

Abstract
FK506-binding protein 22 (FKBP22) from the psychrotophic bacterium Shewanella sp. SIB1 (SIB1 FKBP22) is a homodimeric protein with peptidyl prolyl cis-trans isomerase (PPIase) activity. Each monomer consists of the N-terminal domain responsible for dimerization and C-terminal catalytic domain. To reveal interactions at the dimer interface of SIB1 FKBP22, the crystal structure of the N-domain of SIB1 FKBP22 (SN-FKBP22, residues 1-68) was determined at 1.9 Å resolution. SN-FKBP22 forms a dimer, in which each monomer consists of three helices (α1, α2, and α3N). In the dimer, two monomers have head-to-head interactions, in which residues 8–64 of one monomer form tight interface with the corresponding residues of the other. The interface is featured by the presence of a Val-Leu knot, in which Val37 and Leu41 of one monomer interact with Val41 and Leu37 of the other, respectively. To examine whether SIB1 FKBP22 is dissociated into the monomers by disruption of this knot, the mutant protein V37R/L41R-FKBP22, in which Val37 and Leu41 of SIB1 FKBP22 are simultaneously replaced by Arg, was constructed and biochemically characterized. This mutant protein was indistinguishable from the SIB1 FKBP22 derivative lacking the N-domain in oligomeric state, far-UV CD spectrum, thermal denaturation curve, PPIase activity, and binding ability to a folding intermediate of protein, suggesting that the N-domain of V37R/L41R-FKBP22 is disordered. We propose that a Val-Leu knot at the dimer interface of SIB1 FKBP22 is important for dimerization and dimerization is required for folding of the N-domain.
Keywords: FKBP22, Shewanela sp. SIB1, crystal structure, N-domain, PPIase, dimer interface, Val-Leu knot
Introduction
FK506-binding proteins (FKBPs), cyclophilines, and parvulins are three structurally unrelated families of peptidyl prolyl cis-trans isomerase (PPIase), which catalyzes cis-trans isomerization of Xaa-Pro peptide bonds of proteins.1 This isomerization is intrinsically slow and therefore has been regarded as a rate limiting step of folding reaction of proteins.2 FKBP22 from the psychrotrophic bacterium Shewanella sp. SIB1 (SIB1 FKBP22) is a member of the macrophage infectivity potentiator (MIP)-like FKBP subfamily.3 It shows the amino acid sequence identities of 56% to Escherichia coli FKBP22 (accession no. AAC77164), 20% to Neurospora crassa FKBP22 (accession no. O60046), 43% to E. coli FkpA (accession no. AAC76372), and 41% to Legionella pneumophila MIP (accession no. S42595). Because the cellular content of SIB1 FKBP22 in SIB1 increases at 4°C as compared to that at 20°C, SIB1 FKBP22 is thought to be involved in cold adaptation of SIB1 by accelerating folding rate of proteins, that is, significantly retarded by slow prolyl isomerization at low temperatures.3
SIB1 FKBP22 exists as a homodimer, in which each monomer (205 residues) consists of the N-terminal domain (N-domain) responsible for dimerization and the C-terminal domain (C-domain) responsible for PPIase activity.3,4 According to a tertiary model of SIB1 FKBP22 constructed based on the crystal structures of L. pneumophila MIP5 and E. coli FkpA,6 which highly resemble to each other, SIB1 FKBP22 assumes a non-globular V-shaped homodimeric structure.4,7 In this structure, each monomer forms one “arm” of the V-shaped structure and two monomers interact with each other at their N-domains. The N- and C-domains of each monomer are connected by a 40-residue-long α3-helix, such that the N-terminal one-third and C-terminal one-third of the α3-helix are involved in the N- and C-domains, respectively. SIB1 FKBP22 has an ability to bind to a folding intermediate of protein as well,8 as does E. coli FkpA.6,9 A V-shaped dimeric structure of SIB1 FKBP22 is required for efficient binding to a folding intermediate of protein and maximal PPIase activity for protein substrate.7
The N-domain, which is unique to the MIP-like FKBP subfamily proteins, consists of the α1-helix, α2-helix, and the N-terminal region of the α3-helix in the SIB1 FKBP22 structure model. Of various interactions at the dimer interface, those at the interface between two α2-helices are most intriguing, because these helices are packed together in an antiparallel fashion at the core region of the dimeric structure of the N-domain. A Val-Leu knot, in which Val37 and Leu41 of one monomer interact with Leu41 and Val37 of the other, respectively, is present at this interface. This knot is conserved as a Met knot (Met38 and Met42) in the L. pneumophila MIP structure5 and a Leu-Val knot (Leu55 and Val59) in the E. coli FkpA structure.6 It has been suggested that these knots provide strong hydrophobic interactions and therefore contribute to the stabilization of the dimer interface. However, it remains to be determined whether MIP-like FKBP subfamily proteins with a homodimeric structure are dissociated into monomers by disruption of these knots. It also remains to be determined whether a Val-Leu knot is formed at the dimer interface of SIB1 FKBP22 as seen in the structural model.
In this study, we determined the crystal structure of the N-domain of SIB1 FKBP22 (SN-FKBP22, residues 1–68). The structure resembles those of the N-domains of L. pneumophila MIP and E. coli FkpA and a Val-Leu knot is present at the interface between two α2-helices at the core region of the dimeric structure. To examine whether SIB1 FKBP22 is dissociated into the monomers by disruption of this knot, the double mutant protein of SIB1 FKBP22, V37R/L41R-FKBP22, was constructed. This mutant protein was monomeric and its biochemical properties were indistinguishable from those of the isolated C-domain of SIB1 FKBP22 containing the entire α3-helix, suggesting that a Val-Leu knot at the interface between two α2-helices is important for dimerization of SIB1 FKBP22 and dimerization is required for folding of the N-domain.
Results and Discussion
Protein preparation and crystallization
According to a tertiary model, the N-domain of SIB1 FKBP22 consists of the α1-helix, α2-helix, and N-terminal region of the α3-helix. To reveal interactions at the dimer interface of SIB1 FKBP22 by X-ray crystallographic analyses, a His-tagged form of the isolated N-domain containing the N-terminal region of the α3-helix (SN-FKBP22, residues 1-68) was constructed. This protein was constructed because attempts to crystallize SIB1 FKBP22 and the isolated N-domain containing the entire α3-helix (N-domain+, residues 1–94) have so far been unsuccessful. N-domain+ exists as a dimer and retains an ability to bind to a folding intermediate of protein, suggesting that it is folded into a correct structure.4,8 The primary structure of SN-FKBP22 is schematically shown in Figure 1 in comparison with those of SIB1 FKBP22 and N-domain+. SN-FKBP22 accumulated in E. coli cells in a soluble form upon overproduction and was purified to give a single band on SDS-PAGE (data not shown). The amount of the protein purified from 1 L culture was typically 4.3 mg, which was comparable to that of N-domain+. When SN-FKBP22 was screened for crystallization conditions using the kits available from commercial sources, it gave crystals suitable for X-ray crystallographic analyses.
Figure 1.
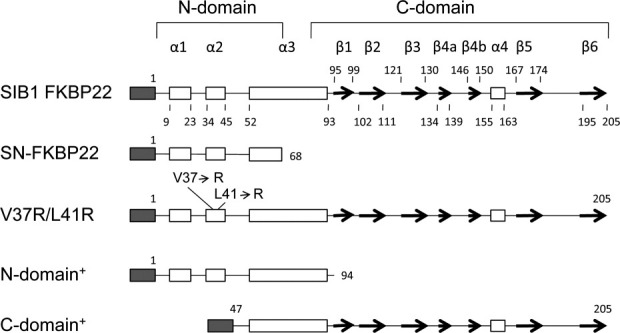
Schematic representations of the primary structures of SIB1 FKBP22 and its derivatives. A His-tag attached to the N-termini of the proteins is represented by shaded box. The α-helices and β-strands are represented by open boxes and arrows, respectively. These secondary structures are arranged based on tertiary models of SIB1 FKBP22 and its derivatives. Numbers indicate the positions of the residues relative to the initiator methionine residue of the proteins without a His-tag. The locations of the mutated residues and the ranges of the N- and C-domains are also shown.
General features of the structure
The crystal structure of SN-FKBP22 was determined at 1.9 Å resolution. SN-FKBP22 crystal contains one monomer per asymmetric unit with solvent content of 49% and Mathews coefficient of 2.4, calculated based on the molecular weight of the protein. The monomer of SN-FKBP22 contains 65 residues (Asp3-Arg67) with a His-tag, Met1-Ser2, and Leu68 missing. These N- and C-terminal residues should be present but were not observed in the structure probably due to a structural disorder. The calculated total surface of monomer was 6009.4 Å2. The monomer of SN-FKBP22 consists of three α-helices, α1 (Thr7-Ala26), α2 (Ile34-Ala46), and α3N (Met54-Arg66) [Fig. 2(A,B)]. The third helix is termed α3N, because this helix is expected to extend to the C-terminal catalytic domain in the entire protein molecule. These helices are connected by seven-residue loops (Asn27-Asp33 between α1- and α2-helices and Gly47-Ser53 between α2- and α3N-helices).
Figure 2.

Three-dimensional structrue of SN-FKBP22. (A) Stereo view of the main chain folding of SN-FKBP22 is shown. One monomer is colored deep green, while the other is colored light green. The amino acid residues that make hydrophobic interactions (23 residues for each monomer) and those that form hydrogen bonds (Gln10, Asp33, Arg18, Lys48) at the dimer interface are indicated by stick models, in which the oxygen, nitrogen, and sulfer atoms are colored red, blue, and yellow, respectively. These hydrophobic residues are determined using PISA (Protein Interfaces, Surfaces, and Assemblies) service.10 The side chains of Val and Leu, which form a Val-Leu knot, are colored deep magenta for one monomer and light magenta for the other. The hydrogen bonds, which are formed between Gln10 (Nɛ1) and Asp33 (Oδ1) and between Arg18 (Nη1, Nη2) and Lys48 (main chain O), are shown by broken lines. N and C represent the N- and C-termini, respectively. (B) The same figure as Figure 2(A) with different view angle [view angle from the top of the structure shown in Fig. 2(A)]. (C) Comparison with tertiary model. Crystal structure of SN-FKBP22 (green) is superimposed onto the tertiary model of SIB1 FKBP22 (red). Inset shows superposition of the crystal structure of SN-FKBP22 and the equivalent region of the tertiary model of SIB1 FKBP22. One monomer is colored dark, while the other is colored light. (D) Stereo view of electron density around the Val-Leu knot. The 2Fo – Fc map contoured at the 1.5 σ level is shown. Val37 and Leu41 are indicated by stick models. One monomer is deeply-colored, while the other is lightly-colored. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Biological assembly
The Protein Interfaces, Surfaces, and Assemblies (PISA) analysis10 showed that SN-FKBP22 forms a biological dimer with calculated free energy for dissociation (ΔGdiss) of 39.7 kcal mol−1, indicating that this dimer cannot be spontaneously dissociated into monomers due to a strong interaction between two monomers. The dimeric structure of SN-FKBP22, in which two monomers have a head-to-head interaction, was generated from two adjacent monomers via the crystallographic twofold symmetric axis [Fig. 2(A,B)]. The dimer interface spans almost the entire region of each monomer (Met8-Ile64), and the α1- and α2-helices of one monomer interact with the α1- and α2-helices of the other, respectively, in an antiparallel manner. The area of the dimer interface is 4132.1 Å2, which is 34% of the total dimer surface. Despite the similarities in the amino acid sequences between SN-FKBP22 and L. pneumophila MIP and between SN-FKBP22 and E. coli FkpA, the root-mean-square deviation (rmsd) values between these two structures are relatively high (3.2 Å between SN-FKBP22 and L. pneumophila MIP and 2.5 Å between SN-FKBP22 and E. coli FkpA).
The dimer interface is stabilized by hydrophobic interactions and hydrogen bonds, but mainly by hydrophobic interactions. Only four direct hydrogen bonds (non water-mediated hydrogen bonds) are formed between Gln10 Nɛ2 of one monomer and Asp33 Oδ1 of the other and between Arg18 Nη1 and Nη2 of one monomer and Lys O of the other [Fig. 2(A,B)]. In contrast, 23 hydrophobic residues interact with one another at the dimer interface [Fig. 2(A,B)]. They are Met8, Ala12, Val16, Met20, Leu24, Ala25, Phe29, Ile32, Ile34, Ala36, Val37, Ala39, Leu41, Ala42, Ala44, Phe45, Ala51, Val52, Leu57, Val59, Ala60, Phe61, and Ile64.
Validity of three-dimensional model
The crystal structure of SN-FKBP22 highly resembles a tertiary model of SIB1 FKBP22 [Fig. 2(C)], suggesting that this model is valid. The N-domain provides a platform for a V-shaped dimeric structure of SIB1 FKBP22. However, the orientations of two α3N-helices in the SN-FKBP22 structure are slightly different from those in the tertiary model. This difference may be caused by removal of the entire C-domain including the C-terminal half of the α3-helix or great flexibility of the α3-helix that controls plasticity of a V-shaped structure. It has been proposed that flexibility of a V-shaped structure is required for binding of various types of protein substrates.11 Further structural studies will be required to understand the role of the α3-helix for SIB1 FKBP22 function.
Val-Leu knot
An interesting feature of the dimer interface is the presence of a Val-Leu knot between two α2-helices. In this knot, Val37 and Leu41 of one monomer interact with Leu41 and Val37 of the other, respectively [Fig. 2(D)]. The accessible surface areas (ASAs) of Val37 and Leu41 are calculated to be 2.71 and 4.69% of their total surface areas, respectively, indicating that these residues are almost fully buried inside the structure. The knot is facilitated by a series of the C-C contacts between Val37 of one monomer and Leu41 of the other. For example, Val37 Cβ of one monomer contacts Leu41 Cδ2 of the other. The distance between these atoms is 4.87 Å. Likewise, Val37 Cγ1 of one monomer contacts the Cδ2, Cγ, and Cδ1 atoms of Leu41 of the other. The distances between Val37 Cγ1 and these atoms are 4.85 Å for Leu41 Cδ2, 4.09 Å for Leu41 Cγ, and 4.34 Å for Leu41 Cδ1. Thus, this knot would greatly contribute to the stabilization of the dimer interface. A series of the similar C-C contacts are also present at both ends of the interface of two α2-helices, where Ile34 of one monomer interacts with Ala42 and Phe45 of the other. These contacts may also contribute to the stabilization of the dimer interface.
Construction of mutant protein
To examine whether SIB1 FKBP22 with a homodimeric structure is dissociated into the monomers by disruption of a Val-Leu knot, the mutant protein V37R/L41R-FKBP22, in which Val37 and Leu41 of SIB1 FKBP22 are simultaneously replaced by Arg, was constructed. These mutations should greatly reduce the stability of the dimer interface, because they not only remove the Val-Leu knot but also introduce electrostatic repulsion into the dimer interface. The primary structure of this mutant protein is schematically shown in Figure 1 in comparison with that of the isolated C-domain containing the entire α3-helix (C-domain+, residues 47–205). C-domain+ exists as a monomer and retains PPIasepep activity, but loses more than 90% of binding ability to a folding intermediate of protein and PPIasepro activity.4,8 V37R/L41R-FKBP22 was overproduced in E. coli at 10°C in a His-tagged form. The protein accumulated in E. coli cells in a soluble form and was purified to give a single band on SDS-PAGE (data not shown). The amount of the protein purified from 1 L culture was typically 3.6 mg, which was comparable to that of SIB1 FKBP22. The molecular mass of V37R/L41R-FKBP22 was estimated to be 22 kDa from gel filtration column chromatography, which was carried out as described previously.4 This value is comparable to the calculated one (24,047), indicating that this mutant protein exists as a monomer as does C-domain+.
CD spectra
To examine whether the mutations affect the protein conformation, the far- and near-UV CD spectra of V37R/L41R-FKBP22 were measured. These spectra are shown in Figure 3 in comparison with those of SIB1 FKBP22 and C-domain+. The far-UV CD spectrum of V37R/L41R-FKBP22 is similar to that of C-domain+ but is considerably different from that of SIB1 FKBP22 [Fig. 3(A)]. The helical content was calculated to be 26% for V37R/L41R-FKBP22, 49% for SIB1 FKBP22, and 21% for C-domain+ from these spectra using the method of Wu et al.12 According to a tertiary model, the N- and C-domains of SIB1 FKBP22 are characterized by the high (53%) and low (17%) helical contents. Therefore, great reduction in the helical content of V37R/L41R-FKBP22 as compared to that of SIB1 FKBP22 strongly suggests that the N-domain of V37R/L41R-FKBP22 is mostly disordered. The helical content of C-domain+ is also greatly reduced as compared to that of SIB1 FKBP22 because it lacks the N-domain.
Figure 3.
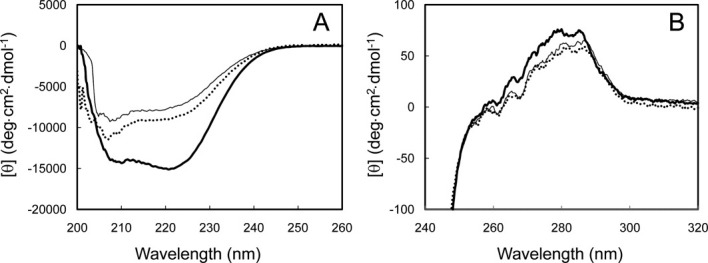
CD spectra of V37R/L41R-FKBP22. The far-UV (A) and near-UV (B) CD spectra of V37R/L41R-FKBP22 (dotted line) are shown in comparison with those of SIB1 FKBP22 (thick line) and C-domain+ (thin line). All spectra were measured at 10°C as described under Materials and methods.
On the other hand, the near-UV CD spectra of V37R/L41R-FKBP22, SIB1 FKBP22, and C-domain+ highly resemble with one another [Fig. 3(B)], suggesting that the conformation around the tryptophan and tyrosine residues of these proteins are similar with one another. SIB1 FKBP22 contains one tryptophan and seven tyrosine residues and all of them, except one tyrosine residue, are located in the C-domain. Therefore, the conformation of the C-domain of SIB1 FKBP22 might not be seriously changed by the mutations at the N-domain or removal of the N-domain.
Thermodynamics of unfolding
Heat induced unfolding of SIB1 FKBP22 and V37R/L41R-FKBP22 was analyzed by differential scanning calorimetry (DSC). These DSC curves were reproduced by repeating thermal scans, indicating that thermal unfolding of these proteins is reversible. The DSC curves of these proteins are shown in Figure 4 in comparison with that of C-domain+ previously reported.4 The DSC curve of SIB1 FKBP22 shows two well separated transitions. The melting temperatures (Tm) of these transitions are 33.0°C for the first one and 46.8°C for the second one, which have been reported to reflect thermal unfolding of the C-domain and N-domain, respectively.4 In contrast, the DSC curve of V37R/L41R-FKBP22 shows only single transition as does C-domain+. The Tm value of this transition (36.8°C) is slightly higher than but comparable to that of the first transition of FKBP22. It is also comparable to that of C-domain+ (35.6°C). These results are consistent with the CD spectra of the protein, which suggests that the C-domain of V37R/L41R-FKBP22 is correctly folded while the N-domain is mostly disordered.
Figure 4.
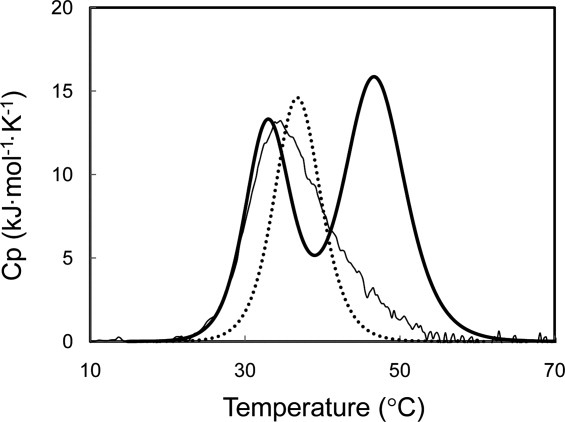
DSC curve of V37R/L41R-FKBP22. The DSC curve of V37R/L41R-FKBP22 (dotted line) is shown in comparison with those of SIB1 FKBP22 (thick line) and C-domain+ (thin line). These curves were measured at a scan rate of 1°C min−1. These proteins were dissolved in 20 mM sodium phosphate (pH 8.0) at approximately 1 mg mL−1.
PPIase activity
The PPIase activity for peptide substrate (PPIasepep) was determined at 10°C by the protease coupling method using N-succinyl-Ala-Leu-Pro-Phe-p-nitroanilide (Suc-ALPF-pNA) as a substrate. The catalytic efficiency (kcat/Km) of V37R/L41R-FKBP22 was estimated to be 1.13 ± 0.034 μM−1 s−1, which is 1.5-fold higher than that of SIB1 FKBP22 [Fig. 5(A)]. In contrast, when the PPIase activity for protein substrate (PPIasepro) was determined at 10°C by the RNase T1 refolding assay, V37R/L41R-FKBP22 exhibited much less activity as compared to that of SIB1 FKBP22 [Fig. 5(B)]. The catalytic efficiency (kcat/Km) of V37R/L41R-FKBP22 was estimated to be 0.05 ± 0.008 μM−1 s−1, which was 10-fold lower than that of SIB1 FKBP22. Interestingly, the PPIasepep and PPIasepro activities of V37R/L41R-FKBP22 were comparable to those of C-domain+ [Fig. 5(A,B)].
Figure 5.
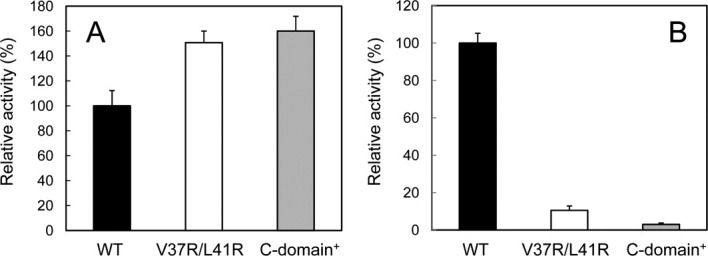
PPIase activities of V37R/L41R-FKBP22. The PPIasepep (A) and PPIasepro (B) activities of V37R/L41R-FKBP22, which were determined at 10°C and pH 7.8 using Suc-ALPF-pNA and RNase T1 as a substrate, respectively, are shown in comparison with those of SIB1 FKBP22 (WT) and C-domain+. Relative activities were calculated by dividing the catalytic efficiency (kcat/Km) of V37R/L41R-FKBP22 or C-domain+ by that of SIB1 FKBP22. The experiment was carried out in duplicate. Each value represents the average value and errors from the average values are shown.
Binding to reduced α-lactalbumin
SIB1 FKBP22 has an ability to bind to reduced α-lactalbumin,8 which has been used as a folding intermediate of protein.9,13 To examine whether V37R/L41R-FKBP22 and SN-FKBP22 retain an ability to bind to reduced α-lactalbumin, binding of reduced α-lactalbumin to SIB1 FKBP22, V37R/L41R-FKBP22, C-domain+, and SN-FKBP22 was analyzed using surface plasmon resonance (Biacore). Reduced α-lactalbumin was injected onto the sensor chip on which either one of these proteins was immobilized. The sensorgrams obtained by injecting 100 μM of reduced α-lactalbumin are shown in Figure 6. Because no increase of resonance unit (RU) was detected when reduced α-lactalbumin was injected onto the sensor chip on which V37R/L41R-FKBP22 or C-domain+ was immobilized, the binding affinity of V37R/L41R-FKBP22 to a folding intermediate of protein is greatly reduced by more than 20-fold compared with that of SIB1 FKBP22, as is that of C-domain+. In contrast, increase of RU was detected when reduced α-lactalbumin was injected onto the sensor chip on which SIB1 FKBP22 or SN-FKBP22 was immobilized. The dissociation constants, KD, of reduced α-lactalbumin for binding to SIB1 FKBP22 and SN-FKBP22, which were determined by measuring the equilibrium-binding responses at various concentrations of reduced α-lactalbumin, were 6.5 and 24.1 μM, respectively. This result indicates that SN-FKBP22 retains binding ability to a folding intermediate of protein, although its binding affinity is reduced by 3.7-fold compared with that of SIB1 FKBP22.
Figure 6.
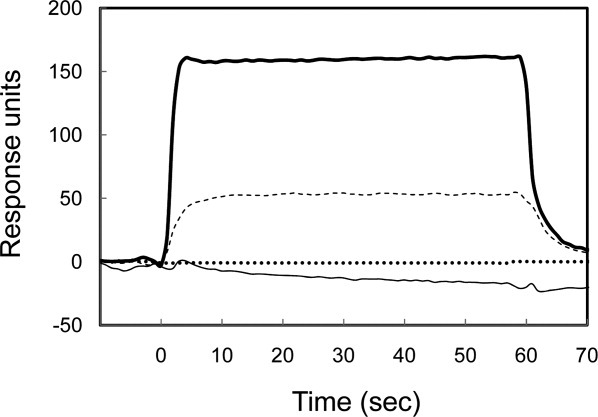
Binding of reduced α-lactalbumin to SIB1 FKBP22 and its derivatives. The sensorgrams from Biacore X showing the binding of reduced α-lactalbumin (100 μM) to immobilized V37R/L41R-FKBP22 (dotted line), SIB1 FKBP22 (thick line), C-domain+ (thin line), and SN-FKBP22 (dashed line) are shown. Injections were done at time zero for 60 s.
Importance of Val-Leu knot for dimerization and folding of N-domain
In this study, we showed that a Val-Leu knot at the hydrophobic core of the dimer interface is important for dimerization of SIB1 FKBP22. This knot is similar to a hydrophobic zipper, if it is compared with a portion of the zipper, in which two hydrophobic residues make a pairwise interaction. Hydrophobic zipper has been reported to be important for dimerization of protein mediated by the interaction of α-helices.14,15 It is also important for folding of globular proteins, which is initiated by the formation of a coiled coil structure at the hydrophobic core.15 Although a variety of hydrophobic residues are found at the zipper position, Leu is most frequently found (Leu zipper).16,17 Leu-Val zipper, in which Val is substituted for Leu in every two zipper positions, has also been reported to be important for dimerization of protein.18,19
The observation that the N-domain of this protein is almost fully disordered suggests that dimerization is required for folding of the N-domain. Association of two N-domains with a partially folded structure may induce native structure, as reported for staphylococcal nuclease.20 Intermolecular association reflects specific interaction between a hydrophobic surface of one partially folded N-domain and that of the other. In this respect, Val-Leu knot of SIB1 FKBP22 is essential for facilitating the specific interaction between two monomers. We have previously shown that a V-shaped structure is required for efficient binding to a folding intermediate of protein and maximal PPIasepro activity.7 However, the monomeric mutant protein of SIB1 FKBP22, NNC-FKBP22, in which N-domain is tandemly repeated through a flexible linker, retains approximately 15% of the binding ability to a folding intermediate of protein and PPIasepro activity of SIB1 FKBP22.7 NNC-FKBP22 is expected to assume a similar structure to that of SIB1 FKBP22, except that one “arm” of V-shaped structure (α3-helix and C-domain from one of the two monomers) is removed. In contrast, V37R/L41R-FKBP22 exhibits much lower binding affinity to a folding intermediate of protein and PPIasepro activity than NNC-FKBP22. These proteins differ in the conformation of the N-domain. The N-domain of V37R/L41R-FKBP22 is mostly disordered, while that of NNC-FKBP22 is folded. We propose that folding of the N-domain is a minimum requirement for binding to a protein substrate, while optimum function of SIB1 FKBP22 is achieved by the formation of a V-shaped dimeric structure. In fact, SN-FKBP22, which is folded into a similar structure to that of the N-domain of SIB1 FKBP22 but does not assume a V-shaped structure, retains 30% of the binding affinity of SIB1 FKBP22 to a folding intermediate of protein.
Materials and Methods
Protein preparation
All proteins were prepared in a His-tagged form. Plasmid pSN-FKBP22, used to overproduce SN-FKBP22, was constructed by ligating the DNA fragment amplified by PCR into the NdeI-BamHI sites of pET28a. Plasmid pSIB1, used to overproduce a His-tagged form of SIB1 FKBP22,3 was used as a template. The sequences of the 5′ and 3′ PCR primers are 5′-AGAGAGAATTCATATGTCAGATTTGTTCAG-3′ (the NdeI site is underlined) and 5′-CGGGATCCCGTTATAAAC GACGACTAAT-3′ (the BamHI site is underlined), respectively. Plasmid pV37R/L41R, used to overproduce V37R/L41R-FKBP22, was constructed using the PCR overlap extension method.21 Plasmid pSIB1 was used as a template. The sequence of the 5′ PCR primer is the same as that used to construct plasmid pSN-FKBP22. The sequence of the 3′ PCR primer is 5′-GGCCACTGGATCCAACTACAGCAATTCTCA-3′ (the BamHI site is underlined). The mutagenic primers were designed such that both codons for Val37 (GTT) and Leu41 (CTT) are changed to CGT for Arg. The DNA fragment amplified by PCR was ligated into the NdeI-BamHI sites of pET28a. PCR was performed with GeneAmp PCR system 2400 (Applied Biosystems) using KOD polymerase (Toyobo). The nucleotide sequences of the genes encoding the mutant proteins were confirmed using a Prism 310 DNA sequencer (Applied Biosystems). All oligonucleotides were synthesized by Hokkaido System Science.
Overproduction and purification of SN-FKBP22 and V37R/L41R-FKBP22 were carried out as described previously for SIB1 FKBP22.3 Selenomethionine-substituted SN-FKBP22 was overproduced using methionine-auxotrophic E. coli strain B834(DE3)pLysS in a defined medium.22 The purification procedures were the same as those for the native protein. SIB1 FKBP22, N-domain+, and C-domain+ were overproduced and purified as described previously.3,4 Production levels of the recombinant proteins in E. coli cells and their purities were analyzed by SDS-PAGE23 using 15% polyacrylamide gel, followed by staining with Coomassie Brilliant Blue R250. Protein concentrations of SIB1 FKBP22 and its derivatives were determined from the UV absorption on the basis that the absorbance at 280 nm of a 0.1% (1 mg mL−1) solution is 0.68 for SIB1-FKBP22 and V37R/L41R-FKBP22, 0.75 for C-domain+, and 0.17 for SN-FKBP22. These values are calculated by using ɛ = 1576 M−1 cm−1 for Tyr and 5225 M−1 cm−1 for Trp at 280 nm.24
Crystallization and data collection
Hexagonal crystals of native SN-FKBP22 were obtained in drops containing 0.1 M Na/K phosphate (pH 6.2) with 20% (w/v) PEG-1000 and 0.2 M NaCl after one-week incubation at 4°C. Native crystal of SN-FKBP22 was first used to collect diffraction data that extended to 1.9 Å resolution. The crystal was cryoprotected by soaking it in the reservoir solution containing 18% (v/v) ethylene glycol. The mounted crystal was flash-frozen in a nitrogen gas stream at 100 K. Diffraction data were collected at BL38B1 beam line in SPring-8, Hyogo, Japan. A total 180 images were collected with an exposure time of 20 s per image and oscillation angle of 1.0°. Nevertheless, we could not obtain the phases by molecular replacement method using the available homologous structures.
Phasing and refinement
To facilitate phasing, selenomethionine-substituted SN-FKBP22 (Se-Met SN-FKBP22) was produced and crystallized in the same condition with that of the native protein. Three-wavelength MAD data extending to 1.9 Å were collected in BL44XU beam line in SPring-8. Data indexing, integration, and scaling were done using HKL2000 suite with anomalous flag, followed by experimental phasing using SHELX suite25 in HKL2MAP GUI.26 Several cycles of structure refinement were done using Refmac27 and Coot.28 The model has been refined to 1.9 Å resolution with Rwork 20.39 and Rfree 26.42%. The crystal belongs to space group P3221 with one dimer in the asymmetric unit. The refinement statistics are given in Table I. The figures were prepared using PyMol.
Table I.
Data Collection and Refinement Statistic
| Data collection | λ1 (peak) | λ2 (inflection) | λ1 (remote) |
|---|---|---|---|
| Resolution range (Å) | 30.00–1.90 | 30.00–1.90 | 30.00–1.90 |
| Outer shell (Å) | 1.93–1.90 | 1.93–1.90 | 1.93–1.90 |
| Wavelength (Å) | 0.97888 | 0.97919 | 0.96500 |
| Reflections | |||
| Total | 87,939 | 90,221 | 97,029 |
| Unique | 7712 | 7739 | 7764 |
| Redudancy | 5.40 (5.30) | 5.40 (5.30) | 5.40 (5.30) |
| Completeness (%) | 98.30 (100) | 98.70 (100) | 98.90 (100) |
| I/σ2 | 18.18 (2.62) | 19.81 (1.92) | 19.02 (1.48) |
| Rmerge (%)a | 11.40 (42.3) | 10.60 (53.0) | 10.90 (73.5) |
| Refinement | |
|---|---|
| Space group | P3221 |
| Unit cell dimensions (Å) | a = b = 39.70 c = 101.90 |
| (°) | α = β = 90.00 γ = 120.00 |
| Resolution range (Å) | 34.38–1.90 |
| Data cutoff (F/σ (F)) | None |
| Number of reflections working | 7299 |
| R-factor/R-free (%)b | 23.39/27.60 |
| Solvent content (%) | 49 |
| Mathews coefficient | 2.4 |
| Number of atoms/asymmetric unit | |
| Protein | 493 |
| Water | 28 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.008 |
| Bond angles (°) | 1.02 |
| B-factor (Å) | |
| Main chain bonded atoms | 27.0 |
| Side chain bonded atoms | 35.4 |
| Ramachandran analysis (%) | |
| Number of residues in favored region | 58 (92.1%) |
| Number of residues in allowed regions | 5 (7.9%) |
| Number of residues in outlier region | 0 (0.0%) |
Rmerge = ∑∴Ihkl — <Ihkl>∴/∑ Ihkl, where Ihkl is the intensity measurement for reflection with indices hkl and <Ihkl> is the mean intensity for multiply recorded reflections.
Rfree was calculated using 5% of the total reflections chosen randomly and omitted from refinement.
Protein data bank accession number
The coordinates and structural factors have been deposited in the Protein Data Bank under accession code 3B09.
Enzymatic activity
The PPIase activity for peptide (PPIasepep) and protein (PPIasepro) substrates were determined at 10°C and pH 7.8 using N-succinyl-Ala-Leu-Pro-Phe-p-nitroanilide (Suc-ALPF-pNA) (Wako Pure Chemical) and RNase T1 (Funakoshi) as a substrate, respectively, as described previously.3
Circular dichroism (CD)
The far-UV and near-UV CD spectra were measured at 10°C and pH 8.0 on a J-725 automatic spectropolarimeter (JASCO), as described previously.4 The mean residue ellipticity, θ, which has units of deg cm2 dmol−1, was calculated by using an average amino acid molecular mass of 110.
Differential scanning calorimetry (DSC)
The DSC measurement was performed on a high-sensitivity VP-DSC controlled by the VPVIEWER™ software package (Microcal) at a scan rate of 1°C min−1. The sample was dissolved in 20 mM sodium phosphate (pH 8.0) at approximately 1.0 mg mL−1. Prior to the measurement, the protein solution was filtered through 0.22 μm pore size membranes and then degassed in a vacuum. The reversibility of thermal denaturation was verified by reheating the sample.
Surface plasmon resonance
Binding of reduced α-lactalbumin to SIB1-FKBP22 and its derivatives was analyzed at 10°C and pH 8.0 by surface plasmon resonance using the Biacore X instrument (Biacore), as described previously.7,8 The dissociation constant, KD, was determined using steady-state affinity program of BIAevaluation Software (Biacore) from the plot of the equilibrium binding responses as a function of the concentrations of reduced α-lactalbumin.
Acknowledgments
The synchrotron radiation experiments were performed at the beam lines BL38B1 and BL44XU in the SPring-8 with the approval of the Japan Synchrotron Radiation Research Institute (Proposal numbers 2010A1158, 2010A6915). The study represents a portion of the dissertation submitted by Cahyo Budiman to Osaka University in partial fulfillment of the requirement for his PhD.
References
- 1.Gothel SF, Marahiel MA. Peptidyl-prolyl cis-trans isomerases, a superfamily of ubiquitous folding catalysts. Cell Mol Life Sci. 1999;55:423–436. doi: 10.1007/s000180050299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jacob RP, Schmid FX. Energetic coupling between native-state prolyl isomerization and conformational protein folding. J Mol Biol. 2008;377:1560–1575. doi: 10.1016/j.jmb.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 3.Suzuki Y, Haruki M, Takano K, Morikawa M, Kanaya S. Possible involvement of an FKBP family member protein from a psychrotrophic bacterium Shewanella sp. SIB1 in cold-adaptation. Eur J Biochem. 2004;271:1372–1381. doi: 10.1111/j.1432-1033.2004.04049.x. [DOI] [PubMed] [Google Scholar]
- 4.Suzuki Y, Takano K, Kanaya S. Stabilities and activities of the N- and C-domains of FKBP22 from a psychrotrophic bacterium overproduced in E. coli. FEBS J. 2005;272:632–642. doi: 10.1111/j.1742-4658.2004.04468.x. [DOI] [PubMed] [Google Scholar]
- 5.Riboldi-Tunnicliffe A, Konig B, Jessen S, Weiss MS, Rahfeld J, Hacker J, Fischer G, Hilgenfeld R. Crystal structure of Mip, a prolylisomerase from Legionella pneumophila. Nat Struct Biol. 2001;8:779–783. doi: 10.1038/nsb0901-779. [DOI] [PubMed] [Google Scholar]
- 6.Saul FA, Arie JP, Vulliez-le Normand NB, Kahn R, Betto NM, Bentley GA. Structure and functional studies of FkpA from Escherichia coli, a cis/trans peptidyl-prolyl isomerase with chaperone activity. J Mol Biol. 2004;335:595–608. doi: 10.1016/j.jmb.2003.10.056. [DOI] [PubMed] [Google Scholar]
- 7.Budiman C, Bando K, Angkawidjaja C, Koga Y, Takano K, Kanaya S. Engineering of monomeric FK506-binding protein 22 with peptidyl prolyl cis-trans isomerase: importance of V-shaped dimeric structure for binding to protein substrate. FEBS J. 2009;276:4091–4101. doi: 10.1111/j.1742-4658.2009.07116.x. [DOI] [PubMed] [Google Scholar]
- 8.Suzuki Y, Win OY, Koga Y, Takano K, Kanaya S. Binding analysis of a psychrotrophic FKBP22 to a folding intermediate of protein using surface plasmon resonance. FEBS Lett. 2005;579:5781–5784. doi: 10.1016/j.febslet.2005.09.067. [DOI] [PubMed] [Google Scholar]
- 9.Ramm K, Pluckthun A. High enzymatic activity and chaperone function are mechanistically related features to the dimeric E. coli peptidyl-prolyl-isomerase FkpA. J Mol Biol. 2001;310:485–498. doi: 10.1006/jmbi.2001.4747. [DOI] [PubMed] [Google Scholar]
- 10.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 11.Hu K, Galius V, Pervushin K. Structural plasticity of peptidyl–prolyl isomerase sFkpA is a key to its chaperone function as revealed by solution NMR. Biochemistry. 2006;45:11983–11991. doi: 10.1021/bi0607913. [DOI] [PubMed] [Google Scholar]
- 12.Wu CS, Ikeda K, Yang JT. Ordered conformation of polypeptides and proteins in acidic dodecyl sulfate solution. Biochemistry. 1981;20:566–570. doi: 10.1021/bi00506a019. [DOI] [PubMed] [Google Scholar]
- 13.Scholz C, Stoller G, Zarnt T, Fischer G, Schmid FX. Cooperation of enzymatic and chaperone functions of trigger factor in the catalysis of protein folding. EMBO J. 1997;16:54–58. doi: 10.1093/emboj/16.1.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crick FC. The packing of alpha-helices: simple coiled-coils. Acta Crystallogr. 1953;6:689–697. [Google Scholar]
- 15.Mason JM, Arndt KM. Coiled coil domains: stability, specifity, and biological implications. Chembiochem. 2004;5:170–176. doi: 10.1002/cbic.200300781. [DOI] [PubMed] [Google Scholar]
- 16.Landshulz WH, Johnson PF, McKnight SL. The leucine zipper: a hypothetical structure common to a new class of DNA binding proteins. Science. 1988;24:1759–1764. doi: 10.1126/science.3289117. [DOI] [PubMed] [Google Scholar]
- 17.O'shea EK, Klemm JD, Kim PS, Alber T. X-ray structure of the GCN4 Leucine zipper, a two-stranded, parallel coiled coil. Curr Biol. 1991;3:658–667. doi: 10.1126/science.1948029. [DOI] [PubMed] [Google Scholar]
- 18.Kirino H, Aoki M, Aoshima M, Hayashi Y, Ohba M, Yamagishi A, Wakagi T, Oshima T. Hydrophobic interaction at the subunit interface contributes to thermostability of 3-isopropylmalate dehydrogenase from an extreme thermophile, Thermus thermophilus. Eur J Biochem. 1994;220:275–281. doi: 10.1111/j.1432-1033.1994.tb18623.x. [DOI] [PubMed] [Google Scholar]
- 19.Imada K, Sato M, Tanaka N, Katsube Y, Matsuura Y, Oshima T. Three-dimensional structure of a highly thermostable enzyme 3-isopropylmalate dehydrogenase of Thermus thermophilus at 2.2 Å resolution. J Mol Biol. 1991;222:725–738. doi: 10.1016/0022-2836(91)90508-4. [DOI] [PubMed] [Google Scholar]
- 20.Uversky VN, Segel DJ, Doniach S, Fink AL. Association-induced folding of globular protein. Proc Natl Acad Sci USA. 1998;95:5480–5483. doi: 10.1073/pnas.95.10.5480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Horton RM, Cai ZL, Ho SN, Pease LR. Escherichia coli and other species of the Enterobacteriaceae encode a protein similar to the family of Mip-like FK506-binding proteins. BioTechniques. 1990;8:528–535. [Google Scholar]
- 22.Doublie S. Preparation of selenomethionyl proteins for phase determination. Methods Enzymol. 1997;276:523–530. [PubMed] [Google Scholar]
- 23.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 24.Goodwin TW, Morton RA. The spectrophotometric determination of tyrosine and tryptophan in proteins. Biochem J. 1946;40:628–632. doi: 10.1042/bj0400628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sheldrick GM. A short history of SHELX. Acta Crystallogr A. 2008;64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 26.Pape T, Schneider TR. HKL2MAP: a graphical user interface for phasing with SHELX programs. J Appl Cryst. 2004;37:843–844. [Google Scholar]
- 27.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 28.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]