

Abstract
The human pregnane X receptor (PXR) is a member of the nuclear receptor superfamily of ligand-regulated transcription factors. PXR responds to a structurally diverse variety of endogenous and xenobiotic compounds, and coordinates the expression of genes central to the metabolism and excretion of potentially harmful chemicals, including human therapeutics. The reverse transcriptase inhibitor PNU-142721 has been designed to treat human immunodeficiency virus (HIV) infection. Although this compound has anti-HIV activity, it was established using cell-based assays that PNU-142721 is an efficacious PXR agonist. We present here the 2.8 Å resolution crystal structure of the human PXR ligand-binding domain in complex with PNU-142721. PXR employs one hydrogen bond and fourteen van der Waals contacts to interact with the ligand, but allows two loops adjacent to the ligand-binding pocket to remain disordered in the structure. These observations highlight the role structural flexibility plays in PXR's promiscuous responses to xenobiotics. The crystal structure also explains why PNU-173575, a thiomethyl metabolite of PNU-142721, exhibits enhanced PXR activation relative to the unmodified compound and why PNU-142721 can also activate rat PXR. Taken together, the results presented here elucidate the structural basis for PXR activation by PNU-142721 and related chemicals.
Keywords: human nuclear xenobiotic receptor, PXR, reverse transcriptase, anti-HIV drug, PNU-142721
Introduction
Nucleoside (or nucleotide) reverse transcriptase inhibitors (NRTIs) and non-nucleoside (or nucleotide) reverse transcriptase inhibitors (NNRTIs) are the two popular types of anti-HIV drugs that target reverse transcriptase (RT).1 NRTIs directly bind to the catalytic site of RT and competitively inhibit enzyme activity, while NNRTIs bind to an allosteric lipophilic site adjacent to the catalytic site of RT and function in a noncompetitive manner.2 Due to their distinct mechanisms, NRIs and NNRTIs are commonly used together in HIV treatment regimen to achieve optimal efficacy.3,4 Like other antiviral drugs, the usefulness of anti-HIV drugs is often compromised by mutations in the drug binding sites of HIV RT, which arise quickly due to the high mutation rate of the virus.5 Over the past two decades, the design of new drugs that can efficiently treat drug-resistant HIV has become a significant challenge.6 Indeed, the combinational use of multiple anti-HIV drugs with distinct mechanisms of action, such as the triple-cocktail treatment, has thus-far been proven to be the most effective anti-HIV regimen available.7
A fundamental problem associated with coadministering multiple drugs is the risk of drug–drug interactions. Cytochrome P450 (CYP) enzymes have been shown to be centrally involved in such interactions due to the principal role these proteins play in eliminating potentially toxic xenobiotics.8 The nuclear receptor pregnane X receptor (PXR) is a master regulator of the expression of CYP enzymes involved in drug metabolism. PXR is the primary sensor of xenobiotic stress in the liver and other first-pass tissues and can be activated by a structurally diverse collection of molecules including many drugs.9,10 Like other nuclear receptors, PXR contains a DNA binding domain (DBD), a ligand-binding domain (LBD), and a flexible linker domain.10 The PXR-LBD consists of a three-layered α-helical sandwich, common to nuclear receptors and a five-stranded antiparallel α-sheet, unique to PXR.11–16 The PXR-DBD binds specifically to the DNA response elements in the regulatory regions of CYP genes as a heterodimer with the retinoid X receptor (RXR).9 Upon activation by ligand binding to the LBD, PXR recruits coactivators that facilitate the formation of the transcriptional initiation complex and the expression of CYP gene products.9 Coactivator proteins bind to the activation function 2 (AF-2) region of the PXR-LBD using a surface formed, in part, by the C-terminal αAF helix in the receptor.17
PNU-142721 was first identified as a broad-spectrum second-generation NNRTI that demonstrated potent efficacy against various type I HIV mutants resistant to first-generation antivirals like delavirdine and zidovudine18 (Fig. 1). Despite its promising in vitro performance, studies showed that PNU-142721 causes strong drug–drug interactions by activating PXR (unpublished data from Dr. Evan Smith). We elucidate here the structure basis for PNU-142721 mediated PXR activation by solving the crystal structure of PXR-LBD in complex with PNU-142721.
Figure 1.
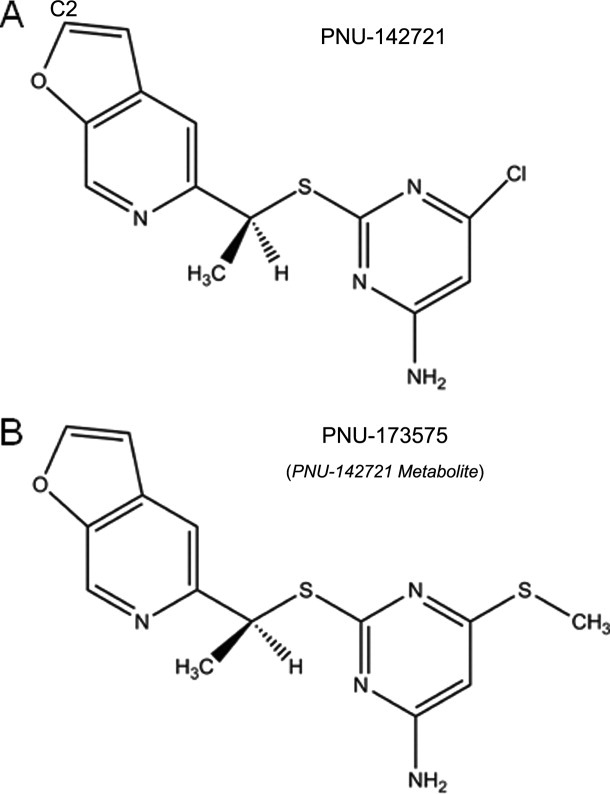
Chemical Structures of PNU-142721 and its metabolite PNU-173575.
Results
PXR-PNU142721 complex crystal structure
To understand the structural basis of receptor activation by this potential therapeutic lead, we determined the crystal structure of the human PXR-LBD in complex with PNU-142721. One protein-ligand complex was observed in each asymmetric unit. The structure was determined by molecular replacement in space group P43212 and refined to 2.8 Å resolution [Fig. 2(A) and Table I]. The PXR-LBD in this complex consists of a three-layered α-helical sandwich (α1/α3, α4/α5/α8, and α7/α10) common to nuclear receptors and a five-stranded antiparallel β-sheet unique to PXR [Fig. 2(A)]. Three regions located adjacent to the ligand-binding pocket (the loops 176–198 and 309–315, as well as residue 217) are disordered and not built in the final model (Fig. 3). The region between residues 176 and around 198 have been reported to be disordered in other PXR-LBD structures resolved to date.11–17 The PXR-PNU142721 complex exhibits the same overall fold observed in previous PXR structures, sharing, for example, 0.22 Å root-mean-square deviation over 254 Cα positions with the structure of the unliganded PXR-LBD.15 Of note, this is the second structure of PXR in which the 309–315 loop of the ligand binding domain remains disordered. The first was the complex with the much larger rifampicin ligand.16 Thus, we find that even relatively small ligands allow PXR's ligand binding pocket to remain flexible.
Figure 2.
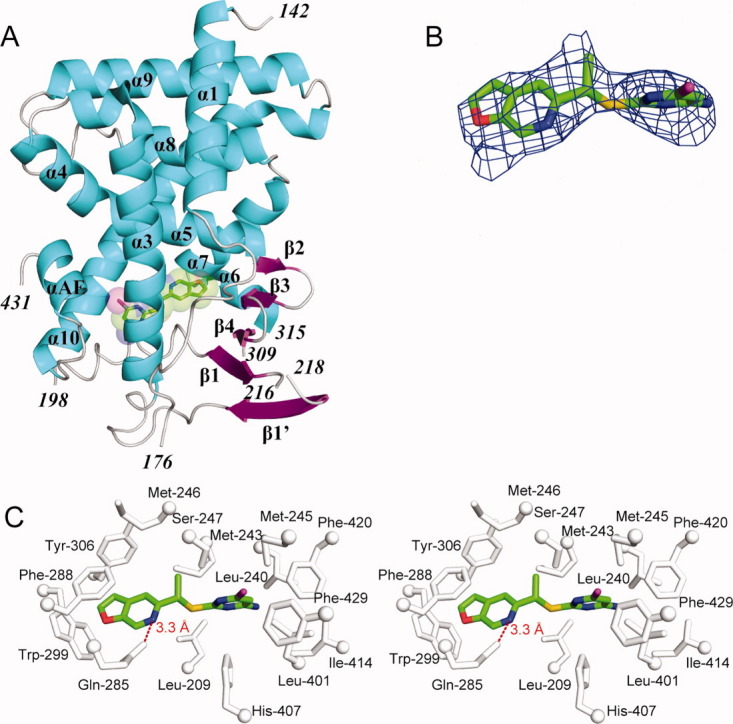
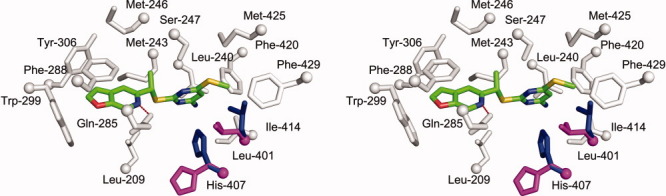
Crystal structure of the human PXR-LBD in complex with PNU-142721. (A) The overall structure of PXR-LBD in complex with PNU-142721. The α-helices, β-strands and loops are rendered in cyan, magenta, and gray, respectively. PNU-142721 is colored by elements, with green, red, blue, yellow and magenta representing carbon, oxygen, nitrogen sulfur and chlorine atoms, respectively. Note the proximity of the PNU-142721 to αAF of the AF-2 surface of PXR. (B) The 2.8 Å simulated annealing omit map contoured at 2.5 σ is shown in blue mesh. PNU-142721 is shown and colored by elements. (C) Stereoview of the ligand-binding pocket of the PXR-PNU142721 complex. Residues that line the ligand-binding pocket of PXR are shown in gray and labeled. The hydrogen bond is represented by a red dash line. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Table I.
Crystallographic Statistics for the PXR-PNU142721 Complex
| Resolution (Å) (highest shell) | 41.3–2.8 (2.87–2.80) |
| Space group | P43212 |
| Unit cell dimension (Å) | a=b= 92.3, c = 84.4 |
| No. of total reflections | 9394 |
| No. of unique reflections | 8944 |
| Rsym (%) (highest shell)a | 9.4 (53.0) |
| Completeness (%) (highest shell) | 99.5 (96.9) |
| Mean I/σ (highest shell) | 42.0 (5.1) |
| Rcryst (%) (highest shell)b | 23.8 (27.6) |
| Rfree (%) (highest shell)c | 28.9 (42.8) |
| RMSD | |
| Bond lengths (Å) | 0.033 |
| Bond angles (°) | 2.5 |
| Dihedral angles (°) | 23.6 |
| Number of atoms | |
| Protein | 2105 |
| Solvent | 15 |
| Ligand | 20 |
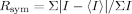 where I is the observed intensity and 〈I〉 is the average intensity of multiple symmetry-related observation of the reflection.
where I is the observed intensity and 〈I〉 is the average intensity of multiple symmetry-related observation of the reflection.
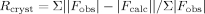 where Fobs and Fcalc are the observed and calculated structure factors, respectively.
where Fobs and Fcalc are the observed and calculated structure factors, respectively.
 for 5% of the data not used at any stage of the refinement.
for 5% of the data not used at any stage of the refinement.
Figure 3.
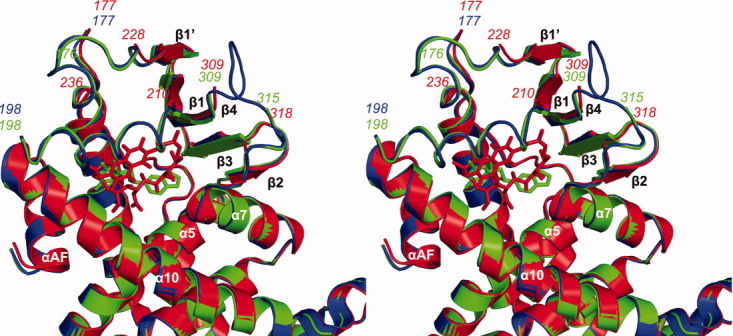
Analysis of the ligand binding pocket of PXR-LBD-PNU complex. Stereoview of a superimposition between the apo-PXR (blue), PXR-PNU142721 (green) and PXR-rifampicin (red) complexes. Major secondary structures around the ligand-binding pocket are labeled. The hydrogen bond is represented by a red dash line. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
PNU-142721 binding to PXR-LBD
A 2.8 Å simulated annealing omit map contoured at 2.5 σ was used to identify the electron density for PNU-142721, which binds to the PXR ligand-binding pocket in a single orientation [Fig. 2(B)]. PNU-142721 forms van der Waals contacts with the side chains of 14 residues that line the ligand-binding pocket [Fig. 2(C)]. In addition, the side-chain amine group of Gln-285 forms a 3.3 Å, presumably weak hydrogen bond with the nitrogen atom of the furopyridine ring of PNU-142721 [Fig. 2(C)]. As observed in other complexes, van der Waals contacts play a principal role in stabilizing PNU-142721 within PXR's ligand-binding pocket. Phe-288 forms a parallel aromatic stacking interaction with the furopyridine ring of PNU-142721, while Trp-299 and Tyr-306 form edge-to-face aromatic stacking contacts with the same ligand ring. The pyrimidine ring of PNU-142721 forms van der Waals interactions with the side chain of eleven residues, including a 3.6 Å interaction with Met-425 and a 5.0 Å interaction with Phe-429, which are located on αAF of the PXR AF-2 surface. These interactions likely help to stabilize the active conformation of the AF-2 surface for coactivator binding.
PNU142721 metabolite binding to PXR-LBD
PNU-173575, a thiomethyl metabolite of PNU-142721, is a more potent activator of PXR than the unmodified compound (unpublished data from Dr. Evan Smith). As shown in Figure 1, the only difference between these two compounds is that the chloro group on the pyrimidine ring of PNU-142721 is replaced by a thiomethyl group in PNU-173575. To understand why this small change improves PNU-173575 PXR activation, we modeled PNU-173575 into PXR's ligand-binding pocket using the structure of PNU142721 as a template (Fig. 4). To accommodate PNU-173575, the side chain of Leu-411 and His-407 were shifted by favored rotamers by alterations of 100.3° and 88.5°, respectively (Fig. 4). Although the rotamer change in His-407 is likely to cause the loss of a 4.8 Å van der Waals contact between the ligand and PXR, two new van der Waals contacts are observed between the thiomethyl group and the side chain of residues that are located on the αAF of the PXR AF-2 surface, including a 3.9 Å contact with Met-425, and a 4.1 Å contact with Phe-429. Thus, these new van der Waals contacts likely explain the improved PXR activation of PNU-173575, as they are better able to stabilize the active conformation of receptor's AF-2 surface.
Figure 4.
Modeled structure of human PXR-LBD in complex with PNU-173575. Residues that line the PXR ligand binding pocket are shown in gray, except for His-407 and Leu-411 which undergo rotamer changes in the presence of PNU-173575. The side-chain position of His-407 and Leu-411 in the presence of PNU-142721 and PNU-173575 are represented in blue and magenta, respectively. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Modeling of the structure of rat PXR-LBD in complex with PNU-142721
PNU-142721 can induce PXR-mediated CYP3A expression in rats (unpublished data from Dr. Evan Smith). Due to the relatively high sequence similarity (76% identity) shared between the rat and human PXRs, the rodent receptor is expected to have a structure highly analogous to human PXR. To understand rat PXR activation by PNU-142721, we modeled the structure of the ligand-binding pocket of rat PXR based on the structure of human PXR. The following five residues that interact with PNU-142721 in human PXR were altered to the corresponding residue (listed second) in the rat sequence: Leu209Met, Met243Leu, Met246Val, Gln285Ile and His407Gln (Fig. 5). Because they are largely conservative with respect to amino acid type, no significant change in the interaction between PNU-142721 and the PXR-LBD was observed in this modeled binding pocket. Indeed, PNU-142721 still forms 14 van der Waals contacts with residues that line the ligand binding pocket of rat PXR. Thus, for this particular lead scaffold, rat PXR served as an excellent model for potential activation of human PXR.
Figure 5.
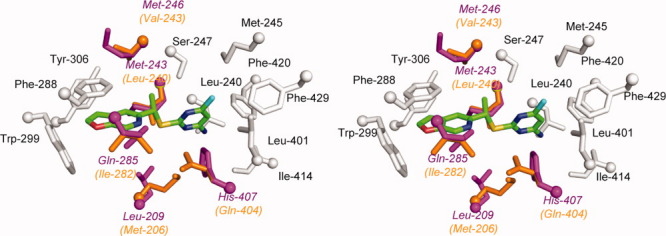
Modeled structure of rat PXR-LBD in complex with PNU-142721. Residues that line rat PXR ligand binding pocket and are conserved between human PXR and rat PXR are labeled and colored in gray, while residues that differ between human PXR and rat PXR are highlighted in orange (rat PXR) and magenta (human PXR), respectively. PNU-142721 is colored by elements, with green, red, blue, yellow and cyan representing carbon, oxygen, nitrogen sulfur and chlorine atoms, respectively. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Discussion
As the use of anti-HIV drug cocktails becomes increasingly necessary, dangerous drug–drug interactions may emerge as a major challenge in the antiretroviral field. Studies showed that potent HIV-1 RT inhibitor PNU-142721 produced drug–drug interactions, although the underlying mechanisms remained elusive. It has been shown that PNU-142721 activates the nuclear xenobiotic receptor PXR in a concentration-dependent manner and PNU-173575, the metabolite of PNU-142721, is a better PXR-agonist than PNU-142721 (unpublished data from Dr. Evan Smith). Altogether, these results established that PNU-142721 is an efficacious PXR agonist and it likely causes drug–drug interactions by activating PXR.
We elucidated the crystal structure of the PXR-LBD in complex with PNU-142721. The mechanism of ligand-induced PXR activation has been well studied by X-ray crystallography and other techniques. To activate PXR, a ligand needs to bind to the ligand-binding pocket of PXR and form direct interactions with the αAF helix, thereby stabilizing the AF-2 surface in the active conformation.10 Due to the large size and flexibility of the PXR's ligand binding pocket, however, it has been difficult to predict the exact binding mode of a novel ligand in PXR's ligand binding pocket by known PXR structures, although recent studies have demonstrated progress in this regard.19 Our PXR-PNU-142721 complex crystal structure reveals that van der Waals interactions play a major role in stabilizing the interaction between drug and receptor [Fig. 2(C)]. PNU-142721 forms van der Waals contacts with 14 residues in PXR's ligand-binding pocket, which covers all of five ligand-binding hot spots recently identified by Ngan et al,20 indicating a strong affinity between PNU-142721 and PXR. As in previous PXR-ligand structures, Gln-285 is found to form a hydrogen bond with the ligand, indicating its conserved role in ligand interaction. PNU-142721 forms direct van der Waals contacts with Met-425 and Phe-429, which are located on the αAF helix. The contact with Met-425 has been commonly observed in previous published PXR-ligand structures, while the contact with Phe-429 only appears in the PXR-estradiol structure to date.
The crystal structure of the PXR-PNU142721 complex was then used to examine two recent observations: (1) the thiomethyl metabolite PNU-173575 exhibits improved PXR activation relative to PNU-142721 and (2) PNU-142721 is also an agonist for rat PXR (unpublished data from Dr. Evan Smith). First, we found that the modeled structure of PNU-173575 in PXR's ligand binding pocket indicates that 173575 can bind to PXR in a similar way as PNU-142721. By means of the longer and more hydrophobic thiomethyl group, PNU-173575 appears capable of forming improved interactions with the αAF helix, and thereby stabilize the AF-2 surface in an active conformation. Second, the ligand-binding pocket of rat PXR can be modeled simply by mutating five residues in human PXR to their counterparts in rat PXR. Despite several changes in relevant residues, key ligand interactions are conserved between human PXR and rat PXR, indicating that rat PXR can bind PNU-142721 in a similar way to human PXR. Our results reinforce the hypothesis that, in this particular case, the rat could be used as a good model for the prediction of potential drug–drug interaction in human.
If one wanted to modify PNU-142721 to eliminate its activation of PXR, the crystal structure of the complex could be used as a guide. The van der Waals interaction between the chloro-group on the pyrimidine ring of PNU-142721 and the αAF helix of PXR appear key to the stabilization of AF-2 surface in the active conformation. Therefore, disrupting the interaction between PNU-142721 and the AF-2 surface could be achieved by removing the chloro-group from the pyrimidine ring. However, previous studies have shown that the pyrimidine thioether moiety is the major anti-HIV pharmacophore of PNU-142721.18 The furopyridine moiety, however, was not as important,18 and thus, it might be safer to disrupt the interaction between PNU-142721 and PXR by modifying this ring. As observed in the complex structure, the furopyridine ring of PNU-142721 interacts with five residues in the ligand-binding pocket including Phe-288, Trp-299, Tyr 306, His-327, and Gln-285. Among these contacts, the π–π interaction between the side-chain of Phe-288 and the furopyridine ring provide the major stabilizing forces, keeping the furopyridine ring in the current orientation. Disruption of this interaction by introducing a bulky group on the C2 atom of the ligand (see Fig. 1), which is spatially closest to Trp-299 (see Fig. 5), may effectively destabilize the interaction of PNU-142721 with PXR. Either PNU-142721 or Trp-299 would have to move to accommodate this bulky group. Trp299 is located on four-stranded β-sheet, which is known for its rigidity and does not change its conformation even upon the binding of large PXR ligands like rifampcin. Therefore, it is not likely that Trp-299 would adjust its position to accommodate the bulky group. Also, there is no observable space around Trp-299 for its side-chain to take alterative rotamers to accommodate the bulky group. Therefore, it would be expected that PNU-142721 might move away from Trp-299 toward the other side of the ligand-binding pocket. This movement will break three important interactions between PXR and the furopyridine ring of PNU-142721, including the π–π interaction with Phe-288, the van der Waals interaction with Tyr-306, and the hydrogen bonding with the side chain of Gln-285. Although several new van der Waals interactions might form meanwhile, they are not likely to compensate for the losses mentioned above. Taken together, the proposed modification here might mitigate PXR agonist activity by disrupting the binding of PNU-142721 to PXR.
In summary, the data presented here reveal the detailed mechanism of PNU-142721-mediated PXR activation. Using the structural model presented here, we explained why the metabolite PNU-173575 is a better PXR agonist and why PNU-142721 can also activate rat PXR. Based on the structural data presented here, several routes toward the redesign of PNU-142721 were proposed that may limit PXR activation while keeping intact the HIV-1 RT inhibitory activity of this compound.
Materials and Methods
Protein expression and purification
The human PXR-LBD (residues 130–434) was coexpressed with an 88-amino acid fragment of human steroid receptor coactivator 1 (SRC-1) (623–710) in E. coli and purified as described previously.15 Purified hPXR-LBD was concentrated to 3 mg/mL in buffer containing 20 mM Tris-HCl (pH 7.8), 250 mM NaCl, 5 mM DTT, 2.5 mM EDTA and 5% Glycerol (v/v) using a Centri-prep 30K unit (Amicon) in the presence of 20-fold molar excess PNU-142721. Purified protein was flash-frozen using liquid nitrogen and stored at−80°C.
Crystallization, data collection, and structural determination
The hPXR-PNU142721 complex was crystallized by hanging-drop vapor diffusion at room temperature against 50 mM imidazole (pH 7.8) and 16% 2-propanol (v/v). Crystals were handled for cryoprotection as described previously.15 Data collection was conducted at SER-CAT at the Advanced Photon Source in Argonne National Labs (Beamline 22-ID). Diffraction data were indexed, integrated and scaled using HKL2000.21 The structure of hPXR-PNU142721 complex was determined by molecular replacement with the MolRep module of the CCP4 suite22,23 using the crystal structure of the apo PXR as the search model. Clear rotational and translational solutions were obtained in space group P43212. The structure was manually adjusted using WinCoot 3.1,24 and refined using the Refmac module of CCP4,25 with 5% of the reflections set aside for Rfree validation before any structural refinement. Simulated annealing omit map was generated using the program CNS.26 All the molecular graphic figures in this work were created using Pymol.27 While the electron density alone was not sufficient to distinguish unambiguously the position of the chlorine and the primary amine of the pyrimidine ring, they were modeled based on the following reasoning. It has been well established that, to activate PXR, a ligand must stabilize the AF-2 surface in the active conformation by forming direct hydrophobic interactions with the αAF helix. Therefore, we placed the more hydrophobic chlorine toward the αAF helix.
Acknowledgments
The authors thank Dr. Evan Smith (Department of Pharmacokinetics, Dynamics and Metabolism, Pfizer Global Research and Development) for providing PNU-142721 and relevant information. They are very grateful to Dr. Yu Xue and Dr. Eric Ortlund for assistance in diffraction data analysis, and Dr. Laurie Betts for advice in manuscript preparation. They also acknowledge the beamline scientists for the assistance in data collection on Beamline 22 at SER-CAT at the Advanced Photon Source in Argonne National Labs.
Glossary
Abbreviations
- AF-2
activation function-2
- AIDS
acquired immune deficiency syndrome
- CYP3A
cytochrome P450 3A
- DBD
DNA-binding domain
- DTT
Dithiothreitol
- EDTA
ethylenediaminetetraacetic acid
- HIV
human immunodeficiency virus
- LBD
ligand-binding domain
- NNRTIs
nonnucleoside reverse transcriptase inhibitors
- NRTIs
nucleoside reverse transcriptase inhibitors
- PXR
pregnane X receptor
- RT
reverse transcriptase
- RXR
retinoid X receptor
- SRC-1
steroid receptor coactivator 1
References
- 1.Jochmans D. Novel HIV-1 reverse transcriptase inhibitors. Virus Res. 2008;134:171–185. doi: 10.1016/j.virusres.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 2.Ren J, Stammers DK. HIV reverse transcriptase structures: designing new inhibitors and understanding mechanisms of drug resistance. Trends Pharmacol Sci. 2005;26:4–7. doi: 10.1016/j.tips.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 3.Dieterich DT. Long-term complications of nucleoside reverse transcriptase inhibitor therapy. AIDS Read. 2003;13:176–84. 187. [PubMed] [Google Scholar]
- 4.Nolan D, Mallal S. Complications associated with NRTI therapy: update on clinical features and possible pathogenic mechanisms. Antivir Ther. 2004;9:849–863. [PubMed] [Google Scholar]
- 5.Shafer RW, Rhee SY, Pillay D, Miller V, Sandstrom P, Schapiro JM, Kuritzkes DR, Bennett D. HIV-1 protease and reverse transcriptase mutations for drug resistance surveillance. AIDS. 2007;21:215–223. doi: 10.1097/QAD.0b013e328011e691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Geretti AM. Clinical implications of HIV drug resistance to nucleoside and nucleotide reverse transcriptase inhibitors. AIDS Rev. 2006;8:210–220. [PubMed] [Google Scholar]
- 7.Bartlett JA, Fath MJ, Demasi R, Hermes A, Quinn J, Mondou E, Rousseau F. An updated systematic overview of triple combination therapy in antiretroviral-naive HIV-infected adults. AIDS. 2006;20:2051–2064. doi: 10.1097/01.aids.0000247578.08449.ff. [DOI] [PubMed] [Google Scholar]
- 8.Pal D, Mitra AK. MDR- and CYP3A4-mediated drug-drug interactions. J Neuroimmune Pharmacol. 2006;1:323–339. doi: 10.1007/s11481-006-9034-2. [DOI] [PubMed] [Google Scholar]
- 9.Kliewer SA, Goodwin B, Willson TM. The nuclear pregnane X receptor: a key regulator of xenobiotic metabolism. Endocr Rev. 2002;23:687–702. doi: 10.1210/er.2001-0038. [DOI] [PubMed] [Google Scholar]
- 10.Carnahan VE, Redinbo MR. Structure and function of the human nuclear xenobiotic receptor PXR. Curr Drug Metab. 2005;6:357–367. doi: 10.2174/1389200054633844. [DOI] [PubMed] [Google Scholar]
- 11.Teotico DG, Bischof JJ, Peng L, Kliewer SA, Redinbo MR. Structural basis of human pregnane X receptor activation by the hops constituent colupulone. Mol Pharmacol. 2008;74:1512–1520. doi: 10.1124/mol.108.050732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xue Y, Moore LB, Orans J, Peng L, Bencharit S, Kliewer SA, Redinbo MR. Crystal structure of the pregnane X receptor-estradiol complex provides insights into endobiotic recognition. Mol Endocrinol. 2007;21:1028–1038. doi: 10.1210/me.2006-0323. [DOI] [PubMed] [Google Scholar]
- 13.Xue Y, Chao E, Zuercher WJ, Willson TM, Collins JL, Redinbo MR. Crystal structure of the PXR-T1317 complex provides a scaffold to examine the potential for receptor antagonism. Bioorg Med Chem. 2007;15:2156–2166. doi: 10.1016/j.bmc.2006.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Watkins RE, Maglich JM, Moore LB, Wisely GB, Noble SM, Davis-Searles PR, Lambert MH, Kliewer SA, Redinbo MR. 2.1 Å crystal structure of human PXR in complex with the St. John's wort compound hyperforin. Biochemistry. 2003;42:1430–1438. doi: 10.1021/bi0268753. [DOI] [PubMed] [Google Scholar]
- 15.Watkins RE, Wisely GB, Moore LB, Collins JL, Lambert MH, Williams SP, Willson TM, Kliewer SA, Redinbo MR. The human nuclear xenobiotic receptor PXR: structural determinants of directed promiscuity. Science. 2001;292:2329–2333. doi: 10.1126/science.1060762. [DOI] [PubMed] [Google Scholar]
- 16.Chrencik JE, Orans J, Moore LB, Xue Y, Peng L, Collins JL, Wisely GB, Lambert MH, Kliewer SA, Redinbo MR. Structural disorder in the complex of human pregnane X receptor and the macrolide antibiotic rifampicin. Mol Endocrinol. 2005;19:1125–1134. doi: 10.1210/me.2004-0346. [DOI] [PubMed] [Google Scholar]
- 17.Watkins RE, Davis-Searles PR, Lambert MH, Redinbo MR. Coactivator binding promotes the specific interaction between ligand and the pregnane X receptor. J Mol Biol. 2003;331:815–828. doi: 10.1016/s0022-2836(03)00795-2. [DOI] [PubMed] [Google Scholar]
- 18.Campiani G, Ramunno A, Maga G, Nacci V, Fattorusso C, Catalanotti B, Morelli E, Novellino E. Non-nucleoside HIV-1 reverse transcriptase (RT) inhibitors: past, present, and future perspectives. Curr Pharm Des. 2002;8:615–657. doi: 10.2174/1381612024607207. [DOI] [PubMed] [Google Scholar]
- 19.Ekins S, Kortagere S, Iyer M, Reschly EJ, Lill MA, Redinbo MR, Krasowski MD. Challenges predicting ligand-receptor interactions of promiscuous proteins: the nuclear receptor PXR. PLoS Comput Biol. 2009;5:e1000594. doi: 10.1371/journal.pcbi.1000594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ngan CH, Beglov D, Rudnitskaya AN, Kozakov D, Waxman DJ, Vajda S. The structural basis of pregnane X receptor binding promiscuity. Biochemistry. 2009;48:11572–11581. doi: 10.1021/bi901578n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Otwinowski Z, Minor W. Methods in Enzymology: Macromolecular Crystallography, part A. Processing of x-ray diffraction data collected in oscillation mode. In: Carter CJ, Sweet RM, editors. Vol. 276. New York: Academic Press; 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 22.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 23.Collaborative Computational Project, Number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 24.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 25.Winn MD, Murshudov GN, Papiz MZ. Macromolecular TLS refinement in REFMAC at moderate resolutions. Methods Enzymol. 2003;374:300–321. doi: 10.1016/S0076-6879(03)74014-2. [DOI] [PubMed] [Google Scholar]
- 26.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 27.Delano WL. The PyMOL molecular graphics system. San Carlos, CA: DeLano Scientific LLC; 2002. [Google Scholar]