

Abstract
Serine proteases are an abundant class of enzymes that are involved in a wide range of physiological processes and are classified into clans sharing structural homology. The active site of the subtilisin-like clan contains a catalytic triad in the order Asp, His, Ser (S8 family) or a catalytic tetrad in the order Glu, Asp and Ser (S53 family). The core structure and active site geometry of these proteases is of interest for many applications. The aim of this study was to investigate the structural properties of different S8 family serine proteases from a diverse range of taxa using molecular modeling techniques. In conjunction with 12 experimentally determined three-dimensional structures of S8 family members, our predicted structures from an archaeon, protozoan and a plant were used for analysis of the catalytic core. Amino acid sequences were obtained from the MEROPS database and submitted to the LOOPP server for threading based structure prediction. The predicted structures were refined and validated using PROCHECK, SCRWL and MODELYN. Investigation of secondary structures and electrostatic surface potential was performed using MOLMOL. Encompassing a wide range of taxa, our structural analysis provides an evolutionary perspective on S8 family serine proteases. Focusing on the common core containing the catalytic site of the enzyme, the analysis presented here is beneficial for future molecular modeling strategies and structure-based rational drug design.
Keywords: serine protease, SB clan, S8 family, homology, threading, modeling
Background:
Serine proteases are among the most abundant enzymes in nature and are involved in a wide range of biological processes, including digestion, blood clotting, embryo development, signal transduction and the immune response [1]. This diverse class of enzymes is characterized by the presence of three critical amino acids in the catalytic site – serine (Ser), histidine (His) and aspartate (Asp), which is referred to as the catalytic triad. The presence of this catalytic triad in at least three structurally unrelated contexts indicates that it has evolved independently at least three times [2]. The MEROPS database ( http://merops.sanger.ac.uk/) is an exceptional resource for information on proteases that employs a useful classification system [3]. Under this system, families of proteases are classified according to statistically significant similarities in the amino acid sequence. These protease families are further grouped into clans that have dissimilar amino acid sequences, but typically have structural homology and/or the same linear order of catalytic triad residues. The subtilisin-like (SB) clan of serine proteases is comprised of two distinct families, S8 (subtilases) and S53 (sedolisins). The catalytic residues are in the order Asp, His, Ser in the S8 family, but the S53 family contains a catalytic tetrad in the order Glu, Asp and Ser. Only the Ser in both families and the His in family S8 and the Glu in family S53 are in equivalent positions. The Ser in both families is found within a Gly-Thr-Ser-Xaa-Xaa-Xbb-Pro motif (where Xaa is an aliphatic amino acid and Xbb is a small amino acid) [3]. This study focuses solely on the S8 family, which contains the bacterial serine endopeptidase subtilisin and homologs such as proteinase K, kexin and furin. In addition to the distinct catalytic triad, the protease structure typically comprises three layers of seven-stranded β sheets between two layers of α helices. As is the case with all classical serine proteases, the catalytic mechanism involves nucleophilic attack by the Ser hydroxyl group on the carbonyl atom of the subtrate, which is catalyzed by the His imidazole group as a general base (supported by a hydrogen bond to the Asp). The resulting tetrahedral intermediate is stabilized by an oxyanion hole. The His imidazole group transfers the proton to the amine leaving group and the tetrahedral intermediate breaks down to an acylenzyme intermediate. In a second addition-elimination reaction, the acylenzyme is attacked by a water molecule to form a second tetrahedral intermediate. With the protonation of the Ser by the His imidazole group, this intermediate breaks down and the C-terminus of the substrate is released. Most S8 family members are non-specific endopeptidases that preferentially cleave after hydrophobic residues, with some exceptions such as kexin and furin that cleave after dibasic amino acids [2]. S8 family serine proteases have a wide variety of biological functions, but they are particularly involved in nutrition and protein processing. Most notably, studies have shown that they are implicated in various diseases and are therefore a potential target for pharmacological intervention. For example, mutations in human proprotein convertase subtilisin-like kexin type 9 (PCSK9) have been associated with hypercholesterolemia [4] and other potential targets include virulence factors such as the Streptococcus pyogenes C5a peptidase [5]. The S8 family serine proteases have also been very popular candidates for protein engineering and directed evolution, which have yielded commercially successful results [6]. A structural analysis of these proteases, particularly the active site geometry, is of interest for many applications such as the above. Indeed, much of the information we have on this abundant class of enzymes has been deduced from experimental three dimensional (3D) structure analysis [7], but for many family members there is no structural data available. In such cases, in silico molecular modeling is a powerful tool to predict the 3D structure of a protein based on the amino acid sequence. The LOOPP homology modelling server ( http://loopp.org/) is used to computationally build atomically detailed models using a set of candidates as structural templates and then it ranks the best models [8]. In the present study, we investigate the structural properties of different S8 family serine proteases using contemporary molecular modeling techniques. Here we present predicted structures from the extremophilic archaeon Pyrococcus furiosus, the malarial protozoan parasite Plasmodium falciparum, and Asian rice (Oryza sativa). By encompassing a wide range of taxa, our structural analysis provides an evolutionary perspective on this protease family. The potential for structure-based rational drug design and protein engineering, and future molecular modeling strategies is also discussed.
Methodology:
Experimental structure data of SB serine proteases (S8 family) for 9 bacteria, 2 fungi, and 1 animal (Table S1, see supplementary material) were obtained from the Protein Data Bank (PDB, http://www.rcsb.org/pdb). Our in-house modeling software MODELYN [9] was used to analyze structural parameters, including the distance between Cα atoms of the catalytic triad. In addition to the experimental structures, amino acid sequences of S8 family SB serine proteases (Table S1, see supplementary material) for 1 archaeon (Pyrococcus furiosus), 1 protozoan (Plasmodium falciparum), and 1 plant (Oryza sativa) were obtained from the MEROPS protease database ( http://merops.sanger.ac.uk) in FASTA format [3]. Upon unsuccessful homology-based structure prediction using SWISS-MODEL (due to less than 30% sequence similarity with known experimental structures), these sequences were submitted to the LOOPP server [8] for threading based structure prediction. This analysis reported a ranked list of 10 possible structure predictions (Tables S2, S3 and S4, see Supplementary material) for each of the protease sequences, including match scores, sequence identity (%) and the extent of sequence coverage (%). Predicted structures were superposed with respect to a selected set of Cα atoms and a suitable starting scaffold was determined. Root mean square deviation (RMSD) values helped to identify the common segments, corresponding to the structurally conserved regions. For further refinement of the model, PROCHECK was used to check the distribution of φ- ψ dihedral angles and eliminate Ramachandran outliers. After side chain regeneration using SCRWL [10], the general structural parameters of the refined model, such as deviations of bond lengths, bond angles from standard values, overall atom clashscores and rotamer outliers were validated against experimental structure data using MOLPROBITY [11] and MODELYN. The ribbon structure and electrostatic potential surface of the structures were determined by MOLMOL [12]. To determine sequence conservation between species, CLUSTALW [13] was used for multiple sequence alignment and PEPSTATS [14] was used to analyze amino acid composition.
Results and Discussion:
For the P. furiosus, P. falciparum and O. sativa proteases (Tables S2, S3 and S4, see supplementary material), the threading based server LOOPP predicted 10 structures for each sequence from 10 different PDB experimental structures (for O. sativa, only the first 5 probable structures were considered). The best matched structures for each showed high confidence scores ranging from 4.7 to 9.0 and sequence identity ranging from 20 to 30%, with best length coverage between 61 and 93%. For P. furiosus, the matched structures were superposed with respect to a selected set of Cα atoms (36%), with the structure 1EA7 having the best score of 4.73 (RMSD values were between 0.332 and 0.746 Å, which helped to identify common segments corresponding to structurally conserved regions). From these superposed structures, the variable loop regions were identified on the starting scaffold derived from 1EA7. For P. falciparum, structures were superposed with respect to selected Cα atoms (41%) with the structure 1MEE having the highest score of 3.07 (RMSD values between 0.298 and 0.642 Å). For O. sativa were superposed with respect to selected Cα atoms (34%), with the structure 1XF1 having the best score of 6.3 (RMSD values were between 0.431 and 0.612 Å). After this regularization, the overall backbone conformations of the predicted structures were measured to identify and eliminate Ramachandran outliers (Figure 1 and Tables S5, see supplementary material). The general structural parameters of experimental and predicted structures were comparable (Tables S6, see supplementary material), which validated the threading based modeling. Superposition of selected SB proteases on the representative X-ray structure (1MEE) of the Bacillus pumilus protease found that 22 to 44% of the Cα atoms superposed with a RMSD below 1Å (Tables S7, see supplementary material). The superposed structures showed highly conserved core structures with large variation in loops outside the core (data not shown). The mean Asp, His, Ser Cα atom distances in the experimentally determined structures were 7.4 ± 0.06, 8.7 ± 0.04 and 10.0 ± 0.03 Å respectively. The small standard deviations indicated that the structural environment around the catalytic triad was highly conserved. The mean values of the predicted structures were 7.3 ± 0.09, 9.0 ± 0.30 and 10.3 ± 0.19 Å respectively, which was in good agreement with the values above. Multiple sequence alignment (Figure 2) confirmed sequence conservation of the catalytic triad residues at Asp32, His64, and Ser221. In addition to the previously described GlyThr-Ser-Xaa-Xaa-Xbb-Pro motif (where Xaa is an aliphatic amino acid and Xbb is a small amino acid) at Ser221 [3], our analysis indicated a Xaa-Xaa-Asp-Xcc-Gly-Xaa motif (where Xcc is a small polar amino acid) at the Asp32 and a His-Gly-ThrXdd-Xcc motif (where Xdd is either His or Arg) at His64. Other highly conserved residues contribute to important structural features, such as the Asn155 (not shown) involved in forming the oxyanion hole. The protease model from P. furiosus had 6 α- helical segments and 7 short β-sheets. The surface electrostatic potentials around the catalytic site were mostly negative with a patch of positive potential above the catalytic Ser residue (Figure 3). In comparison with other species, the P. furiosus protease had a higher proportion of aromatic residues (12.5%) and less smaller amino acids (59.3%). These distinctive features may be associated with increased stabilization and hyperthermophilic adaptation [15, 16], which could be utilized for protein engineering strategies. The protease model from P. falciparum had 9 α-helical segments and 6 β-sheets. The pattern of surface electrostatic potential was very different from others analyzed, with the surface containing mostly neutral regions around the catalytic site (Figure 4). This malarial protease potentially mediates one or more of the serine protease activities associated with parasite erythrocyte invasion [17] and therefore the unique neutral regions around the catalytic site could be utilized in rational anti-malarial drug design. The Oryza sativa SB protease model had 6 α-helical segments and 7 β-sheets. The electrostatic potentials around the catalytic triad residues were mostly negative with a positive region near the His of the catalytic triad (Figure 5). This protease is expressed in the seed and seedling shoots of the rice plant [18] and could potentially have a role in signal transduction or a role in fungal resistance like its tobacco ortholog [19]. This protease is a potential candidate for protein engineering strategies to improve rice crops.
Figure 1.
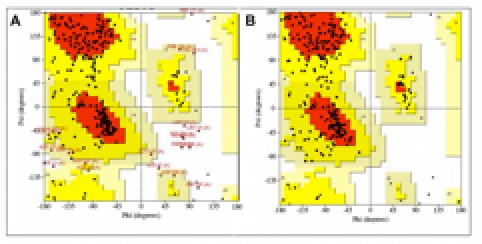
Representative Ramachandran plot of φ-ψ dihedral angles of a modeled SB serine protease structure before and after backbone refinement. PROCHECK was used to check the distribution of φ-ψ dihedral angles and eliminate Ramachandran outliers in each modeled protease structure (A, before; B, after refinement). Residues whose φ-ψ pairs fell outside the most favourable (red) and additional allowed (yellow) zones are annotated in red. Representative Ramachandran plots from the P. falciparum modeling are shown.
Figure 2.
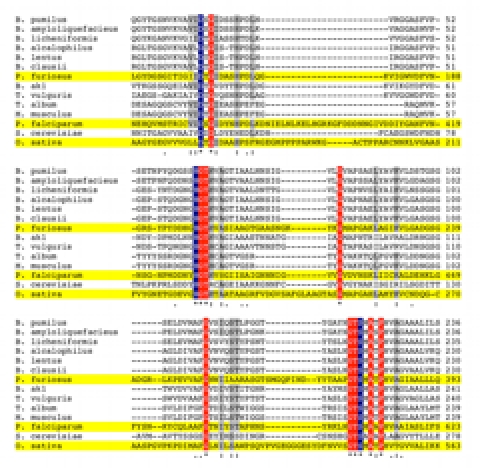
Multiple amino acid sequence alignment of SB serine proteases. CLUSTALW was used to align amino acid sequences of SB serine proteases with experimentally determined and predicted 3D structures (highlighted in yellow). Only the regions showing the conserved catalytic residues Asp (D), His (H), and Ser (S) are shown. Amino acid residues with 100% conservation (*) between aligned sequences are either highlighted in blue (catalytic residues) or red (other). Other residues showing high (:) conservation (highlighted in gray) or medium (.) conservation are also indicated.
Figure 3.
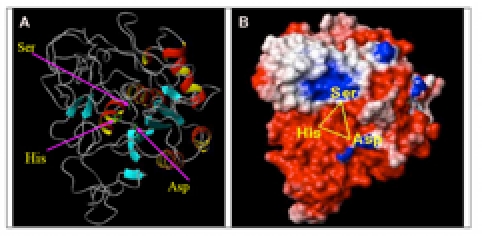
Modeled SB protease structure from Pyrococcus furiosus (PMDB ID: PM0075943). A) Ribbon model showing beta-sheets are shown in (light blue) and with arrow directed to C-terminus, alpha-helices (red and yellow), turn/loops (gray), and catalytic triad residue side chains (green sticks). B) Surface electrostatic potential model showing negative (red), positive (blue), and neutral (white) charges.
Figure 4.
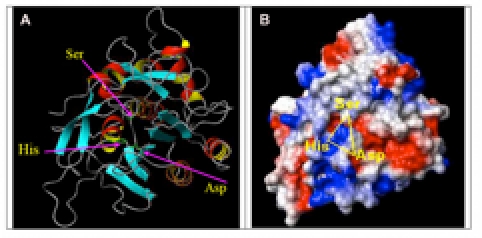
Modeled SB protease structure from Plasmodium falciparum (PMDB ID: PM0075941). A) Ribbon model showing beta-sheets are shown in (light blue) and with arrow directed to C-terminus, alpha-helices (red and yellow), turn/loops (gray), and catalytic triad residue side chains (green sticks). B) Surface electrostatic potential model showing negative (red), positive (blue), and neutral (white) charges.
Figure 5.
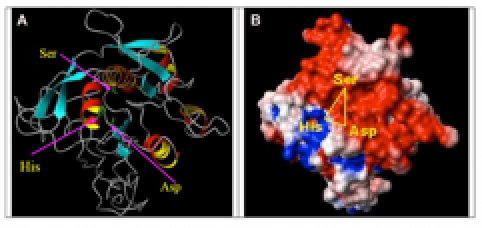
Modeled SB protease structure from Oryza sativa (PMDB ID: PM0075942). A) Ribbon model showing beta-sheets are shown in (light blue) and with arrow directed to Cterminus, alpha-helices (red and yellow), turn/loops (gray), and catalytic triad residue side chains (green sticks). B) Surface electrostatic potential model showing negative (red), positive (blue), and neutral (white) charges.
The following predicted structures are available in the Protein Model Database (PMDB) ( http://mi.caspur.it/PMDB/):
1. SB serine protease from Pyrococcus furiosus (PMDB ID: PM0075943) 2. SB serine protease from Plasmodium falciparum (PMDB ID: PM0075941) 3.SB serine protease from Oryza sativa (PMDB ID: PM0075942)
Conclusion:
The structural geometry of the catalytic core was highly conserved across the diverse range of taxa analyzed and this was reflected in the sequence conservation motifs flanking the catalytic triad residues. Evolutionary divergence was exhibited by large variation in secondary structure features outside the core, differences in overall amino acid distribution, and unique surface electrostatic potential patterns between species. These features are probably associated with environmental adaptation, subcellular localisation, and functional diversity of the different protease orthologs. Indeed, the higher proportion of aromatic residues in the extremophilic archaeon P. furiosus protease potentially provides stabilization [16] and the negatively charged residues around the catalytic site could also confer stabilization [15]. The mostly neutral surface electrostatic potential pattern around the catalytic site of the P. falciparum protease was very different from others analyzed. Significantly, as this protease is potentially associated with malarial erythrocyte invasion [17], the unique neutral regions around the catalytic site could be utilized in rational anti-malarial drug design [20].
Supplementary material
Acknowledgments
Authors A.L. and C.M. are grateful for the funding and infrastructural support provided by the Indian Institute of Chemical Biology, Kolkata, West Bengal, India. Authors E.J.R. and A.C. acknowledge the support given by the Department of Pathology, University of Otago, Dunedin, the Health Research Council (E.J.R.), and the National Research Centre for Growth and Development (A.C.), New Zealand.
Footnotes
Citation:Laskar et al, Bioinformation 7(5): 239-245 (2011)
References
- 1.L Hedstrom. Curr Protoc Protein Sci. 2002;21:10. doi: 10.1002/0471140864.ps2110s26. [DOI] [PubMed] [Google Scholar]
- 2.MJ Page, E Di Cera. Cell Mol Life Sci. 2008;65:1220. doi: 10.1007/s00018-008-7565-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.ND Rawlings, et al. Nucleic Acids Res. 2010;38:D227. doi: 10.1093/nar/gkp971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.M Abifadel, et al. Nat Genet. 2003;34:154. [Google Scholar]
- 5.Q Cheng, et al. Infect Immun. 2002;70:2408. doi: 10.1128/IAI.70.5.2408-2413.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.PN Bryan. Biochim Biophys Acta. 2000;1543:203. doi: 10.1016/s0167-4838(00)00235-1. [DOI] [PubMed] [Google Scholar]
- 7.CS Wright, et al. Nature. 1969;221:235. [Google Scholar]
- 8.J Meller, R Elber. Proteins. 2001;45:241. [Google Scholar]
- 9.C Mandal, et al. 1998 MODELYN: A molecular modelling program, version PC-1.0. Indian copyright No. 9/98. [Google Scholar]
- 10.AA Canutescu, et al. Protein Sci. 2003;12:2001. [Google Scholar]
- 11.IW Davis, et al. Nucleic Acids Res. 2004;32:W615. [Google Scholar]
- 12.R Koradi, et al. J Mol Graph. 1996;14:29. [Google Scholar]
- 13.JD Thompson, et al. Nucleic Acids Res. 1994;22:4673. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.P Rice, et al. Trends Genet. 2000;16:276. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 15.O Dym, et al. Science. 1995;267:1344. [Google Scholar]
- 16.RJ Siezen, et al. Protein Eng. 1991;4:719. doi: 10.1093/protein/4.7.719. [DOI] [PubMed] [Google Scholar]
- 17.F Hackett, et al. Mol Biochem Parasitol. 1999;103:183. doi: 10.1016/s0166-6851(99)00122-x. [DOI] [PubMed] [Google Scholar]
- 18.H Yamagata, et al. Biosci Biotechnol Biochem. 2000;64:1947. doi: 10.1271/bbb.64.1947. [DOI] [PubMed] [Google Scholar]
- 19.H Kinal, et al. Plant Cell. 1995;7:677. [Google Scholar]
- 20.B Turk, et al. Nat Rev Drug Discov. 2006;5:785. doi: 10.1038/nrd2092. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.