

Abstract
We previously showed that widespread expression of Nras G12D/G12D from its endogenous locus in mice leads to an acute myeloproliferative disease (MPD) with a complete penetrance, whereas bone marrow-specific expression of Nras G12D/G12D in recipient mice did not result in sustained MPD phenotypes but 100% penetrant acute T-cell lymphoblastic leukemia/lymphoma (TALL). Such a phenotypic switch also is seen in the case of endogenous oncogenic Kras. Two possibilities might account for this observation and they are not necessarily mutually exclusive. First, the MPD phenotypes in primary Nras G12D/G12D mice might be a transient phenomenon attributable to microenvironmental factors that do not necessarily imply the potential for long-term maintenance in a hematopoietic-cell autonomous manner. Second, it is likely that MPD phenotypes are maintained by genetically altered hematopoietic stem cells (HSCs). Nras G12D/G12D signaling might substantially alter HSC behaviors (e.g., induce their proliferative exhaustion) so that these HSCs no longer sustain MPD phenotypes to a lethal stage in recipient mice. Here, we show some preliminary results to support the second hypothesis. Our results suggest that different lineages of hematopoietic cells might have differential requirements of HSC activity and Nras G12D signaling during leukemogenesis.
Key words: oncogenic Nras, HSCs, leukemogenesis, MPD, TALL, BALL
Nras G12D/G12D Mice Develop a Lethal MPD without pIpC Injections
We first hypothesized that the MPD phenotypes in primary Nras G12D/G12D mice are likely complicated by systemic interferon (IFN)-mediated responses, induced Nras G12D/G12D expression in non-hematopoietic cells, and the simultaneous expression of Nras G12D/G12D in 80–90% of myeloid cells. To test this hypothesis, we generated compound Cre-Lox mice (LSL Nras G12D/G12D; Mx1-Cre; abbreviated as Nras G12D/G12D) as described previously in reference 1. Some of these mice were treated with polyinosinicpolycytidylic acid (pIpC) to induce widespread expression of oncogenic Nras from its endogenous locus (n = 10), while the others were left alone without pIpC treatment (n = 5). It is known that endogenous IFN production induces leaky Cre expression from the IFNα/β-inducible Mx1 promoter without pIpC stimulation.2,3 Because bone marrow cells are most sensitive to IFN, leaky Cre expression leads to oncogene expression primarily in a fraction of bone marrow cells. Such cells show a growth advantage over uninduced cells. They quickly become dominant in bone marrow and drive MPD development, as described in the case of oncogenic Kras.3 However, the MPD phenotypes developed in Kras G12D mice without pIpC induction are significantly weaker than those injected with pIpC, indicating that systemic interferon-mediated responses, induced Kras G12D expression in non-hematopoietic cells, and/or the simultaneous expression of Kras G12D in 80–90% of myeloid cells play a positive role in promoting MPD development.
We analyzed Nras G12D/G12D mice with or without pIpC treatment four weeks after pIpC treatment and at a moribund stage (Fig. 1). At the four-week time point, peripheral blood samples were collected and complete blood count was performed (Fig. 1A). The average white blood cell count in Nras G12D/G12D mice with pIpC injectionselevated ∼two-fold over that in controls, whereas this number increased ∼four-fold in Nras G12D/G12D mice without pIpC injections. At a moribund stage, the control and diseased mice were sacrificed humanely and hematopoietic tissues were collected for analysis (Fig. 1B and C). We observed marked splenomegaly in Nras G12D/G12D mice with pIpC injections (∼seven-fold over controls), and this phenotype was more pronounced in Nras G12D/G12D mice without pIpC injections (∼24-fold over controls) (Fig. 1B). Extramedullary hematopoiesis was evident with varying proportions of granulocytic, monocytic, erythroid, and megakaryocytic lineage cells in both groups of animals (data not shown). Flow cytometric analysis using myeloid specific markers revealed a predominantly granulocytic/monocytic myeloid hyperplasia in the spleen and bone marrow of diseased mice (Fig. 1C and reviewed in ref. 1). Although the percentage of CD4- CD8- cells significantly increased in the thymi of Nras G12D/G12D mice without pIpC injections, the thymic size was indistinguishable from that of controls (data not shown). Unlike Kras G12D mice, systemic IFN-mediated responses and/or induced Nras G12D/G12D expression in non-hematopoietic cells via pIpC injections appear to play a negative role in MPD development. Therefore, our results suggest that the MPD phenotypes in primary Nras G12D/G12D mice are likely due to the cell-autonomous expression of Nras G12D/G12D in hematopoietic cells. We believe that Nras G12D/G12D mice without pIpC injections serve as a more physiological model of MPD and provide a powerful system to assess thetherapeutic efficacy of novel molecular agents.
Figure 1.

Nras G12D/G12D mice develop a lethal MPD without pIpC injections. Compound mice (LSL Nras G12D/G12D; Mx1-Cre; abbreviated as Nras G12D/G12D) were injected with or without pIpC (+pIpC or w/o pIpC) as described previously in reference 1. Mx1-Cre mice served as controls. (A) Four weeks after pIpC treatment, peripheral blood was collected for complete blood count. Results are presented as the average of white blood cell (WBC) count + standard deviation. *p < 0.05. (B and C) At a moribund stage, diseased mice were sacrificed humanely for analysis. (B) Results are presented as the average of spleen weights + standard deviation. *p < 0.01. (C) Flow cytometric analysis of bone marrow and spleen cells isolated from a representative Nras G12D/G12D mouse without pIpC treatment using myeloid lineage specific markers. Debris and unlysed red blood cells (low forward scatter) and dead cells (propidium iodide positive) were excluded from analysis. Percentages of cells in top two quadrants are indicated on the density plots.
Recipient Mice Transplanted with a Higher Dose of Nras G12D/G12D Cells Develop Lethal Malignancies in Several Lineages of Hematopoietic Cells
We previously found that when transplanted with 2.5 × 105 bone marrow cells (CD45.2+) isolated from Nras G12D/G12D mice along with an equal number of wild-type competitor cells (CD45.1+), 0 out of 64 recipient mice developed a sustained MPD.1 Moreover, we noticed that the transient MPD phenotypes always diminished after Nras G12D/G12D HSC activity disappeared as demonstrated by the disappearance of donor-derived CD45.2 cells in peripheral blood over time (data not shown). Thus, we hypothesized that MPD phenotypes are maintained by genetically altered HSCs and Nras G12D/G12D signaling might substantially alter HSC behaviors (e.g., induce their proliferative exhaustion) so that these HSCs no longer sustain MPD phenotypes to a lethal stage in recipient mice. To test this hypothesis, we increased donor cell number to 1 × 106 Nras G12D/G12D bone marrow cells with an equal number of wild-type competitor cells. If our hypothesis is correct, the increased number of Nras G12D/G12D HSCs may be able to sustain the development of a lethal MPD in recipient mice.
At this higher dose of HSCs, all the recipient mice died of lethal hematopoietic malignancies (n = 18) 4–8.5 mo after transplant (Fig. 2A). This disease latency is not significantly different from that reported at a lower cell dose.1 We analyzed hematopoietic phenotypes in diseased animals. Fourteen mice developed MPD (Fig. 2B), similar to the chronic MPD developed in recipient mice transplanted with Nras G12D/+ bone marrow cells (Fig. 2C). Clearly, at its endogenous level, Nras G12D/G12D does not promote the transformation of chronic MPD to acute myeloid leukemia. It does, however, significantly shorten the disease latency compared with Nras G12D/+ (6–24 mo after transplant).1 Four recipient mice developed a thymic T-cell disease closely resembling TALL (Fig. 2B). Interestingly, unlike the TALL developed in recipient mice with a lower cell dose, the TALL developed at a higher cell dose were CD44 negative (Fig. 2D). Three recipient mice developed a B-cell disease in thymus and lymph nodes, closely resembling acute B-cell lymphoblastic leukemia/lymphoma (BALL) (Fig. 2B and E). This disease has not been found in recipient mice transplanted with either Nras G12D/+ cells4 or Nras G12D/G12D cells at a lower cell dose,1 suggesting that BALL requires both Nras G12D/G12D signaling and sustained HSC activity. We examined the immunophenotypes of the malignant B cells and found that they were CD43+/− CD19+ CD25− IgM+/− (data not shown). The development of BALL is not a total surprise as we previously observed abnormal B-cell development in 1 out of 10 recipient mice transplanted with Kras G12D cells.5 However, this mouse died of a lethal MPD closely resembling juvenile myelonomocytic leukemia and TALL ∼3.5 mo after transplant and did not develop a lethal B-cell disease in a timely manner. In contrast, recipient mice transplanted with Nras G12D/G12D cells developed a lethal MPD and/or TALL significantly more slowly, which allowed BALL development. It is notable that some of the MPD animals simultaneously developed TALL or BALL.
Figure 2.

Recipient mice transplanted with a higher dose of Nras G12D/G12D cells develop lethal malignancies in several lineages of hematopoietic cells. Lethally irradiated mice (CD45.1+) were transplanted with 1 × 106 total bone marrow cells of Nras G12D/G12D mice along with same number of competitor cells. (A) Kaplan-Meier survival curves of reconstituted mice. Cumulative survival is plotted against days after transplantation. The range of lethality for each malignancy is labeled above the time scale. MPD, myeloproliferative disease; TA LL, acute T-cell lymphoblastic leukemia/lymphoma; BALL, acute B-cell lymphoblastic leukemia/lymphoma. (B) Disease distribution patterns in recipient mice transplanted with Nras G12D/G12D cells. Note: some MPD animals simultaneously developed TA LL or BALL. (C–E) Flow cytometric analysis of different hematopoietic tissues isolated from various diseased animals using lineage specific markers. Debris and unlysed red blood cells (low forward scatter) and dead cells (propidium iodide positive) were excluded from analysis. Percentages of defined populations of cells are indicated on the density plots.
The development of highly penetrant MPD in recipient mice transplanted with a higher dose of Nras G12D/G12D cells supports our second hypothesis. Currently, we are actively investigating how Nras G12D/G12D signaling affects HSC function.
Different Lineages of Hematopoietic Cells Show Distinct Requirements of HSC Activity and Nras G12D Signaling during Leukemogenesis
Based on our results, we propose a model to illustrate a distinct requirement of genetically altered HSCs and oncogene dose in different lineages of hematopoietic cells during leukemogenesis (Fig. 3). In myeloid cells, development of MPD requires sustained HSC activities but Nras G12D/+ is sufficient to initiate MPD. Doubling this oncogene dose to Nras G12D/G12D at its endogenous level accelerates MPD development but does not promote its transformation to an acute phase. In the T-cell compartment, development of TALL does not require sustained HSC activities but does need stronger oncogene signaling. Thus, Nras G12D/G12D promotes TALL much more efficiently than Nras G12D/+ and in rare incidences of TALL initiated by Nras G12D/+, the oncogene signaling is often upregulated through unknown mechanisms.1 In the B-cell lineage, development of BALL requires both sustained HSC activities and strong oncogene signaling. Thus, only a fraction of recipient mice transplanted with a higher dose of Nras G12D/G12D cells develop BALL. Finally, we would like to point out that our model does not exclude the possibility that the interactions among different lineages of hematopoietic cells might also play an important role during leukemogenesis.
Figure 3.
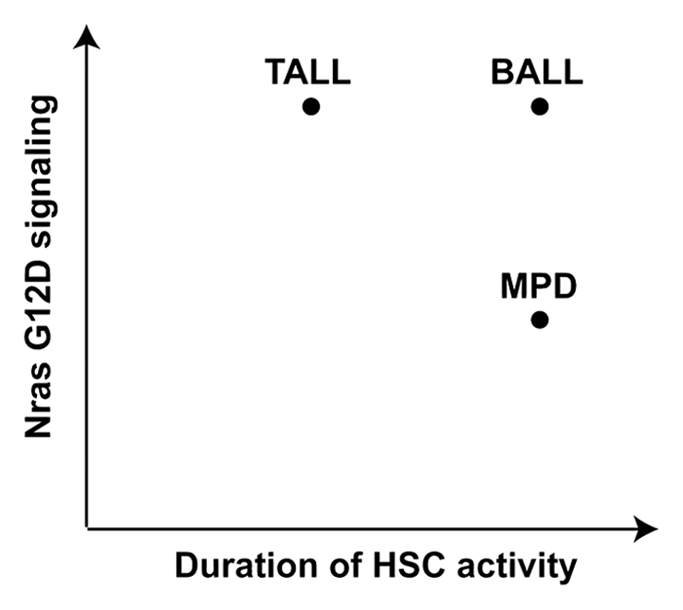
Different lineages of hematopoietic cells have distinct requirements of HSC activity and Nras G12D signaling during leukemogenesis.
Acknowledgments
We are grateful to Drs. Kevin Haigis and Tyler Jacks for providing us the conditional oncogenic Nras mice. We declare no competing financial interests. This work was supported by a Howard Temin Award from the National Cancer Institute, a Shaw Scientist Award from the Greater Milwaukee Foundation, a research grant from Wendy Will Case Cancer Fund, an ASH Scholar Award from the American Society of Hematology, a V Scholar Award from the V Foundation for Cancer Research, and a pilot project grant from the American Cancer Society Institutional Research Grant to J.Z.
Abbreviations
- BALL
acute B-cell lymphoblastic leukemia/lymphoma
- HSC
hematopoietic stem cell
- IFN
interferon
- MPD
myeloproliferative disease
- TALL
acute T-cell lymphoblastic leukemia/lymphoma
References
- 1.Wang JY, Liu YG, Li ZY, Wang ZD, Tan LX, Ryu MJ, et al. Endogenous oncogenic Nras mutation initiates hematopoietic malignancies in a dose- and cell type-dependent manner. Blood. 2011;118:368–379. doi: 10.1182/blood-2010-12-326058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kühn R, Schwenk F, Aguet M, Rajewsky K. Inducible gene targeting in mice. Science. 1995;269:1427–1429. doi: 10.1126/science.7660125. [DOI] [PubMed] [Google Scholar]
- 3.Chan IT, Kutok JL, Williams IR, Cohen S, Kelly L, Shigematsu H, et al. Conditional expression of oncogenic K-ras from its endogenous promoter induces a myeloproliferative disease. J Clin Invest. 2004;113:528–538. doi: 10.1172/JCI20476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang JY, Liu YG, Li ZY, Du J, Ryu MJ, Taylor PR, et al. Endogenous oncogenic Nras mutation leads to aberrant GM-CSF signaling in granulocytic/monocytic precursors in a murine model of chronic myelomonocytic leukemia. Blood. 2010;116:5991–6002. doi: 10.1182/blood-2010-04-281527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang J, Wang J, Liu Y, Sidik H, Young KH, Lodish HF, et al. Oncogenic Kras-induced leukemogeneis: hematopoietic stem cells as the initial target and lineage-specific progenitors as the potential targets for final leukemic transformation. Blood. 2009;113:1304–1314. doi: 10.1182/blood-2008-01-134262. [DOI] [PMC free article] [PubMed] [Google Scholar]