a 1:2:1 ratio for the +0, +4, and +8 Da triplet. A crosslink that has been exclusively formed due to disulfide bond scrambling will have the 1:2:1 signature as a hallmark, and should be discarded as a false positive. D: Example of an isotope pattern with a 1:X:1 ratio, where in this case X = 0.38. This signal is predominantly originating from a crosslink that reflects a crosslinking event between the lysine residues in the peptides DK155IK and TK173VER that are in close proximity in the native protein, but is overlaid with a false-positive crosslink that has formed by scrambling. All peaks displaying an isotope pattern with less of the +4 Da peak than 1:2:1 reflect true crosslinks. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
However, if there are peptides present in solution that are modified with DTSSP, and if the disulfide moiety gets reduced and randomly recombines with any other peptide bearing such a reduced DTSSP modification, the resulting crosslink will show an additional diagnostic isotopic peak, between the 8 Da doublets, with twice the intensity of each of the doublet peaks [Fig. 3(C), false-positive crosslink]. This novel isotopic peak is due to crosslinking reagent that, as a result of the thiol-exchange, contains four hydrogen and four deuterium atoms. If a crosslinked peptide has formed exclusively by scrambling it will show a 1:2:1 ratio of the +0, +4, and +8 Da isotopic peaks, and it should be discarded as a false-positive.
Peaks showing neither a 1:0:1 nor a 1:2.1 ratio in the isotope pattern represent a mixture of a native crosslink overlaid with a false-positive, that is, a true crosslink between two peptides in the native protein structure and the same two peptides having formed by scrambling. This will yield a 1:X:1 ratio of the +0, +4, and +8 Da isotopic peaks, where X is smaller than 2 and bigger than 0 [Fig. 3(D), native crosslink overlaid with false-positive crosslink]. The proportion of the signal originating from scrambling is then
The thiol-exchange in peptides crosslinked with isotope-labeled DTSSP can thus be precisely quantified by the changed isotope pattern. When X is zero there is no scrambling, when X = 2 there is 100% scrambling and every value of X below 2 represents true crosslinks. In the example in Figure 3(D), X = 0.38 means that 32% of the signal is noise from scrambling but 68% of the signal can be safely trusted as a true, native crosslinking event.
Disulfide bond scrambling of DTSSP crosslinks is proportional to free cysteine content
We analyzed a number of different proteins and protein mixtures according to the crosslinking mass spectrometry workflow described earlier. We found that for all proteins, processed by the same standard procedure, the degree of scrambling clearly correlated with the cysteine content of the protein(s) (Table I). No scrambling was observed for αB-crystallin, which has no cysteines. Of the proteins in Table I, malate dehydrogenase (MDH) has the highest cysteine content (2.5%), and in four independent experiments, no native interpeptide crosslinks could be detected in MDH. A cysteine content of 2.5% is quite high for an intracellular protein, but proteins may have much higher cysteine contents; up to more than 10%,23,24 stressing the importance of being able to control the degree of scrambling during a crosslinking mass spectrometry experiment and to distinguish native from false-positive crosslinks. When using unlabeled DTSSP to probe protein structure, it is impossible to assess the validity of interpeptide crosslinks detected.
Table I.
Disulfide Bond Scrambling Dependence on Cysteine Content and Experimental Conditions
| Protein(s), experiment | Cys (%) | No.of expts | Valid crosslinks | Types (%) | False-positive crosslinks | Types (%) | Scrambling (%) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 0 | 1 | 2 | ||||||
| αB-crystallin | 0 | 3 | 28 | 32 | 11 | 57 | 0 | 0 | 0 | 0 | 0 |
| Hsp21, short trypsin | 0.5 | 1 | 42 | 31 | 14 | 55 | 0 | 0 | 0 | 0 | 0 |
| αB-crystallin + C8/α7 | 0.9 | 4 | 51 | 46 | 10 | 42 | 1 | 0 | 0 | 2 | 2 |
| Hsp21 | 0.5 | 8 | 42 | 37 | 5 | 56 | 1 | 0 | 0 | 2 | 2 |
| Hsp21 + CS | 0.8 | 3 | 38 | 28 | 13 | 56 | 1 | 0 | 0 | 3 | 3 |
| MDH, short trypsin | 2.5 | 1 | 15 | 48 | 14 | 10 | 6 | 0 | 5 | 24 | 29 |
| Hsp21 + MDH | 1.8 | 1 | 27 | 37 | 11 | 24 | 11 | 8 | 3 | 18 | 29 |
| CS | 0.9 | 4 | 11 | 39 | 6 | 17 | 7 | 0 | 6 | 33 | 39 |
| MDH | 2.5 | 4 | 2 | 0 | 6 | 0 | 29 | 48 | 6 | 39 | 94 |
Proteins were crosslinked and analyzed according to the workflow described in Figure 1, or with a modification in the experiment as indicated in the left-most column: short trypsin = digestion for 1 hr at a protease:protein ration of 1:10 instead of 1:100. Crosslinks were designated “valid” or “false-positive,” depending on their isotope pattern. If the peak at +4 Da was lower than the “light” and “heavy” peaks (X < 1), the crosslink was considered valid; if it was higher than the “light” and “heavy” peaks (X > 1), it was considered a false-positive. Crosslink type 0 indicates a dead-end, 1 an intrapeptide, and 2 an interpeptide crosslink. The scrambling percentage is the percentage of false-positive crosslinks of the total (valid and false-positive crosslinks).
The disulfide bond scrambling of DTSSP crosslinks is likely due to recombination of dead-end crosslinks and can be reduced by shorter trypsin incubation
True crosslink (ratio 1:0:1) and false-positive (ratio 1:2:1) isotope patterns could be detected for different crosslink peaks in the same experiment, indicating that scrambling to form false-positive crosslinks occurs more frequently for certain peptide combinations. The false-positive interpeptide crosslinks appear to be due to recombination of dead-end crosslinks, some of which are very abundant according to their high peak intensities in the mass spectra and broad elution profiles in chromatography. To decrease the degree of scrambling during a typical crosslinking mass spectrometry experiment, we tested modifications in the crosslinking protocol. The most promising strategy to reduce disulfide scrambling seems to shorten the trypsin digestion time. The degree of scrambling was greatly reduced for MDH in this way (Table I).
Native crosslinks in protein structure
In the two homologous molecular chaperone heat shock proteins, Hsp21 and αB-crystallin, the total number of detected and validated DTSSP crosslinks was large, 26 and 24 crosslinks, respectively (Tables II and III), and if dead-end crosslinks were included 42 and 35, respectively (Supporting information Tables S-I and S-II). These numbers are much higher than what was previously detected in Hsp2120 and αB-crystallin19 (8 and 4, respectively, as indicated in Tables S-I and S-II) showing that the two-step LC-MALDI MSMS workflow outlined in Figure 1 is an efficient procedure to detect and validate crosslinks.
Table II.
Interpeptide and Intrapeptide Crosslinks for Hsp21
| Theoretical [M+H]+ | Matched sequence(s) and modification | Modified amino acid residues | Distance Cα–Cα (Å) | Mono-, di-, or oligomer | ||
|---|---|---|---|---|---|---|
| 1527.58 | * | M1QDQR5 + M1QDQR5 | 1 | 1 | — | unstr. |
| 3209.43 | * | M1QDQR5 + E6NSIDVVQQGQQKGNQGSSVEK27 | 1 | 18 | — | unstr. |
| 2421.08 | * | M1QDQR5 + G19NQGSSVEKRPQQR32 | 1 | 27 | — | unstr. |
| 2474.08 | * | M1QDQR5 + A84PWDIKEEEHEIK96 | 1 | 89 | — | unstr. |
| 2050.85 | M1QDQR5 + E90EEHEIKMR98 | 1 | 96 | — | unstr. | |
| 2215.95 | * | M1QDQR5 + F99DMPGLSKEDVK110 | 1 | 106 | — | unstr. |
| 2521.21 | * | M1QDQR5 + I111SVEDNVLVIKGEQK125 | 1 | 121 | — | unstr. |
| 2131.81 | * | M1QDQR5 + K126EDSDDSWSGR136 | 1 | 126 | — | unstr. |
| 1551.73 | M1QDQR5 + I156KAELK161 | 1 | 157 | — | unstr. | |
| 2393.20 | * | M1QDQR5 + A158ELKNGVLFITIPK171 | 1 | 161 | — | unstr. |
| 1482.65 | * | M1QDQR5 + T172KVER176 | 1 | 173 | — | unstr. |
| 1792.84 | * | M1QDQR5 + K177VIDVQIQ184 | 1 | 177 | — | unstr. |
| 3198.50 | E6NSIDVVQQGQQKGNQGSSVEKRPQQR32 Intra | 18 | 27 | — | unstr. | |
| 3164.50 | * | E6NSIDVVQQGQQKGNQGSSVEK27 + T172KVER176 | 18 | 173 | — | unstr. |
| 2333.11 | G19NQGSSVEKRPQQR32 + G122EQKK126 | 27 | 125 | — | unstr. | |
| 2376.15 | * | G19NQGSSVEKRPQQR32 + T172KVER176 | 27 | 173 | — | unstr. |
| 2084.92 | * | A84PWDIKEEEHEIKMR98 Intra | 89 | 96 | 5 | m |
| 3078.31 | * | A84PWDIKEEEHEIK96 + K126EDSDDSWSGR136 | 89 | 126 | 12 | d |
| 3627.73 | * | A84PWDIKEEEHEIKMR98 + L145QLPDNCEKDKIK157 | 89/96 | 153/155 | 18 | m |
| 2916.47 | E90EEHEIKMR98 + A158ELKNGVLFITIPK171 | 96 | 161 | 15 | m | |
| 3125.44 | * | I111SVEDNVLVIKGEQK125 + K126EDSDDSWSGR136 | 121 | 126 | 20 | m |
| 2476.28 | * | I111SVEDNVLVIKGEQK125 + T172KVER176 | 121 | 173 | 28 | o |
| 2086.88 | K126EDSDDSWSGR136 + T172KVER176 | 126 | 173 | 25 | d | |
| 1308.67 | * | D154KIK157 + T172KVER176 | 155 | 173 | 14 | m |
| 1506.80 | I156KAELK161 +T172KVER176 | 157 | 173 | 18 | m | |
| 1747.91 | * | T172KVER176 + K177VIDVQIQ184 | 173 | 177 | 12 | m |
Crosslinks indicated with an asterisk (*) were also detected in a crosslinking experiment using BS3. Intra, intrapeptide crosslink; m, monomers; d, dimers; o, oligomers; unstr., unstructured regions.
Table III.
Interpeptide and Intrapeptide Crosslinks for αB-crystallin
| Theoretical [M+H]+ | Matched sequence(s) and modification | Modified amino acid residues | Distance Ca–Ca (Å) | Mono-, di-, or oligomer | ||
|---|---|---|---|---|---|---|
| 2222.07 | * | M1DIAIHHPWIR11 + L70EKDR74 | 1 | 72 | — | unstr. |
| 2954.52 | * | M1DIAIHHPWIR11 + V91KVLGDVIEVHGK103 | 1 | 92 | — | unstr. |
| 2027.98 | * | M1DIAIHHPWIR11 +K121YR123 | 1 | 121 | — | unstr. |
| 2462.19 | * | M1DIAIHHPWIR11+ K150QVSGPER157 | 1 | 150 | — | unstr. |
| 2830.42 | M1DIAIHHPWIR11 + E164EKPAVTAAPKK175 | 1 | 166/174/175 | — | unstr. | |
| 1493.71 | * | L70EKDR74 + L70EKDR74 | 72 | 72 | — | o |
| 2047.00 | L70EKDR74 + H83FSPEELKVK92 | 72 | 90 | — | o | |
| 1299.62 | * | L70EKDR74 + K121YR123 | 72 | 121 | 24 | m |
| 1733.83 | * | L70EKDR74 + K150QVSGPER157 | 72 | 150 | 10 | m |
| 2102.06 | L70EKDR74 + E164EKPAVTAAPKK175 | 72 | 166/174/175 | — | m | |
| 2294.05 | * | L70EKDR74 + E164EKPAVTAAPKK175 Hydr | 72 | 166/174/175 | — | m |
| 1973.97 | * | L70EKDR74 + E164EKPAVTAAPK174 | 72 | 166 | — | m |
| 3103.53 | * | H83FSPEELKVK92 + V93LGDVIEVHGKHEER107 | 90 | 103 | 9 | m |
| 2118.05 | * | V91KVLGDVIEVHGKHEER107 Intra | 92 | 103 | 14 | m |
| 2032.07 | * | V91KVLGDVIEVHGK103 + K121YR123 | 92 | 121 | 10 | m |
| 2466.28 | * | V91KVLGDVIEVHGK103 + K150QVSGPER157 | 92 | 150 | 22 | m |
| 2356.15 | * | V93LGDVIEVHGKHEER107 + K121YR123 | 103 | 121 | 16 | m |
| 1539.74 | * | K121YR123 +K150QVSGPER157 | 121 | 150 | 22 | m |
| 1779.88 | * | K121YR123 +E164EKPAVTAAPK174 | 121 | 166 | — | m |
| 1973.95 | * | K150QVSGPER157 + K150QVSGPER157 | 150 | 150 | — | o |
| 2342.18 | * | K150QVSGPER157 + E164EKPAVTAAPKK175 | 150 | 166/174/175 | — | m |
| 2534.18 | * | K150QVSGPER157 + E164EKPAVTAAPKK175 Hydr | 150 | 166/174/175 | — | m |
| 2214.09 | * | K150QVSGPER157 + E164EKPAVTAAPK174 | 150 | 166 | — | m |
| 1442.70 | * | E164EKPAVTAAPKK175 Intra | 166/174 | 174/175 | — | m |
Crosslinks indicated with an asterisk (*) were also detected in a crosslinking experiment using BS3. Intra, intrapeptide crosslink; Hydr, hydrolyzed dead-end crosslinker modification; m, monomers; d, dimers; o, oligomers; unstr., unstructured regions.
All interpeptide crosslinks could be mapped into the structures of Hsp2122 and αB-crystallin.25–28 Hsp21 is a dodecamer, and αB-crystallin is a 24-mer, but both are made up of structurally similar dimers [Fig. 4(B–E)]. The lysine residues occur in several parts of the sequences [Fig. 4(A)], and most crosslinks fit well into the dimeric structures [Fig. 4(B–E)], although crosslinks in the unstructured N-terminal region and the C-terminal extension29 are difficult to map. Except for the 7–8 intramonomeric crosslinks presented in Figure 4, there were crosslinks in Table II that appear to be intermonomeric, within the dimers or within the oligomer. Both lysine and sequence coverages were high, indicating a high solvent accessibility of all lysines and complete digestion by trypsin, respectively (Table S-III).
Figure 4.
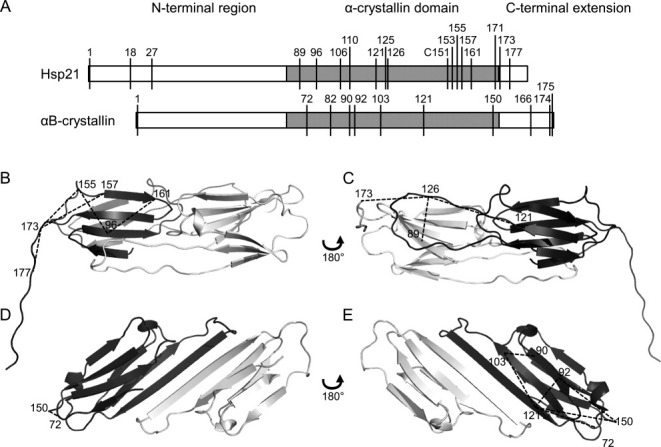
Mapping crosslink distance constraints into structures. Crosslinks listed in Tables II and III are shown here in the structures of two homologous molecular chaperone heat shock proteins, Hsp21 and αB-crystallin. A: Both protein sequences consist of an N-terminal region (white), the α-crystallin domain (gray), and the C-terminal extension (white), with the position of lysine residues indicated. B–E: Hsp21 and αB-crystallin dimers consist of two monomers (dark and light gray), of which only the α-crystallin domain is well-structured. B and C: Dimeric homology model of Hsp2122 with mapped crosslink distance constraints. D and E: Dimeric crystal structure of αB-crystallin25 (PDB ID 2WJ7) with mapped crosslink distance constraints. Each dimer is shown from two sides; one side showing the β2-β3-β8-β9 face of the dark-gray monomer (B, D), and the other side showing the β4-β5-β7 face of the dark-gray monomer (C, E). Crosslinks are indicated by dashed lines and the numbers indicate the lysines crosslinked.
Crosslinking between the same lysine residues was also independently confirmed with another lysine-specific crosslinker, bis(sulfosuccinimidyl) suberate (BS3) (Tables II and III, marked with asterisks). Altogether, numerous detected and validated crosslinks fitted well to protein structure data, demonstrating that the current workflow with DTSSP crosslink detection by two-step LC-MALDI MSMS is an efficient way to obtain informative distance restraints in proteins with unknown structure.
Materials and Methods
Proteins and reagents
Recombinantly expressed Hsp21 from Arabidopsis thaliana (UniProtKB P31170) and human αB-crystallin (UniProtKB P02511) and the α-type 20S proteasomal subunit C8/α7 (C8/α7) (UniProtKB P25788) were obtained as previously described.20,30 Sus scrofa (pig) citrate synthase (CS) and MDH (UniProtKB P00889 and P00346, respectively) were from Roche (Basel, Switzerland). All proteins were desalted and buffer exchanged into 50 mM HEPES (4-(2-hydroxyethyl)-1-piperazine ethanesulfonic acid) at pH 8.0 and 5 mM MgCl2 using disposable PD-10 protein desalting columns (GE Healthcare, Little Chalfont, UK). Protein concentrations are those of monomeric Hsp21 (21 kDa), αB-crystallin (20 kDa), C8/α7 (28 kDa), CS (49 kDa), and MDH (33 kDa), and were determined with the Bradford assay,31,32 using Bos taurus (bovine) serum albumin (UniProtKB P02769) as a standard. The isotope-labeled crosslinking reagents DTSSP, consisting of a 1:1 molar ratio mixture of DTSSP-H8 and DTSSP-D8, and BS3, consisting of a 1:1 molar ratio mixture of BS3-H12 and BS3-D12, were obtained from Creative Molecules (Victoria, Canada).
Chemical crosslinking
In each experiment, the following three samples were prepared: a control ‘reference’ sample to which no crosslinker was added, the crosslinked sample to which DTSSP was added, and a control sample to which DTSSP was added and after digestion DTT, to reduce the disulfide bond in the crosslinker. In the experiments involving a mixture of two proteins, the proteins were mixed in a 1:1 molar ratio to a final total protein concentration of 50 μM. DTSSP was dissolved in distilled water to a concentration of 30 mM immediately before use. Samples containing 50 μM protein (in 50 mM HEPES, pH 8.0, and 5 mM MgCl2) in 20 μL were incubated with DTSSP (final concentration 3 mM) at 25°C. After 15 min the crosslinking reaction was quenched by adding 1 M tris-(hydroxymethyl)-aminomethane (Tris) to a final concentration of 20 mM. To remove excess reagent and Tris and to concentrate the proteins, the samples were precipitated with ice-cold acetone.
Trypsin digestion
The protein pellets from acetone-precipitation were carefully redissolved in 20 μL 25 mM NH4HCO3 at pH 7.8. The samples were digested with sequencing-grade modified trypsin (Promega, Madison, WI) at 37°C at a protease:protein (w/w) ratio of 1:100 for 1 h, followed by a ratio of 1:50 overnight. Half of the digest of the crosslinked sample was subsequently reduced by adding 50 mM DTT and incubating at 37°C for 30 min. All samples were acidified by adding 2 μL 10% trifluoroacetic acid (TFA) and stored at −20°C until further analysis. In case of short-time trypsin digestion, the samples were digested at 37°C for 1 h at a protease:protein (w/w) ratio of 1:10.
Reversed phase liquid chromatography
The samples were separated by reversed phase liquid chromatography using an 1100 Series Nanoflow LC system (Agilent Technologies, Waldbronn, Germany). The mobile phases used for separation were composed of A: 1% (v/v) acetonitrile and 0.1% (v/v) TFA, and B: 90% (v/v) acetonitrile and 0.1% (v/v) TFA. For each sample, 8 μL was injected, which was estimated to contain ∼8 μg peptides. Samples were loaded onto a Zorbax 300SB-C18 0.3 mm precolumn (Agilent Technologies) in buffer A at a flow rate of 0.040 mL/min, delivered by the isocratic pump. By switching the micro 6-port/2-position module, the nanopump delivering a linear gradient from 0 to 100% buffer B (flow rate 1.4 μL/min) was then connected to the precolumn to move the sample onwards to the separation column, a PepSwift Monolithic Capillary Column (200 μm i.d. × 5 cm) (Dionex, Amsterdam, The Netherlands). Peptides eluted between 5 and 45% buffer B, and were collected in 64 fractions directly on the MALDI target plate.
MALDI-TOF/TOF mass spectrometry
Matrix solution consisting of 5 mg/mL α-cyano-4-hydroxy cinnamic acid, 50% acetonitrile, 0.1% TFA, 25 mM citric acid, and standard peptides (Angiotensin II, m/z 1046.541 Da; Neurotensin, m/z 1672.918 Da and ACTH (adrenocorticotropic hormone) 18–39, m/z 2465.199 Da) for internal calibration, was manually applied to the dried peptide fractions and allowed to dry. All mass spectrometric data were acquired using a 4700 Proteomics Analyzer (Applied Biosystems/MDS SCIEX). MS spectra were recorded in the Positive Reflector mode with 2000 laser shots per spectrum. The spectra were internally calibrated using the standard peptides and/or abundant confidently identified peaks from the samples. After MS-data analysis by our program FINDX, inclusion lists with crosslink peaks were submitted for the selection of crosslink precursors for MSMS fragmentation. MSMS spectra were recorded in the MS–MS 1 kV Positive mode with 3000 laser shots per spectrum.
Data analysis by FINDX
The program FINDX was written in the Python 2.7 programming language and can be used to analyze MS and MSMS data from crosslinking experiments with the crosslinking reagents DTSSP and BS3.
In MS mode, peaklist files from the reference and DTT control samples can optionally be included in the analysis, which means that any peaks in those samples are not allowed to be considered as crosslinks. When using isotope-labeled forms of either crosslinking reagent, MS peaks can be required to be an 8 (DTSSP) or a 12 (BS3) Da doublet. The program starts by merging all MS spectra from the LC fractions into one peaklist file. Subsequently, the peaklist is filtered by removing masses that are found in either reference or DTT control samples, as well as masses lacking the 8 or 12 Da doublet. The masses on the filtered peaklist are then compared against a theoretical list including all dead-end, intrapeptide, and interpeptide crosslinks, as well as optionally any of those with one extra dead-end crosslinker. The definition of a dead-end crosslink is that one end of the crosslinker has reacted with an amine from the protein, and the other end has either been hydrolyzed or reacted with Tris, which was used to quench the reaction. The DTSSP modification resulting in an interpeptide or dead-end crosslink corresponds to a mass difference of 173.98 Da or 191.99 Da, respectively. Using BS3, the mass differences are 138.07 Da and 156.08, respectively. The MS precursor tolerance, the enzyme used, the number of missed cleavages, the allowance of methionine oxidations, and the minimum decrease in peak intensity after DTT treatment can all be specified prior to analysis.
In MSMS mode, the precursor masses are matched and the fragment experimental masses are subsequently matched to the theoretical fragments, including the 66 Da doublet typical for DTSSP crosslinks.15 More information concerning data analysis can be found in Supporting Information. During the development of FINDX, data were also analyzed using the program GPMAW.33 The program FINDX is available on request via e-mail: findxlink@gmail.com.
Conclusions
The full potential offered by offline LC-MALDI-TOF/TOF mass spectrometry is used in this workflow, where MS data are analyzed to select precursors for MSMS data acquisition, in a two-step procedure by which numerous crosslinks could be detected and validated. We have shown here that the degree of thiol-exchange in DTSSP crosslinked peptides is proportional to the protein cysteine content and that it can be reduced by shortened trypsin incubation and evaluated by the isotope pattern and the new 4 Da peak that appears between the 8 Da doublet peaks in isotope-labeled DTSSP crosslinked peptides. True crosslinks, originating from two lysine residues located in crosslinkable distance within the native protein three-dimensional (3D)-structure, give a 1:0:1 isotope pattern which is overlaid to varying extent depending on the extent of thiol-exchange by the 1:2.1 isotope pattern, the hallmark of false positives derived solely from thiol-exchange. The proportion of native versus false positive crosslinking can therefore be precisely quantified, and isotope-labeled DTSSP safely used to detect true crosslinks between lysine residues in the 3D-structure.
Acknowledgments
Prof. Ulf Nilsson is acknowledged for valuable input concerning the influence of thiols on disulfide bonds.
Supplementary material
References
- 1.Leitner A, Walzthoeni T, Kahraman A, Herzog F, Rinner O, Beck M, Aebersold R. Probing native protein structures by chemical cross-linking, mass spectrometry, and bioinformatics. Mol Cell Proteomics. 2010;9:1634–1649. doi: 10.1074/mcp.R000001-MCP201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Singh P, Panchaud A, Goodlett DR. Chemical cross-linking and mass spectrometry as a low-resolution protein structure determination technique. Anal Chem. 2010;82:2636–2642. doi: 10.1021/ac1000724. [DOI] [PubMed] [Google Scholar]
- 3.Sinz A. Chemical cross-linking and mass spectrometry to map three-dimensional protein structures and protein–protein interactions. Mass Spectrom Rev. 2006;25:663–682. doi: 10.1002/mas.20082. [DOI] [PubMed] [Google Scholar]
- 4.Lee YJ. Mass spectrometric analysis of cross-linking sites for the structure of proteins and protein complexes. Mol Biosyst. 2008;4:816–823. doi: 10.1039/b801810c. [DOI] [PubMed] [Google Scholar]
- 5.Dihazi GH, Sinz A. Mapping low-resolution three-dimensional protein structures using chemical cross-linking and Fourier transform ion-cyclotron resonance mass spectrometry. Rapid Commun Mass Spectrom. 2003;17:2005–2014. doi: 10.1002/rcm.1144. [DOI] [PubMed] [Google Scholar]
- 6.Chowdhury SM, Du XX, Tolic N, Wu S, Moore RJ, Mayer MU, Smith RD, Adkins JN. Identification of cross-linked peptides after click-based enrichment using sequential collision-induced dissociation and electron transfer dissociation tandem mass spectrometry. Anal Chem. 2009;81:5524–5532. doi: 10.1021/ac900853k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen ZA, Jawhari A, Fischer L, Buchen C, Tahir S, Kamenski T, Rasmussen M, Lariviere L, Bukowski-Wills JC, Nilges M, Cramer P, Rappsilber J. Architecture of the RNA polymerase II-TFIIF complex revealed by cross-linking and mass spectrometry. EMBO J. 2010;29:717–726. doi: 10.1038/emboj.2009.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Muller DR, Schindler P, Towbin H, Wirth U, Voshol H, Hoving S, Steinmetz MO. Isotope tagged cross linking reagents. A new tool in mass spectrometric protein interaction analysis. Anal Chem. 2001;73:1927–1934. doi: 10.1021/ac001379a. [DOI] [PubMed] [Google Scholar]
- 9.Tang XT, Munske GR, Siems WF, Bruce JE. Mass spectrometry identifiable cross-linking strategy for studying protein–protein interactions. Anal Chem. 2005;77:311–318. doi: 10.1021/ac0488762. [DOI] [PubMed] [Google Scholar]
- 10.Zhang HZ, Tang XT, Munske GR, Tolic N, Anderson GA, Bruce JE. Identification of protein–protein interactions and topologies in living cells with chemical cross-linking and mass spectrometry. Mol Cell Proteomics. 2009;8:409–420. doi: 10.1074/mcp.M800232-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Muller MQ, Dreiocker F, Ihling CH, Schafer M, Sinz A. Cleavable cross-linker for protein structure analysis: reliable identification of cross-linking products by tandem MS. Anal Chem. 2010;82:6958–6968. doi: 10.1021/ac101241t. [DOI] [PubMed] [Google Scholar]
- 12.Lauber MA, Reilly JP. Novel amidinating cross-linker for facilitating analyses of protein structures and interactions. Anal Chem. 2010;82:7736–7743. doi: 10.1021/ac101586z. [DOI] [PubMed] [Google Scholar]
- 13.Petrotchenko EV, Serpa JJ, Borchers CH. Use of a combination of isotopically coded cross-linkers and isotopically coded N-terminal modification reagents for selective identification of inter-peptide crosslinks. Anal Chem. 2010;82:817–823. doi: 10.1021/ac901637v. [DOI] [PubMed] [Google Scholar]
- 14.Back JW, Notenboom V, de Koning LJ, Muijsers AO, Sixma TK, de Koster CG, de Jong LZ. Identification of cross-linked peptides for protein interaction studies using mass spectrometry and O-18 labeling. Anal Chem. 2002;74:4417–4422. doi: 10.1021/ac0257492. [DOI] [PubMed] [Google Scholar]
- 15.King GJ, Jones A, Kobe B, Huber T, Mouradov D, Hume DA, Ross IL. Identification of disulfide-containing chemical cross-links in proteins using MALDI-TOF/TOF-mass spectrometry. Anal Chem. 2008;80:5036–5043. doi: 10.1021/ac702277q. [DOI] [PubMed] [Google Scholar]
- 16.Bennett KL, Kussmann M, Bjork P, Godzwon M, Mikkelsen M, Sorensen P, Roepstorff P. Chemical cross-linking with thiol-cleavable reagents combined with differential mass spectrometric peptide mapping—a novel approach to assess intermolecular protein contacts. Protein Sci. 2000;9:1503–1518. doi: 10.1110/ps.9.8.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Back JW, Sanz MA, De Jong L, De Koning LJ, Nijtmans LJ, De Koster CG, Grivell LA, Van der Spek H, Muijsers AO. A structure for the yeast prohibitin complex: structure prediction and evidence from chemical crosslinking and mass spectrometry. Protein Sci. 2002;11:2471–2478. doi: 10.1110/ps.0212602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Swaim CL, Smith DL, Smith JB. The reaction of alpha-crystallin with the cross-linker 3,3′-dithiobis(sulfosuccinimidyl propionate) demonstrates close proximity of the C termini of alpha A and alpha B in the native assembly. Protein Sci. 2004;13:2832–2835. doi: 10.1110/ps.04910004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peterson JJ, Young MM, Takemoto LJ. Probing alpha-crystallin structure using chemical cross-linkers and mass spectrometry. Mol Vis. 2004;10:857–866. [PubMed] [Google Scholar]
- 20.Ahrman E, Lambert W, Aquilina JA, Robinson CV, Emanuelsson CS. Chemical cross-linking of the chloroplast localized small heat-shock protein, Hsp21, and the model substrate citrate synthase. Protein Sci. 2007;16:1464–1478. doi: 10.1110/ps.072831607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gorman JJ, Wallis TP, Pitt JJ. Protein disulfide bond determination by mass spectrometry. Mass Spectrom Rev. 2002;21:183–216. doi: 10.1002/mas.10025. [DOI] [PubMed] [Google Scholar]
- 22.Lambert W, Koeck PB, Ahrman E, Purhonen P, Cheng K, Elmlund D, Hebert H, Emanuelsson C. Subunit arrangement in the dodecameric chloroplast small heat shock protein Hsp21. Protein Sci. 2011;20:291–301. doi: 10.1002/pro.560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fahey RC, Hunt JS, Windham GC. Cysteine and cystine content of proteins—differences between intracellular and extracellular proteins. J Mol Evol. 1977;10:155–160. doi: 10.1007/BF01751808. [DOI] [PubMed] [Google Scholar]
- 24.Miseta A, Csutora P. Relationship between the occurrence of cysteine in proteins and the complexity of organisms. Mol Biol Evol. 2000;17:1232–1239. doi: 10.1093/oxfordjournals.molbev.a026406. [DOI] [PubMed] [Google Scholar]
- 25.Bagneris C, Bateman OA, Naylor CE, Cronin N, Boelens WC, Keep NH, Slingsby C. Crystal structures of alpha-crystallin domain dimers of alpha B-crystallin and Hsp20. J Mol Biol. 2009;392:1242–1252. doi: 10.1016/j.jmb.2009.07.069. [DOI] [PubMed] [Google Scholar]
- 26.Laganowsky A, Benesch JP, Landau M, Ding LL, Sawaya MR, Cascio D, Huang QL, Robinson CV, Horwitz J, Eisenberg D. Crystal structures of truncated alphaA and alphaB crystallins reveal structural mechanisms of polydispersity important for eye lens function. Protein Sci. 2010;19:1031–1043. doi: 10.1002/pro.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jehle S, Rajagopal P, Bardiaux B, Markovic S, Kuhne R, Stout JR, Higman VA, Klevit RE, van Rossum BJ, Oschkinat H. Solid-state NMR and SAXS studies provide a structural basis for the activation of alpha B-crystallin oligomers. Nat Struct Mol Biol. 2010;17:1037–1042. doi: 10.1038/nsmb.1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peschek J, Braun N, Franzmann TM, Georgalis Y, Haslbeck M, Weinkauf S, Buchner J. The eye lens chaperone alpha-crystallin forms defined globular assemblies. Proc Natl Acad Sci USA. 2009;106:13272–13277. doi: 10.1073/pnas.0902651106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carver JA, Lindner RA. NMR spectroscopy of alpha-crystallin. Insights into the structure, interactions and chaperone action of small heat-shock proteins. Int J Biol Macromol. 1998;22:197–209. doi: 10.1016/s0141-8130(98)00017-8. [DOI] [PubMed] [Google Scholar]
- 30.Boelens WC, Croes Y, de Jong WW. Interaction between alpha B-crystallin and the human 20S proteasomal subunit C8/alpha 7. Biochem Biophys Acta. 2001;1544:311–319. doi: 10.1016/s0167-4838(00)00243-0. [DOI] [PubMed] [Google Scholar]
- 31.Bradford MM. Rapid and sensitive method for quantitation of microgram quantities of protein utilizing principle of protein–dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 32.Stoscheck CM. Quantitation of protein. Method Enzymol. 1990;182:50–68. doi: 10.1016/0076-6879(90)82008-p. [DOI] [PubMed] [Google Scholar]
- 33.Peri S, Steen H, Pandey A. GPMAW—a software tool for analyzing proteins and peptides. Trends Biochem Sci. 2001;26:687–689. doi: 10.1016/s0968-0004(01)01954-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.