

Abstract
Bacteriophage Qβ is a small RNA virus that infects Escherichia coli. The virus particle contains a few copies of the minor coat protein A1, a C-terminally prolonged version of the coat protein, which is formed when ribosomes occasionally read-through the leaky stop codon of the coat protein. The crystal structure of the read-through domain from bacteriophage Qβ A1 protein was determined at a resolution of 1.8 Å. The domain consists of a heavily deformed five-stranded β-barrel on one side of the protein and a β-hairpin and a three-stranded β-sheet on the other. Several short helices and well-ordered loops are also present throughout the protein. The N-terminal part of the read-through domain contains a prominent polyproline type II helix. The overall fold of the domain is not similar to any published structure in the Protein Data Bank.
Keywords: Leviviridae, allolevivirus, small RNA phages, bacteriophage Qβ, minor coat protein, read-through protein, polyproline helix
Introduction
Bacteriophages of the Leviviridae family are among the smallest and simplest known viruses. They have a single-stranded, positive-sense RNA genome, which is about 3500–4200 nucleotides long and encodes a maturation protein, a coat protein, and a subunit of the replicase complex.1 The capsid is built from 90 dimers of coat protein that assemble in an icosahedral shell with T = 3 symmetry.2 In addition to the coat protein, each virion contains a single copy of the maturation protein.3 The maturation protein is bound to the genomic RNA4 and mediates the attachment of the phage to the sides of bacterial pili,5 which is the cellular receptor for all known Leviviridae phages. After attachment to the pili, the RNA-maturation protein complex leaves the capsid and enters the cell through an unknown mechanism.
Many of the known Leviviridae phages are further divided into two genera, leviviruses and alloleviviruses. A marked difference between the two genera is how the phages achieve cell lysis: leviviruses encode a small lysis protein that overlaps with coat and replicase genes in a different reading frame, whereas alloleviviruses mediate lysis using the maturation protein.6,7 The other unique feature of alloleviviruses is the presence of a minor coat protein species A1 in their capsid. The A1 protein is produced when ribosomes occasionally read-through the leaky UGA termination codon of the coat protein gene8 and translation continues for another 600 nucleotides, resulting in a C-terminal extension of the coat protein. The A1 protein is incorporated in 3–10 copies per virion1 and is essential for producing infectious virus particles,9 but its precise function is not known. To gain new insights about this protein, we solved the crystal structure of the read-through extension from bacteriophage Qβ A1 protein.
Results and Discussion
Structure determination and quality of the models
Because of the low number and presumed random orientation in the capsid, the read-through extensions were not visible in the crystal structure of bacteriophage Qβ.10 The A1 protein alone is insoluble and cannot assemble into particles without the assistance of the coat protein,11 and the amount of A1 protein that can be incorporated into the particles seems to be limited to about 15%.1 To make the A1 protein amenable to structural analysis, we expressed the read-through domain separately. The complete read-through extension starting from the end of the coat protein was largely insoluble (data not shown), but a hexahistidine tagged variant starting 11 amino acids away from the coat protein part (residues 144–328 of full-length A1 protein) was highly soluble, could be readily purified, and was chosen to proceed with crystallization. The protein was crystallized in two crystal forms, monoclinic and hexagonal, which diffracted to 1.8 and 2.9 Å resolutions, respectively. The structure of the monoclinic form was solved by multiple isomorphous replacement with anomalous scattering using two derivatives. Except for the expression tag and the first two residues of the crystallized domain, the polypeptide chain could be traced unambiguously, without breaks, from residue 146 (the numbering of residues is as of full-length A1 protein) to the end of the chain. In the hexagonal form, another seven N-terminal residues could not be located in the electron density, and the chain was traced starting from residue 153. The domain adopts an almost identical conformation in the two crystal forms, with an rms deviation of 0.76 Å for the main chain atoms.
Overall structure
The overall fold of the read-through domain [Fig. 1(A)] is not similar to any other published structure in the Protein Data Bank, according to the DALI server.13 Except for the N-terminal region, the domain has a compact, roughly globular shape with a mixed α/β architecture. The core of the domain is built of β-sheets: strands β2, β3, β6, β7, and β8 form a heavily deformed, five-stranded β-barrel on one side of the protein, whereas β1 and β4 and β5, β9, and β10 form two antiparallel sheets on the other side. There are three α-helices and two 310-helices in the protein, which are all short and are located predominantly on the surface. A remarkably long loop (23 residues) connects the first 310-helix and strand β5, but it is well ordered and kept in place by extensive hydrogen bonding involving main chain and side chain atoms. Eight of the first 15 residues that are visible in the electron density map are prolines. These residues form a polyproline type II helix that stretches for about 45 Å before turning 90° toward the rest of the protein [Fig. 1(B)]. The polyproline helix is held in position by two crystal contacts with the globular part of neighboring molecules in the monoclinic crystal form but not in the hexagonal form. Consequently, the distant part of the helix is not visible in the hexagonal form, which suggests that it is flexible in solution. It should be noted that, although poplyproline type II helices are not uncommon in proteins, the vast majority of them are shorter than six residues14 and long helices are rare. The polyproline helix is A1 is quite remarkable in this aspect, since, according to a statistical survey of polyproline helices in protein structures in 2006,14 the longest such helix observed in a crystal structure was that of the benzoylformate decarboxylase from Pseudomonas putida15 (PDB ID 1BFD), which is 14 residues long and contains three prolines. The helix connects two subdomains of the enzyme but otherwise does not seem to have a specific function.
Figure 1.
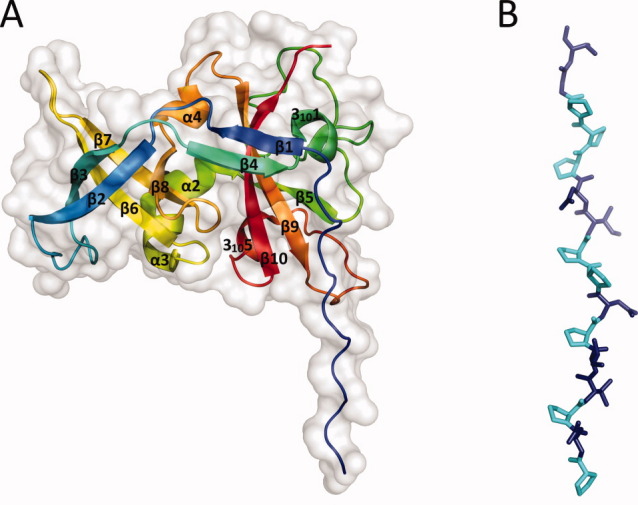
Structure of the read-through domain. (A) Overall structure of the domain. The protein is represented as a cartoon model rainbow-colored blue (N-terminus) to red (C-terminus) and overlaid with a surface representation of the domain (light grey). (B) A detailed view of the polyproline helix. In the first 16 residues of the model, prolines are represented in cyan and other residues in deep blue. Figures 1(A,B) and 2(B) were prepared using PyMol.12
Currently, there is no structural information about residues 133–145, which separate the coat and read-through domains. Secondary structure prediction by JPred16 suggests that this region is unstructured except for the coat protein-proximal six residues, which, together with the last three residues of the coat protein, may be involved in a short α-helix.
Conserved regions
On the basis of phylogenetic and serological criteria, alloleviviruses cluster into two groups denoted III and IV.17,18 Up to date, there are 15 allolevivirus genome sequences available, of these eight are from Group III and seven from Group IV. When all of the sequences are aligned, coat proteins are the most conserved (∼64% sequence identity), followed by the replicase (∼44% identity) and maturation proteins (∼29% identity). When sequences of all of the known A1 extensions are aligned, the total identity is only 26%, making them the most divergent part of all phage proteins. However, in a sequence alignment of A1 extensions from representative phages from Group III (Qβ and MX1) and Group IV (FI and SP) several conserved regions emerge [Fig. 2(A)]. First, in the N-terminal part (residues 146–159), ∼50% of the residues are prolines in all alloleviviruses, suggesting that the polyproline helix is present in all allolevivirus A1 proteins and is probably important for their function. A short stretch of amino acids immediately following the helix is also conserved. The most prominent conserved regions are located at residues 207–219 and 228–238, which form part of the long loop between helix 3101 and β5 and extend to strand β5 and the beginning of helix α2. The C-terminal region of the domain is also relatively conserved. Interestingly, the majority of conserved residues cluster on one side of the protein closer to the polyproline helix [Fig. 2(B)], suggesting that this part of the domain is the most critical for performing its function.
Figure 2.

Conserved regions of the read-through domains. (A) Sequence alignment of the read-through domains from different alloleviviruses. Conserved residues are colored red; of these, identical residues are shaded yellow and nonidentical light yellow. Assigned secondary structure elements are presented below the alignment. A dashed line represents the portion for which no experimental data are available; the α-helix from secondary structure prediction is drawn as a pale blue cylinder. (B) Mapping of the conserved regions on the three-dimensional structure of the read-through domain. Identical and nonidentical but conserved residues as of Figure 2(A) are colored red and yellow-orange, respectively.
Possible function of the A1 protein
The actual function of the read-through domain has remained enigmatic. The amino acid sequence and the three-dimensional (3D) structure of the A1 extension are not similar to other known proteins, leaving no clues about its evolutionary origin. The A1 protein is a landmark of the rather small group of alloleviviruses, which all infect Escherichia coli, whereas all other known Leviviridae phages suffice with just the coat and maturation proteins in the virion. However, the A1 protein is essential for producing viable phage particles, as shown by in vitro virus reassembly assays9 and in vivo plasmid complementation studies.19 The C-termini of coat proteins with some minor structural rearrangements could reach both the inner and outer surface of the capsid. However, current evidence suggests that as structural components of the virion, the read-through extensions are located on the exterior of the capsid. First, Qβ virions form a diffuse band in native polyacrylamide gel electrophoresis,20 and their mobility in the gel and in sucrose density gradients depends on how many copies of the A1 protein are present in the capsid.21 Additionally, when recombinant Qβ capsids contained A1 extensions with an engineered internal epitope tag from hepatitis B virus preS1 region, the tags were accessible to antibodies in an ELISA assay and immunogold electron microscopy confirmed that the antibodies were indeed bound to the capsid surface.11 The five-residue tag was inserted after residue 204, which is now known to be located in the short 3101-helix on the surface of the protein and likely did not disturb the structure of the domain.
An interesting feature of the A1 protein undoubtedly is the long polyproline type II helix at the N-terminal part of the read-through domain. Polyproline helices and proline-rich regions in general are relatively abundant in proteins and have different functions,22 but they frequently serve as ligands for various protein–protein interaction domains, resulting in formation of protein complexes that are often involved in signaling and regulatory pathways in eukaryotic cells (reviewed in Ref.23). In other proteins, proline-rich regions have a structural role and act as relatively rigid spacers to keep protein domains apart. For example, a 68 residue long proline-rich segment of the bacterial protein TonB was recently shown to adopt a polyproline II conformation that spans the periplasm.24
The linker between the coat and read-through domains would stretch for estimated 35 Å, and is then followed by the 45-Å-long polyproline helix, which is apparently also somewhat flexible. The logical explanation for such a long linker is that the read-through domain in the virion is positioned far away from the viral quasi-threefold symmetry axis (relating the three quasi-equivalent subunits A, B, and C) where the C-termini of coat proteins are located. A recent study localized the maturation protein from the distantly related phage MS2 on one of the viral fivefold symmetry axes and suggested that the Qβ maturation protein is localized similarly.25 Because both maturation and A1 proteins are required for infectivity, it seems possible that the two proteins might interact with each other and that the long linker would allow the read-through domain to reach viral fivefold and threefold symmetry axes that are ∼45 Å away from the C-termini of coat proteins. Experiments to test the association of the read-through domain with the maturation protein are underway in our laboratory.
Conclusions
We have shown that the read-through domain of Qβ A1 protein adopts a previously unseen protein fold and has some intriguing structural features, such as a 15 residue-long polyproline type II helix which is one of the best examples of this kind of helix in globular proteins for which the 3D structures have been determined. Although the structure of the read-through domain does not provide immediate answers about its function, it gives a good starting point for further studies that could eventually lead to the understanding of the molecular mechanism by which the small RNA phages infect the bacterial host.
Materials and Methods
Cloning, expression, and purification
The coding sequence of Qβ A1 extension was amplified from plasmid pQβ1026 using forward primer 5′-TACCATGGGGCACCATCATCACCATCATTCAAAC CCGATCCGGTTATTCC-3′ and reverse primer 5′-ATCTGCAGTTAAGCACGAGGAACGACTATCACG-3′. The resulting fragment, encoding an N-terminally 6xHis-tagged A1 extension (denoted His-A1 hereafter) was cloned into a modified pBAD/Thio vector (Invitrogen). For protein production, the plasmid was transformed in E.coli strain TOP10, and cells were grown in LB medium containing 50 μg/mL ampicillin at 37°C until OD590 of the culture reached 1.0. Arabinose was then added to a final concentration of 0.2%, and cells were grown for another 4 h and harvested by centrifugation.
Cells were resuspended in a lysis buffer containing 40 mM Tris-HCl pH 8.0, 200 mM NaCl, 20 mM MgSO4, 0.1% Triton-X100, 0.1 mg/mL DNAse, and 1 mg/mL lysozyme and lysed by three freeze-thaw cycles. The lysate was centrifuged, supernatant was loaded on a HIS-Select cartridge (Sigma), and bound His-A1 protein was eluted with buffer containing 40 mM Tris-HCl pH 8.0, 300 mM NaCl, and 300 mM imidazole. The sample buffer was then exchanged to 20 mM Tris-HCl pH 8.0 and 50 mM NaCl using Amicon spin filters (Millipore), and the preparation was applied to a HiPrep 16/10 Q FF ion exchange column (GE Healthcare), which was equilibrated with the same buffer. The His-A1 protein did not bind to the column under these conditions, whereas the majority of contaminants did. Finally, fractions containing His-A1 were pooled, concentrated, and loaded on a Superdex 200 10/300 GL gel filtration column (GE Healthcare), which was equilibrated with 20 mM Tris-HCl pH 8.0. Fractions containing His-A1 were pooled, concentrated to 10 mg/mL, and stored at −20°C until use.
Crystallization and data collection
The His-A1 protein was initially crystallized using the sitting drop vapor diffusion technique by mixing 1 μL of the protein solution (10 mg/mL) with 1μL of the well solution (0.1M Tris-HCl pH 8.5, 40% PEG 300). Plate-shaped crystals (the monoclinic form) appeared after 3–6 days at room temperature (298 K) and reached maximum dimensions of 0.3 × 0.1 mm. For data collection, crystals were flash-frozen in liquid nitrogen without additional cryoprotectant.
After optimization of crystallization conditions, slightly thicker crystals were obtained using 0.1M Tris-HCl pH 8.5, 20% PEG 300, and 10% PEG 2000 MME as the well solution. These crystals were less fragile, had less anisotropic diffraction and were used for heavy atom compound soaks. To prepare the mercury derivative, crystals were soaked in a mother liquor containing 20 mM Hg(NO3)2 for 30 min, followed by backsoaking in the original mother liquor for 10 s. For iodine derivatization, mother liquor containing 0.1M I2 in 0.1M KI was prepared, the undissolved iodine was removed by centrifugation, and the resulting iodine-saturated solution was used for soaking the crystals overnight. Crystals were flash-frozen without backsoaking.
When the structure of the A1 domain in the monoclinic crystal form was already solved, a hexagonal crystal form was discovered when any buffer was omitted from the crystallization drop (40% PEG 300 in water). Crystals appeared after 2–3 days at room temperature and grew bigger for about 1 week, reaching maximum size of 0.2 mm.
Crystal diffraction data were collected at European Synchrotron Radiation Facility (ESRF) and MAX-lab as indicated in Table I and Supporting Information Table 1.
Table I.
Crystallographic data collection, scaling, and refinement statistics
| Dataset | Monoclinic | Hexagonal |
|---|---|---|
| Data collection and scaling | ||
| Beamline | ESRF ID29 | MAX-Lab I911–2 |
| Spacegroup | P21 | P6322 |
| Cell parameters | a = 44.01 Å | a = 69.11 Å |
| b = 49.12 Å | c = 167.30 Å | |
| c = 44.26 Å | ||
| β = 118.41° | ||
| Wavelength (Å) | 0.9762 | 1.0387 |
| Resolution (Å) | 49.15–1.76 | 40.79–2.90 |
| Highest resolution bin (Å) | 1.86–1.76 | 3.06–2.90 |
| Rmerge | 0.078 (0.334) | 0.098 (0.543) |
| Total number of observations | 55321 | 54994 |
| Number of unique reflections | 16414 | 5773 |
| I/σ(I) | 10.0 (3.2) | 17.5 (3.8) |
| Completeness (%) | 99.2 (99.5) | 100.0 (100.0) |
| Multiplicity | 3.4 (3.3) | 9.5 (10.0) |
| Refinement | ||
| Rwork | 0.174 | 0.213 |
| Rfree | 0.242 | 0.297 |
| Average B factor (Å2) | 17.153 | 41.116 |
| Number of atoms | ||
| Protein | 1463 | 1411 |
| Solvent | 154 | 19 |
| RMS deviations from ideal | ||
| Bond lengths (Å) | 0.023 | 0.012 |
| Bond angles (°) | 1.925 | 1.368 |
| Ramachandran plot27 | ||
| Residues in favored regions (%) | 97.8 | 95.4 |
| Residues in allowed regions (%) | 100.0 | 99.4 |
Values in parentheses are given for the highest resolution shell.
Structure determination
Data were indexed with MOSFLM28 and scaled using SCALA.29 For the monoclinic crystal form, native and derivative datasets were scaled with SCALEIT30 and merged using CAD from the CCP4 suite.31 The position of the first mercury atom was calculated manually from the strongest peak in the Harker section of the isomorphous difference Patterson map. The coordinates of the mercury atom were input into MLPHARE32 and used to locate the remaining mercury and iodine atoms. Heavy atom refinement and phasing was performed in SHARP33 and was followed by solvent flattening in SOLOMON.34 From the resulting map, a partial model was built by BUCANEER35 that was included to provide extra phase information in a second SHARP and SOLOMON run. The resulting map was used to build an improved model with BUCANEER that served as a starting point for manual model building in COOT36 using the high-resolution native data. Refinement was performed using REFMAC.37 The structure of the hexagonal form was solved by molecular replacement with MOLREP38 using coordinates of the A1 domain in the monoclinic crystal form as a search model, followed by model building in COOT and refinement with REFMAC. Scaling and refinement statistics for native datasets are presented in Table I; detailed phasing statistics are given in Supporting Information Table 1.
Atomic coordinates were deposited in the Protein Data Bank under accession codes 3RLK (monoclinic crystal form) and 3RLC (hexagonal crystal form).
Acknowledgments
The authors thank the staff at ESRF and MAX-Lab for their help during data collection and Anna Janson for collecting the mercury derivative datasets.
Supplementary material
References
- 1.Weber K, Konigsberg W, Proteins of the RNA phages . In: RNA phages. Cold Spring Harbor. Zinder ND, editor. New York: Cold Spring Harbor Laboratory; 1975. pp. 51–84. [Google Scholar]
- 2.Valegård K, Liljas L, Fridborg K, Unge T. The three-dimensional structure of the bacterial virus MS2. Nature. 1990;345:36–41. doi: 10.1038/345036a0. [DOI] [PubMed] [Google Scholar]
- 3.Steitz JA. Identification of the A protein as a structural component of bacteriophage R17. J Mol Biol. 1968;33:923–936. doi: 10.1016/0022-2836(68)90328-8. [DOI] [PubMed] [Google Scholar]
- 4.Kozak M, Nathans D. Fate of maturation protein during infection by coliphage MS2. Nat New Biol. 1971;234:209–211. doi: 10.1038/newbio234209a0. [DOI] [PubMed] [Google Scholar]
- 5.Brinton CC, Gemski P, Carnahan J. A new type of bacterial pilus genetically controlled by the fertility factor of E. coli K 12 and its role in chromosome transfer. Proc Natl Acad Sci USA. 1964;52:776–783. doi: 10.1073/pnas.52.3.776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Winter RB, Gold L. Overproduction of bacteriophage Qβ maturation (A2) protein leads to cell lysis. Cell. 1983;33:877–885. doi: 10.1016/0092-8674(83)90030-2. [DOI] [PubMed] [Google Scholar]
- 7.Karnik S, Billeter M. The lysis function of RNA bacteriophage Qβ is mediated by the maturation (A2) protein. EMBO J. 1983;2:1521–1526. doi: 10.1002/j.1460-2075.1983.tb01617.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weiner AM, Weber K. Natural read-through at the UGA termination signal of Qβ coat protein cistron. Nat New Biol. 1971;234:206–209. doi: 10.1038/newbio234206a0. [DOI] [PubMed] [Google Scholar]
- 9.Hofstetter H, Monstein HJ, Weissmann C. The readthrough protein A1 is essential for the formation of viable Qβ particles. Biochim Biophys Acta. 1974;374:238–251. doi: 10.1016/0005-2787(74)90366-9. [DOI] [PubMed] [Google Scholar]
- 10.Golmohammadi R, Fridborg K, Bundule M, Valegård K, Liljas L. The crystal structure of bacteriophage Qβ at 3.5 Å resolution. Structure. 1996;4:543–554. doi: 10.1016/s0969-2126(96)00060-3. [DOI] [PubMed] [Google Scholar]
- 11.Vasiljeva I, Kozlovska T, Cielens I, Strelnikova A, Kazaks A, Ose V, Pumpens P. Mosaic Qβ coats as a new presentation model. FEBS Lett. 1998;431:7–11. doi: 10.1016/s0014-5793(98)00716-9. [DOI] [PubMed] [Google Scholar]
- 12.DeLano WL. The PyMOL molecular graphics system. San Carlos, CA: DeLano Scientific; 2002. [Google Scholar]
- 13.Holm L, Rosenström P. Dali server: conservation mapping in 3D. Nucl Acids Res. 2010;38:W545–W549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berisio R, Loguercio S, De Simone A, Zagaria A, Vitaglianoa L. Polyproline helices in protein structures: a statistical survey. Protein Pept Lett. 2006;13:847–854. doi: 10.2174/092986606777841154. [DOI] [PubMed] [Google Scholar]
- 15.Hasson MS, Muscate A, McLeish MJ, Polovnikova LS, Gerlt JA, Kenyon GL, Petsko GA, Ringe D. The crystal structure of benzoylformate decarboxylase at 1.6 Å resolution: diversity of catalytic residues in thiamin diphosphate-dependent enzymes. Biochemistry. 1998;37:9918–9930. doi: 10.1021/bi973047e. [DOI] [PubMed] [Google Scholar]
- 16.Cole C, Barber JD, Barton GJ. The Jpred 3 secondary structure prediction server. Nucl Acids Res. 2008;36:W197–W201. doi: 10.1093/nar/gkn238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bollback JP, Huelsenbeck JP. Phylogeny, genome evolution, and host specificity of single-stranded RNA bacteriophage (family Leviviridae) J Mol Evol. 2001;52:117–128. doi: 10.1007/s002390010140. [DOI] [PubMed] [Google Scholar]
- 18.Furuse K, Distribution of coliphages in the environment: general considerations . In: Phage ecology. Goyal SM, Gerba CP, Bitton G, editors. New York: Wiley; 1987. pp. 87–124. [Google Scholar]
- 19.Priano C, Arora R, Butke J, Mills DR. A complete plasmid-based complementation system for RNA coliphage Qβ: three proteins of bacteriophages Qβ (group III) and SP (group IV) can be interchanged. J Mol Biol. 1995;249:283–297. doi: 10.1006/jmbi.1995.0297. [DOI] [PubMed] [Google Scholar]
- 20.Strauss EG, Kaesberg P. Acrylamide gel electrophoresis of bacteriophage Qβ: electrophoresis of the intact virions and of the viral proteins. Virology. 1970;42:437–452. doi: 10.1016/0042-6822(70)90287-4. [DOI] [PubMed] [Google Scholar]
- 21.Radloff RJ, Kaesberg P. Electrophoretic and other properties of bacteriophage Qβ: the effect of a variable number of read-through proteins. J Virol. 1973;11:116–128. doi: 10.1128/jvi.11.1.116-128.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Williamson MP. The structure and function of proline-rich regions in proteins. Biochem J. 1994;297:249–260. doi: 10.1042/bj2970249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kay BK, Williamson MP, Sudol M. The importance of being proline: the interaction of proline-rich motifs in signaling proteins with their cognate domains. FASEB J. 2000;14:231–241. [PubMed] [Google Scholar]
- 24.Köhler SD, Weber A, Howard SP, Welte W, Drescher M. The proline-rich domain of TonB possesses an extended polyproline II-like conformation of sufficient length to span the periplasm of Gram-negative bacteria. Protein Sci. 2010;19:625–630. doi: 10.1002/pro.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Toropova K, Stockley PG, Ranson NA. Visualising a viral RNA genome poised for release from its receptor complex. J Mol Biol. 2011;408:408–419. doi: 10.1016/j.jmb.2011.02.040. [DOI] [PubMed] [Google Scholar]
- 26.Kozlovska TM, Cielens I, Dreilinna D, Dislers A, Baumanis V, Ose V, Pumpens P. Recombinant RNA phage Qβ capsid particles synthesized and self-assembled in Escherichia coli. Gene. 1993;137:133–137. doi: 10.1016/0378-1119(93)90261-z. [DOI] [PubMed] [Google Scholar]
- 27.Chen VB, Arendall WB, III, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leslie AW. Recent changes to the MOSFLM package for processing film and image plate data. Joint CCP4 + ESF-EAMCB. Newslett Protein Crystallogr. 1992:26. [Google Scholar]
- 29.Evans PR. Scala. Joint CCP4 + ESF-EAMCB. Newslett Protein Crystallogr. 1997;33:22–24. [Google Scholar]
- 30.Howell L, Smith D. Normal probability analysis. J Appl Cryst. 1992;25:81–86. [Google Scholar]
- 31.Collaborative Computational Project Number 4. The CCP4 suite: programs for protein crystallography (1994) Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 32.Otwinowski Z, Maximum likelihood refinement of heavy atom parameters . In: Proceedings of the CCP4 study weekend. Wolf W, Evans PR, Leslie AW, editors. Warrington, UK: Science and Engineering Research Council, Daresbury Laboratory; 1991. pp. 80–85. [Google Scholar]
- 33.Bricogne G, Vonrhein C, Flensburg C, Schiltz M, Paciorek W. Generation, representation and flow of phase information in structure determination: recent developments in and around SHARP 2.0. Acta Crystallogr D Biol Crystallogr. 2003;59:2023–2030. doi: 10.1107/s0907444903017694. [DOI] [PubMed] [Google Scholar]
- 34.Abrahams JP, Leslie AG. Methods used in the structure determination of bovine mitochondrial F1 ATPase. Acta Crystallogr D Biol Crystallogr. 1996;52:30–42. doi: 10.1107/S0907444995008754. [DOI] [PubMed] [Google Scholar]
- 35.Cowtan K. The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr D Biol Crystallogr. 2006;62:1002–1011. doi: 10.1107/S0907444906022116. [DOI] [PubMed] [Google Scholar]
- 36.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 38.Vagin A, Teplyakov A. MOLREP: an automated program for molecular replacement. J Appl Cryst. 1997;30:1022–1025. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.