

Abstract
Brain-derived neurotrophic factor (BDNF) and its receptor tropomyosin-related kinase B (TrkB) are widely expressed in the vertebrate nervous system and play a central role in mature neuronal function. In vitro BDNF/TrkB signaling promotes neuronal survival and can help neurons resist toxic insults. Paradoxically, BDNF/TrkB signaling has also been shown, under certain in vitro circumstances, to render neurons vulnerable to insults. We show here that in vivo conditional deletion of TrkB from mature motor neurons attenuates mutant superoxide dismutase 1 (SOD1) toxicity. Mutant SOD1 mice lacking motor neuron TrkB live a month longer than controls and retain motor function for a longer period, particularly in the early phase of the disease. These effects are subserved by slowed motor neuron loss, persistence of neuromuscular junction integrity and reduced astrocytic and microglial reactivity within the spinal cord. These results suggest that manipulation of BDNF/TrkB signaling might have therapeutic efficacy in motor neuron diseases.
INTRODUCTION
Brain-derived neurotrophic factor (BDNF) is a member of the neurotrophin family of growth factors and promotes neuron survival by activating the receptor tyrosine kinase TrkB (tropomyosin-related kinase B) (1). BDNF is secreted in an activity-dependent manner (2) and has been shown to play a critical role in synaptogenesis (3), dendrite growth (4,5), synaptic plasticity (6,7), learning (8) and in the acquisition of other differentiated features of neurons (9,10). Exogenously provided BDNF can protect neurons from insults relevant to neurological diseases (11–14), raising the possibility that it may have potential as a therapeutic agent in humans.
In addition to its undisputed beneficial actions on neurons, several groups have shown that BDNF-TrkB signaling in vitro can render neurons vulnerable to insults. Administration of BDNF to cortical neuron cultures increases cell death evoked by oxygen–glucose deprivation (15), nitric oxide toxicity (16) and insults relevant to amyotrophic lateral sclerosis (ALS) (17–19). TrkB forms a receptor for BDNF as part of a hetero-dimeric complex with p75 and ligand binding to p75 has been shown to induce cell death (20,21). This raises the possibility that BDNF renders neurons vulnerable to insults by binding to p75. Although a variety of in vitro studies support this contention (22–24), placing the G93A mutant SOD1 (superoxide dismutase 1) mouse (a commonly employed mouse model of familial ALS) on the p75−/− background has only modest beneficial effects (25,26). There is evidence favoring the view that activation of TrkB by BDNF can render neurons vulnerable to insults: (i) cortical neuron cultures from TrkB−/− mice resist the toxic actions of BDNF (27) and (ii) when grown in a cocktail of trophic factors including BDNF, pharmacological inhibition of TrkB activation protects against insults (17). The relevance of these in vitro observations to animals is entirely unknown.
In the present study, we examined the in vivo role of TrkB expression by adult motor neurons in mutant SOD1-induced motor neuron disease. We find that elimination of TrkB from a subpopulation of adult motor neurons and segmental spinal cord interneurons has beneficial effects on animal survival and locomotor/strength function. These beneficial effects are due to preservation of motor neurons and the neuromuscular junctions (NMJs) in mutant SOD1 expressing animals with motor neurons lacking TrkB.
RESULTS
Cre-mediated recombination of TrkB in spinal cord motor neurons and interneurons
We use the Cre-LoxP system to eliminate TrkB from motor neurons. To avoid developmental effects that could complicate interpretation of these experiments, we chose a Cre driver line in which recombination begins around the first month of postnatal life. The vesicular acetylcholine transporter (VAChT) promoter driving Cre expression has been used previously to disable LoxP-flanked genes specifically in motor neurons (28). Using the ROSA 26 reporter line, we confirmed that VAChT-Cre-medicated recombination begins at around 4 weeks of age and reaches its maximum by 6 months of age (Fig. 1A). Based on size, shape and location, it appears that Cre-mediated recombination is occurring mostly in motor neurons (∼50% of all motor neurons) (29) but also in a small population of interneurons. The LoxP-flanked TrkB allele has been shown to readily undergo recombination (30). We chose the G85R mutant SOD1 mice for study because in this mouse strain, the first pathological features are identified at 6 months of age (31). Thus, motor neuron development is likely to be normal in the G85R-SOD1+/−; TrkBLoxP/LoxP; VAChT-Cre+/− mice and any effects of TrkB signaling in mature motor neurons of mice expressing mutant SOD1 could be assessed. Through a five-step breeding strategy (see Materials and Methods), we generated mice in our prime comparison groups with genotypes: G85R-SOD1+/−; TrkBLoxP/LoxP; VAChT-Cre+/− (deleted TrkB in motor neurons from mutant SOD1 mice) or G85R-SOD1+/−; TrkBLoxP/LoxP; VAChT-Cre−/− (intact TrkB in motor neurons from mutant SOD1 mice). Prior to generation of trigenic mice, each primary strain was backcrossed into the C57BL/6 background for at least five generations. All animals discussed from this point forward are homozygous for the floxed TrkB allele (TrkBLoxP/LoxP). In light of this, unless otherwise specified, we will simplify the notation of the genetic background of the mice we studied by eliding this piece of data. Thus, in this short hand, G85R-SOD1+/−; VAChT-Cre+/− mice have the genotype G85R-SOD1+/−; TrkBLoxP/LoxP; VAChT-Cre+/−.
Figure 1.

Cre-mediated TrkB recombination in spinal cord motor neurons. (A) Lumbar spinal cord frozen sections from postnatal day 10, day 26 and 6-month ROSA 26 reporter mice expressing Cre under the control of the VAChT promoter were stained with X-gal and showed increased expression of Cre in adult ventral horn motor neurons. (B) Lumbar spinal cord frozen sections were stained with Neutral Red, and large neurons in the anterior horn were microdissected with PALM system (50×). (C) The same anterior horn was shown to indicate large spinal cord neurons before microdissection (200×). (D) The same anterior horn was shown after microdissection (200×). (E) Single-cell RT-PCR and nested PCR analyses of microdissected spinal cord neurons for TrkB (upper panel), Cre recombinase (middle panel) and ChAT (lower panel) expression. Cre-mediated recombination was seen in ChAT-positive (motor neuron) cells (lanes 1, 3, 12 and 15) as well as ChAT-negative cells (lanes 16, 17 and 19). (F) Summary of ChAT-positive specimens from RT-PCR/nested PCR.
Each LoxP-flanked gene undergoes Cre-mediated recombination with variable efficiency, and effect likely related to chromatin structure. To show that TrkB was eliminated from motor neurons in G85R-SOD1+/−; VAChT-Cre+/− mice, we began with immunological approaches. None of the antibodies we used provided high-quality images in immunological assays. Immunoblots for TrkB using spinal cord lysates from G85R-SOD1+/−; VAChT-Cre+/− versus G85R-SOD1+/−; VAChT-Cre−/− animals were indistinguishable and this is likely to be due to TrkB expression in non-Cre-expressing cells in the TrkBLoxP/LoxP mice. Given these problems, we turned to laser capture microscopy. We dissected individual large neurons in the ventral horn (putative motor neurons) from frozen sections of 8-month-old G85R-SOD1+/−; VAChT-Cre+/− mice using a laser microdissector (Fig. 1B–D). We extracted RNA from lysates of each dissected neuron and performed RT-PCR and nested PCR for TrkB, Cre and choline acetyltransferase (ChAT, a biosynthetic enzyme enriched in motor neurons) (Fig. 1E).
Among 99 specimens examined, 16 specimens were ChAT-positive, Cre-positive and TrkB-negative (Fig. 1E, lanes 1, 3, 12 and 15; 1F), and these cells probably represent motor neurons (expressing ChAT) in which TrkB expression is eliminated when Cre recombinase is expressed. Twelve specimens were ChAT-positive, Cre-positive and TrkB-positive (Fig. 1E, lane 2; 1F) and these cells probably represent motor neurons in which TrkB is expressed even though Cre is also expressed. Eight specimens were ChAT-positive, Cre-negative and TrkB-negative (Fig. 1F) and these cells probably represent motor neurons in which neither TrkB nor Cre is expressed. Finally, 14 neurons were ChAT-positive, Cre-negative and TrkB-positive and these neurons probably represent motor neurons that express TrkB when Cre is lacking (Fig. 1E, lanes 4, 6, 14, 18; 1F). Assuming that the ChAT-expressing, large ventral horn cells studied by laser capture single-cell PCR are motor neurons, we estimate that ∼57% of Cre (+) cells have no demonstrable TrkB mRNA expression and ∼37% of Cre (−) cells have no demonstrable TrkB expression. These observations lead us to conclude that Cre expression in motor neurons promotes deletion of the LoxP-flanked TrkB allele.
Twenty-six specimens were ChAT-negative (putative non-motor neurons) but positive for Cre and/or TrkB. Among them, 7 cells were ChAT-negative, Cre-positive and TrkB-negative (Fig. 1E, lanes 16, 17 and 19), 8 cells were ChAT-negative, Cre-positive and TrkB-positive (Fig. 1E, lanes 10) and 11 cells were ChAT-negative, Cre-negative and TrkB-positive (Fig. 1E, lanes 5 and 8). We suggest that these specimens are derived from interneurons and that TrkB undergoes Cre-mediated recombination in the presence of Cre recombinase although with less than perfect efficiency. We did not detect expression of ChAT, Cre recombinase and TrkB in other 23 specimens (Fig. 1E, lanes 7, 9, 11 and 13). Together, these data demonstrate that Cre-mediated recombination of the LoxP-flanked TrkB allele occurs in subsets of both motor neurons and a subpopulation of interneurons in ventral horn of G85R-SOD1+/−; VAChT-Cre+/− mice.
Elimination of TrkB increases lifespan, slows disease progression and improves locomotor function in G85R SOD1 mice
We began by assessing survival and health of mice with a single genetic manipulation (i.e. VAChT-Cre+/− or TrkBLoxP/LoxP) or mice with two genetic manipulations (VAChT-Cre+/−; TrkBLoxP/LoxP). Both the uni- and bi-genic mice strains lived at least for 18 months without evidence of an adverse effect on health monitored as normal weight gain, fecundity and activity levels (data not shown). We next turned to the trigenic mice. Male G85R-SOD1+/−; VAChT-Cre+/− mice lived for an average 26 days longer than male G85R-SOD1+/−;VAChT-Cre−/− mice (384 ± 7 (n = 15) versus 358 ± 8 days (n = 15), P = 0.033, Fig. 2A). Female G85R-SOD1+/−; VAChT-Cre+/− mice lived for an average of 28 days longer than female G85R-SOD1+/−; VAChT-Cre−/− [363 ± 7 days (n = 21) versus 335 ± 9 days (n = 14), P = 0.037, Fig. 2B]. Thus, conditional deletion of TrkB from adult motor neurons, and a subpopulation of adjacent interneurons, is associated with lifespan extension of mutant SOD mice of both sexes. For the remainder of this report, we focus on male mutant SOD1 mice.
Figure 2.
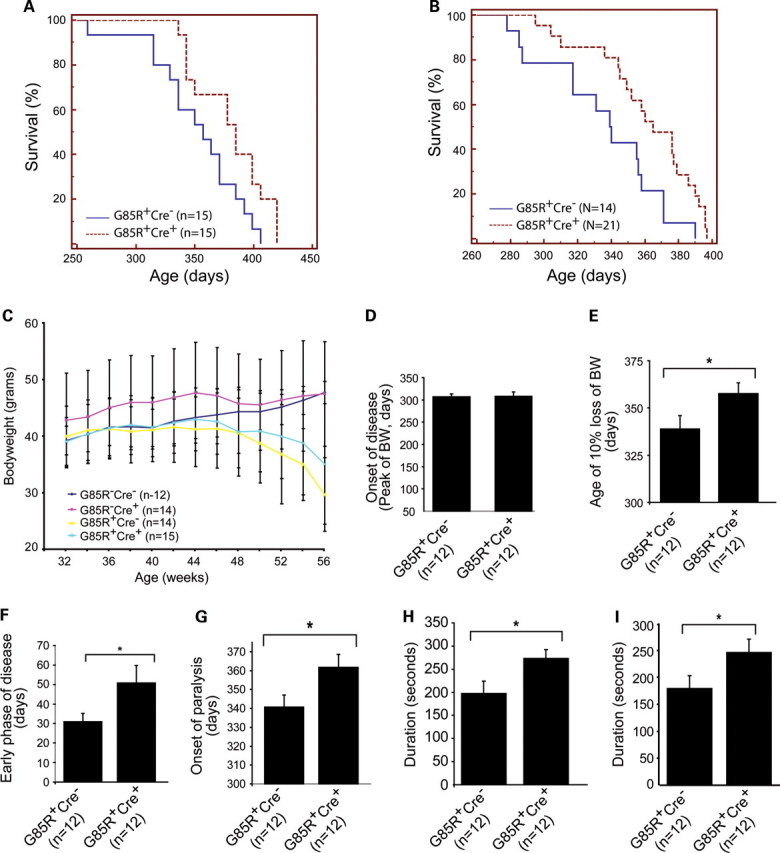
Elimination of TrkB in motor neurons increases lifespan, slows disease progression, delays the onset of paralysis and improves locomotor function in G85R SOD1 mice. (A) Cre-mediated deletion of TrkB in adult motor neurons increases lifespan of male G85R-SOD1+/−; VAChT-Cre+/− mice (384 ± 7 days) compared with G85R-SOD1+/−; VAChT-Cre−/− mice (358 ± 8 days) (P < 0.05). (B) Deletion of TrkB in motor neurons increases lifespan of female G85R-SOD1+/−; VAChT-Cre+/− mice (363 ± 7 days) compared with G85R-SOD1+/−; VAChT-Cre−/− mice (335 ± 9 days) (P < 0.05). (C and D) Conditional deletion of TrkB in motor neurons did not influence age of disease onset of G85R mutant SOD1 mice. (E and F) Progression through (D) and duration (E) of an early disease phase was slower and longer in G85R-SOD1+/−; VAChT-Cre+/− mice. (G) The onset of paralysis was delayed in G85R-SOD1+/−; VAChT-Cre+/− mice (361 ± 7 days) compared with G85R-SOD1+/−; VAChT-Cre−/− mice (340 ± 6 days) (P < 0.05). (H) At the peak of bodyweight, G85R-SOD1+/−;VAChT-Cre+/− mice stayed significantly longer on rotarod beam (272 ± 19 s) than G85R-SOD1+/−; VAChT-Cre−/− mice (197 ± 27 s) (P < 0.05). (I) At the age of 2 weeks after the peak of bodyweight, G85R-SOD1+/−; VAChT-Cre+/− mice continued to stay significantly longer on rotarod beam (246 ± 25 s) than G85R-SOD1+/−; VAChT-Cre−/− mice (179 ± 24 s) (P < 0.05).
We monitored body weight of four groups of male animals: (i) G85R-SOD1−/−; VAChT-Cre−/−, (ii) G85R-SOD1−/−; VAChT-Cre+/−, (iii) G85R-SOD1+/−; VAChT-Cre−/− and (iv) G85R-SOD1+/−; VAChT-Cre+/− (Fig. 2C). By ANOVA, we found no group differences at 46, 48, 50 or 52 weeks of life. Group differences were observed at week 54 (F(3,26) = 3.888, P = 0.02) and at week 56 (F(3.25) = 5.871, P = 0.0035). At week 54, all mouse groups in which G85R SOD1 was expressed were significantly lighter than mice that did not express G85R SOD1 (post hoc analysis with significance set at P<0.05). The same was true at week 56 time point. It is noteworthy that no statistically significant difference in weight was seen between G85R-SOD1+/−; VAChT-Cre−/− and G85R-SOD1+/−; VAChT-Cre+/− mice.
The onset of disease has been defined by the peak of bodyweight curve (32), and the early phase of the clinical motor neuron disease has been defined as the time from the peak of bodyweight to 10% weight loss (33). Previous work suggests that events occurring within motor neurons themselves underlie the ‘initiation phase’ of mutant SOD1 disease (33). We next compared the disease onset and progression between male G85R-SOD1+/−; VAChT-Cre+/− mice and G85R-SOD1+/−;VAChT-Cre−/− mice (n = 12 in each group). When we analyzed our data this way, we found that these two groups of mice had similar disease onset (308 ± 10 versus 306 ± 6 days, G85R-SOD1+/−; VAChT-Cre+/− and G85R-SOD1+/−;VAChT-Cre−/−, respectively, P = 0.460, Fig. 2D). On the other hand, the early phase of the disease was 19 days longer in the G85R-SOD1+/−; VAChT-Cre+/− mice versus the G85R-SOD1+/−; VAChT-Cre−/− mice (358 ± 6 versus 339 ± 7 days, P = 0.027, Fig. 2E and F). We also monitored the onset of paralysis by measuring grip strength. We found that the cumulative probability of the onset of paralysis, as defined by 30% decline of either forelimb or hindlimb grip strength, was significantly delayed by 21 days in G85R-SOD1+/−; VAChT-Cre+/− mice in comparison with the G85R-SOD1+/−; VAChT-Cre−/− mice (361 ± 7 versus 340 ± 6 days, P = 0.037, Fig. 2G). Finally, we evaluated locomotor function using the accelerating rotarod test. When assessed at peak bodyweight, G85R-SOD1+/−; VAChT-Cre+/− mice stayed on the device significantly longer than the G85R-SOD1+/−; VAChT-Cre−/− mice (272 ± 19 versus 197 ± 27 seconds, P = 0.035, Fig. 2H). We also assessed the locomotor function 2 weeks after the peak of bodyweight and again found that G85R-SOD1+/−; VAChT-Cre+/− mice stayed on the device significantly longer than the G85R-SOD1+/−; VAChT-Cre−/− mice (246 ± 25 versus 179 ± 24 s, P = 0.033, Fig. 2I). Together, these observations indicate that the early phase of mutant SOD1 disease is more prolonged and milder in mice with the conditional deletion of TrkB. These observations are consistent with the notion that activation of TrkB regulates biological processes within motor neurons themselves that play a crucial role in the earliest phases of clinical mutant SOD1 disease.
Deletion of TrkB reduces the formation of mutant SOD1 inclusions and ubiquitination in G85R SOD1 mice
The presence of SOD1 immunoreactive inclusions in neuronal cell bodies and processes had been shown as early hallmarks of disease in G85R transgenic mice (31). We next compared the development of SOD1 aggregation in G85R-SOD1+/−;VAChT-Cre+/− and G85R-SOD1+/−; VAChT-Cre−/− mice. We found that prior to the clinical onset, intense and diffusely localized human SOD1 immunoreactivity were seen in cell bodies of ventral motor neurons of both G85R-SOD1+/−;VAChT-Cre+/− and G85R-SOD1+/−; VAChT-Cre−/− mice (Fig. 3A and B, arrows). At this stage, more neuronal processes showing intense immunoreactivity to human SOD1 were seen in G85R-SOD1+/−; VAChT-Cre−/− mice (Fig. 3A, arrowheads) than in G85R-SOD1+/−; VAChT-Cre+/− mice (Fig. 3B). As disease progresses, prominent neuronal processes with intense human SOD1 immunoreactivity increased in abundance in the ventral area of G85R-SOD1+/−; VAChT-Cre−/− (Fig. 3C and E, arrowheads) and G85R-SOD1+/−; VAChT-Cre+/− mice (Fig. 3D and F, arrowheads). Western blot analysis showed similar expression level of human SOD1 in Triton-soluble supernatant of spinal cord lysates from presymptomatic G85R-SOD1+/−; VAChT-Cre−/− and G85R-SOD1+/−;VAChT-Cre+/− mice (Fig. 3G). However, more G85R SOD1 was found in Triton-insoluble pellet in G85R-SOD1+/−;VAChT-Cre−/− mice than in G85R-SOD1+/−;VAChT-Cre+/− mice (Fig. 3H), suggesting that conditional deletion of TrkB decreases aggregation of mutant SOD1 in the early stage of mutant SOD1 disease.
Figure 3.
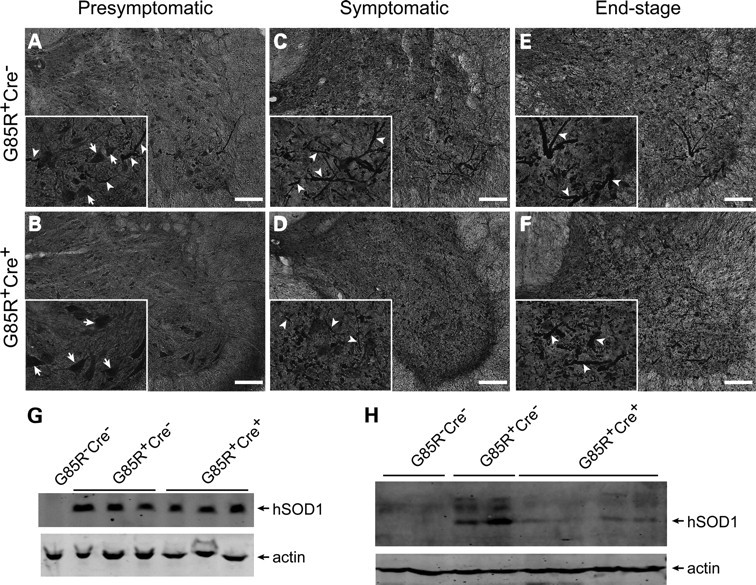
Decreased abundance of mutant SOD1 immunoreactive inclusions in presymptomatic G85R SOD1/TrkB KO mice. (A and B) Immunostaining of presymptomatic spinal cord sections using human SOD1 antibody showed diffuse SOD1 immunoreactivity in ventral horn motor neurons (inset, arrows). Less SOD1 immunoreactive inclusions were seen in presymptomatic G85R-SOD1+/−; VAChT-Cre+/− mice (B) than in G85R-SOD1+/−; VAChT-Cre−/− mice (A and inset, arrowheads). (C–F) Increased amount of SOD1 inclusions were seen in neuronal processes of spinal cord sections from both symptomatic (C and D and insets, arrowheads) and end-stage (E and F and inset, arrowheads) animals. No significant difference in abundance of SOD1 inclusions between G85R-SOD1+/−; VAChT-Cre−/− and G85R-SOD1+/−; VAChT-Cre+/− mice. Scale bars, 100 μm. (G and H) Western blot analysis showed similar expression levels of mutant SOD1 in Triton-soluble lumbar spinal cord lysates of G85R-SOD1+/−; VAChT-Cre−/− and G85R-SOD1+/−; VAChT-Cre+/− mice (G). Less mutant SOD1 was found in Triton-insoluble lumbar spinal cord pellets from G85R-SOD1+/−; VAChT-Cre+/− mice (H).
Another pathological hallmark of ALS is the presence of ubiquitinated inclusions in the perikaryon and proximal axons of surviving spinal motor neurons (34). Next we set out to assess the effects of elimination of motor neuron TrkB on deposition of ubiquitinated proteins in spinal motor neurons. Immunohistochemical staining of lumbar spinal cord sections from mice at different disease stages with ubiquitin antibodies showed that G85R-SOD1−/−; VAChT-Cre−/− and G85R-SOD1−/−;VAChT-Cre+/− control mice had diffuse distribution of ubiquitin immunoreactivity in motor neurons at all ages examined (Fig. 4A–C, D–F, arrows). In presymptomatic G85R-SOD1+/−; VAChT-Cre−/− mice, most ventral horn motor neurons show diffuse ubiquitin staining (Fig. 4G, arrows). Intense ubiquitin inclusions were readily found in some neurons and neuronal processes (Fig. 4G, arrowheads). In presymptomatic G85R-SOD1+/−; VAChT-Cre+/− mice, diffuse ubiquitin staining was seen in spinal motor neurons (Fig. 4J, arrows) with fewer neuronal processes containing ubiquitin inclusions (Fig. 4J, arrowheads). At later disease stage, more neuronal processes containing ubiquitin inclusions were seen in ventral horn area of both G85R-SOD1+/−; VAChT-Cre−/− and G85R-SOD1+/−;VAChT-Cre+/− mice (Fig. 4H and I, K and L, arrowheads).
Figure 4.
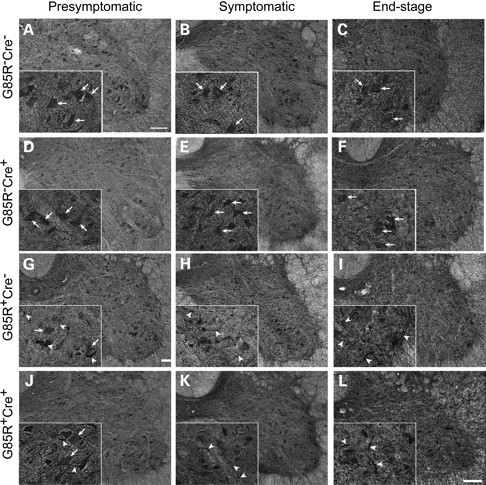
Ubiquitin immunoreactive inclusions in G85R SOD1/TrkB KO mice. (A–F) Immunohistochemical staining of spinal cord sections showed diffuse ubiquitin immunoreactivity in ventral motor neurons from all age groups of G85R-SOD1−/−; VAChT-Cre−/− (A–C and insets, arrows) and G85R-SOD1−/−; VAChT-Cre+/− mice (E and F and insets, arrows). (G–L) More neuronal ubiquitination abnormalities were seen in presymptomatic G85R-SOD1+/−; VAChT-Cre−/− mice (G and inset, arrowheads) than in G85R-SOD1+/−; VAChT-Cre+/− mice (J and inset, arrowheads). No significant difference of ubiquitin-positive inclusions was found at later disease stage (H and I, K and L, insets, arrowheads). Scale bars, 100 μm.
Elimination of TrkB in motor neurons reduces inflammation in G85R SOD1 mice
A number of potential processes could count for the effects of conditional deletion of TrkB on the early phase of mutant SOD1 disease. The cell non-autonomous features of mutant SOD1-induced motor neuron disease have received increasing attention recently. During the clinically manifest phase of the disease, reactive astrocytosis and inflammatory responses are evident. Does the elimination of TrkB from motor neurons influence these cellular responses? To assess this, we immunohistologically examined spinal cords from mice in presymptomatic phase (8-month-old), symptomatic (11-month-old) and end stage of disease for glial fibrillary astrocytic protein (GFAP—a marker for reactive astrocytosis) as an indicator of astroglial response. In both the presymptomatic and symptomatic phase of the disease, the G85R-SOD1+/−;VAChT-Cre+/− mice displayed significantly less GFAP-immunopositive astrocytes in the spinal cord compared with G85R-SOD1+/−; VAChT-Cre−/− mice (Fig. 5A–C, P = 0.0062; Fig. 5D–F, P = 0.026). This was evident by lower power viewing of spinal cord sections and was confirmed by counting the number of GFAP+ cells in the two study groups. Western blot analysis of spinal cord lysates from presymptomatic 8-month-old mice further showed higher expression level of GFAP in G85R-SOD1+/−;VAChT-Cre−/− mice (Fig. 5I).
Figure 5.
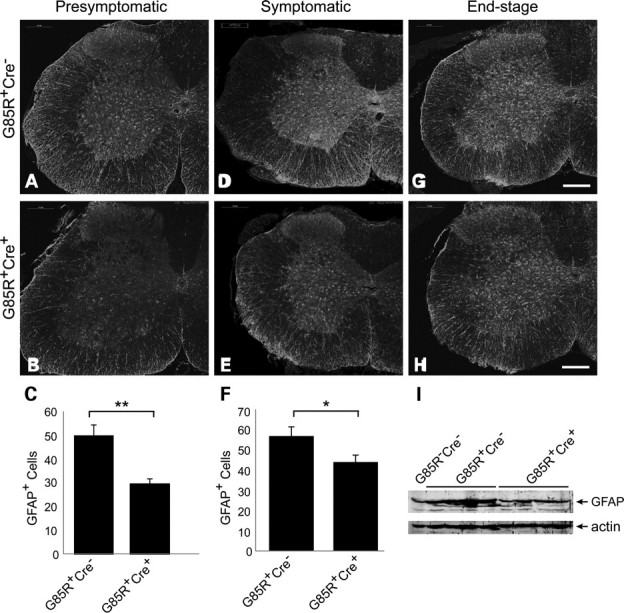
Elimination of TrkB in motor neurons reduces mutant SOD1-associated gliosis. (A–H) Immunostaining of spinal cord sections using GFAP antibody indicated reduced astrocytosis in presymptomatic (B) and symptomatic (D) G85R-SOD1+/−; VAChT-Cre+/− mice compared with age-matched G85R-SOD1+/−; VAChT-Cre−/− mice (A and E). Image analysis confirmed the significance of GFAP signal difference at presymptomatic (C, P < 0.01) and symptomatic (F, P < 0.05) stage. Scale bars, 200 μm. (I) Western blot analysis showed higher level of GFAP in lumbar spinal cords from presymptomatic G85R-SOD1+/−; VAChT-Cre−/− mice.
We used immunoreactivity against ionized calcium-binding adaptor molecule (Iba)-1, a specific marker for activated microglial cells, to detect microglia in the spinal cord. We found that the numbers of Iba1-immunopositive cells were significantly decreased in spinal cords of presymptomatic (8-month-old) (Fig. 6B and C, P = 0.009) and symptomatic G85R-SOD1+/−;VAChT-Cre+/− mice (Fig. 6E and F, P = 0.016) compared with G85R-SOD1+/−; VAChT-Cre−/− mice (Fig. 6A and D). In agreement with this observation, western blot analysis of presymptomatic spinal cord lysates showed lower expression level of Iba1 in G85R-SOD1+/−; VAChT-Cre+/− mice compared with G85R-SOD1+/−; VAChT-Cre−/− mice (Fig. 6I). This result probably indicates that mutant SOD1 expressing motor neurons that lack TrkB survive longer than those that express TrkB, and as a by product of this, cellular responses within the spinal cord parenchyma to damaged motor neurons are delayed.
Figure 6.
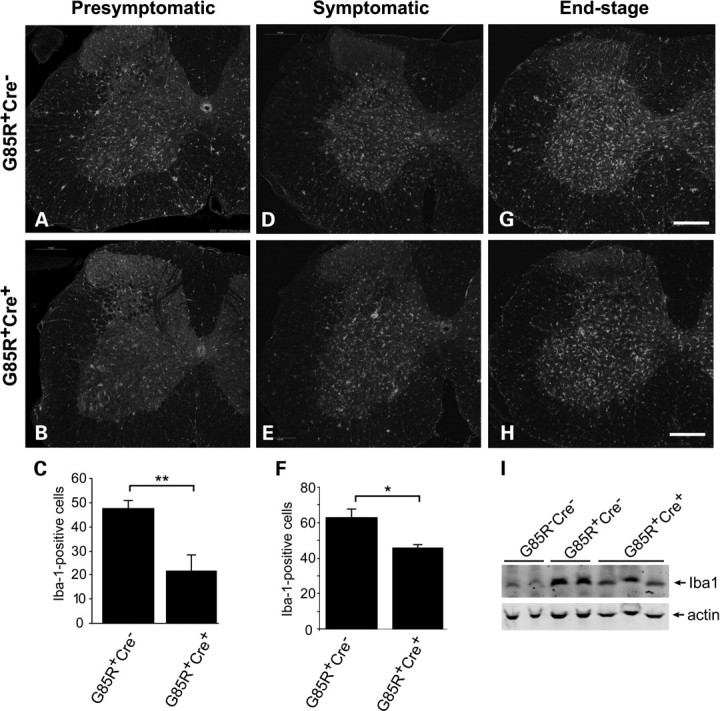
Decreased mutant SOD1-associated neuroinflammation in G85R SOD1/TrkB KO mice. (A–F) Immunostaining of spinal cord sections using Iba-1 antibody showed reduced inflammation in presymptomatic (B) and symptomatic (E) G85R-SOD1+/−; VAChT-Cre+/− mice compared with age-matched G85R-SOD1+/−; VAChT-Cre−/− mice (A and D). Image analysis confirmed the significance of Iba-1 signal difference at presymptomatic (C, P < 0.01) and symptomatic (F, P < 0.05) stage. Scale bars, 200 μm. (I) Western blot analysis showed lower expression level of Iba1 in presymptomatic G85R-SOD1+/−;VAChT-Cre+/− mice compared with G85R-SOD1+/−; VAChT-Cre−/− mice.
Elimination of TrkB delays motor axon degeneration in G85R SOD1 mice
To assess the effects of these manipulations on progression of ventral motor neuron axonal degeneration, we examined and counted axons in the L5 ventral roots. At the 11-month time point, we found obvious loss of axons and degenerating profiles in G85R-SOD1+/−; VAChT-Cre−/− mice (Fig. 7C) in comparison with control groups (Fig. 7A and B). The predominant degenerative features were increased interaxonal space, presence of macrophages and greater variation in axon size (Fig. 7C). In marked distinction, analysis of L5 ventral root from G85R-SOD1+/−; VAChT-Cre+/− mice showed a substantial retention of large-diameter axons (Fig. 7D). Their axons are tightly packed, similar to control G85R-SOD1−/−;VAChT-Cre−/− (Fig. 7A) and G85R-SOD1−/−;VAChT-Cre+/− mice (Fig. 7B). We measured the diameter of axons and then generated size distribution histograms by plotting the number of axons against diameter of axon subdivided into bins. L5 roots from G85R-SOD1−/−; VAChT-Cre−/− animals showed a clear separation of the histograms into two peaks (Fig. 7E). One peak of myelinated fibers is at between 1 and 2 μm diameter (likely represents the axons from γ-motor neurons) and a second peak is in the range of 5–10 μm (likely represents α-motor neuron axons) (Fig. 7E). Elimination of TrkB alone has no effect on axon morphology (Fig. 7B and F). The histograms were shifted to the left, and the total number of large myelinated axons was reduced in G85R-SOD1+/−; VAChT-Cre−/− mice (Fig. 7C and G). In G85R-SOD1+/−; VAChT-Cre+/− mice, there were two clear peaks with size range similar to that of G85R-SOD1−/−;VAChT-Cre−/− animal (Fig. 7H). We counted the remaining axons from L5 ventral roots in three G85R-SOD1−/−;VAChT-Cre−/− mice, three G85R-SOD1+/−; VAChT-Cre−/− mice and six G85R-SOD1+/−; VAChT-Cre+/− mice, and found that there was a significant reduction of large axons (diameter >4 μm) in G85R-SOD1+/−; VAChT-Cre−/− mice (Fig. 7I, 345 ± 19 for G85R-SOD1+/−; VAChT-Cre−/−, 717 ± 14 for G85R-SOD1−/−; VAChT-Cre−/− mice, P < 0.001). Significant preservation of large axons (diameter >4 μm) in L5 ventral roots was seen in G85R-SOD1+/−; VAChT-Cre+/− mice in comparison with G85R-SOD1+/−; VAChT-Cre−/− mice (Fig. 7I, 552 ± 104, P = 0.0039). There is no alteration in the number of small-diameter axons (<4 μm) in L5 ventral roots (Fig. 7J). Examination of L5 ventral roots from 9-month-old animals showed that <1% of large-diameter axons underwent degeneration in both G85R-SOD1+/−; VAChT-Cre−/− and G85R-SOD1+/−; VAChT-Cre+/− mice (Fig. 7K and L). These results indicate that elimination of TrkB from motor neurons in mutant SOD1 mice delays large-diameter putative α-motor neuron axon degeneration.
Figure 7.
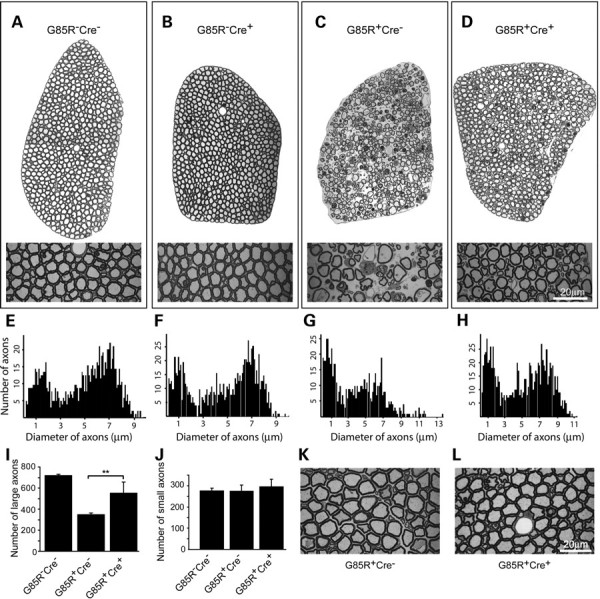
Delayed motor axon degeneration in G85R SOD1/TrkB KO mice. (A–D) Toluidine blue staining of thin sections of L5 ventral root axons from 11-month-old animals showed substantial retention of large-diameter axons in G85R-SOD1+/−; VAChT-Cre+/− mice (D) compared with age- and sex-matched G85R-SOD1+/−; VAChT-Cre−/− mice (C). Elimination of TrkB alone (B) has no effects on axon morphology compared with wild-type control (A). (E–H) Nerve fiber diameter histograms from all the myelinated axons in L5 lumbar ventral roots showed preservation of two clear peaks and no consistent reduction in numbers of large-diameter fibers in the histogram from G85R-SOD1+/−; VAChT-Cre+/− mice (H) compared with G85R-SOD1+/−; VAChT-Cre−/− Mice (G). (I and J) Quantification of remaining axons from L5 lumbar ventral roots showed significant retention of large-diameter axons (>4 μm) in G85R-SOD1+/−; VAChT-Cre+/− mice (I, P < 0.01) compared with G85R-SOD1+/−; VAChT-Cre−/− mice. No change in the numbers of small-diameter axons (<4 μm) was seen (J). (K and L) Examination of 9-month-old animals showed normal appearance of axons in L5 lumbar ventral roots in G85R-SOD1+/−; VAChT-Cre−/− (K) and G85R-SOD1+/−; VAChT-Cre+/− (L) mice. Scale bars, 20 μm.
Elimination of TrkB reduces denervation of NMJs in G85R SOD1 mice
Finally, we examined the NMJs in gastrocnemius, extensor digitorum longus (EDL) and soleus from 10-month-old animals by immunofluorescence labeling. We found that >95% of endplates were innervated in gastrocnemius, EDL and soleus of G85R-SOD1−/−; VAChT-Cre−/− control mice (Fig. 8A–C, J–L). There was no evidence of the NMJ denervation in G85R-SOD1−/−; VAChT-Cre+/− animals (data not shown). At this age, we found that denervation of endplates was 45.9% ± 7.7, 48.7% ± 3.8 and 39.4% ± 8.2 for gastrocnemius, EDL and soleus, respectively, in G85R-SOD1+/−;VAChT-Cre−/− mice (Fig. 8D–F, J–L), and in G85R-SOD1+/−; VAChT-Cre+/− mice, denervation of endplates in gastrocnemius, EDL and soleus was significantly reduced (24% ± 6.2 for gastrocnemius, 32% ± 2% for EDL and 19% ± 5 for soleus) (Fig. 8G–I, J–L). These results indicate that elimination of TrkB from adult motor neurons in mutant SOD1 mice is associated with preservation of the NMJ structure.
Figure 8.

Reduced denervation of NMJs in G85R-SOD1+/−; VAChT-Cre+/− mice. (A–I) Representative images of NMJs from gastrocnemius of 10-month-old animals showed three innervated junctions in wild-type control (A–C), two denervated/one innervated junctions in G85R-SOD1+/−; VAChT-Cre−/− mice (D–F) and one denervated/three innervated junctions in G85R-SOD1+/−; VAChT-Cre+/− (G–I) mice. Scale bars, 50 μm. (J–L) Quantification analysis showed significant reduction of denervated junctions in gastrocnemius (J, P < 0.05), EDL (K, P < 0.01) and soleus (L, P < 0.05) from five G85R-SOD1+/−; VAChT-Cre+/− mice compared with age-matched five G85R-SOD1+/−;VAChT-Cre−/− mice.
VAChT-Cre reduces TrkB in pre-ganglionic sympathetic neurons
In addition to motor neurons, the pre-ganglionic sympathetic neurons in the intermediolateral (IML) column of the thoracic spinal cord are cholinergic. The sympathetic nervous system controls many biological processes, including energy expenditure. This is of interest because mutant SOD1 mice have a hypermetabolic phenotype (35) and part of the control of resting energy expenditure (REE) is mediated by the sympathetic nervous system (36). Pre-ganglionic sympathetic neurons regulate REE through the melanocortin 4 receptor (MC4R) (37), and BDNF/TrkB can be a downstream effector of MC4R signaling (38). These observations raise the possibility that VAChT-Cre-mediated elimination of TrkB from pre-ganglionic sympathetic neurons might correct the metabolic defect in the mutant SOD1 mice and be the basis of the beneficial effects we have described above. In light of this, we asked whether VAChT-Cre was expressed in pre-ganglionic sympathetic neurons. We bred VAChT-Cre mice to the ROSA26-LacZ reporter mice, and performed X-gal histochemistry on slices of the thoracic spinal cord. In 2-month-old animals, we found no cell labeling in the IML (data not shown)—this is consistent with the original observations of Misawa et al. (29). However, when we looked at 8-month-old animals, intense labeling of cells was evident in the IML of the thoracic spinal cord (Fig. 9A). Based on location and polygonal shape, these cells are putative pre-ganglionic sympathetic neurons.
Figure 9.
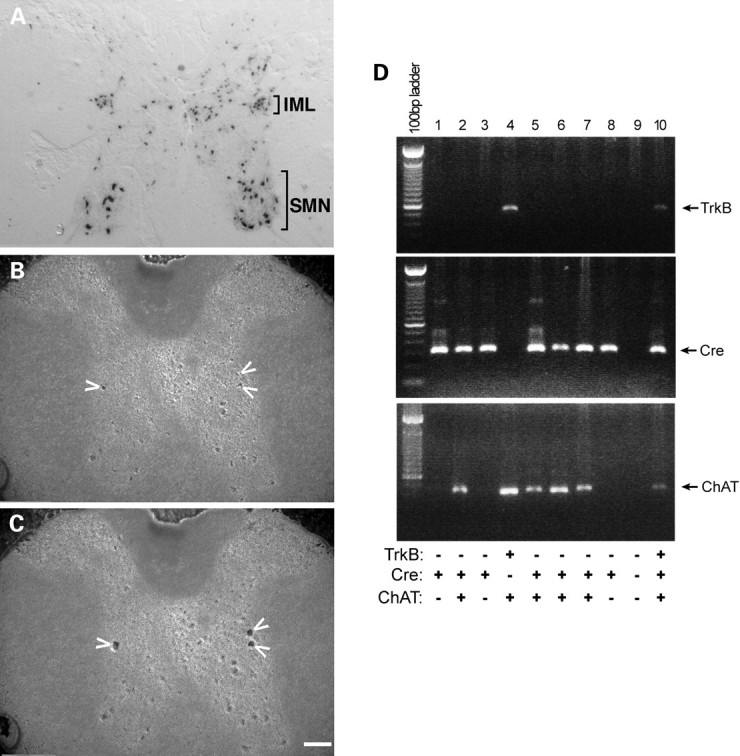
VAChT-Cre reduces TrkB in pre-ganglionic sympathetic neurons. (A) Thoracic spinal cord frozen sections from 8-month-old ROSA 26 reporter mice expressing Cre under the control of the VAChT promoter were stained with X-gal, which showed intense labeling of cells was evident in the IML and in somatic motor neurons (SMN). (B and C) Thoracic spinal cord frozen sections were stained with Cresyl Violet, and neurons in IML (arrowheads) were microdissected with PALM system (50×). Scale bars, 100 μm. (D) Single-cell RT-PCR and nested PCR analyses of microdissected IML neurons for TrkB (upper panel), Cre (middle panel) and ChAT (lower panel) expression.
To determine whether pre-ganglionic sympathetic neurons express TrkB and whether Cre recombinase can lead to its ablation, we performed laser capture single-cell RT-PCR of cells in the IML column (Fig. 9B–D). Analyses of 60 cells led to the identification of 6 that were cholinergic (i.e. they expressed ChAT) (Fig. 9D, lanes 2, 4, 5, 6, 7, 10). Of these six cells, five expressed Cre (Fig. 9D, lanes 2, 5, 6, 7, 10) and four-fifths did not express TrkB (Fig. 9D, lanes 2, 5, 6, 7). A single ChAT(+) Cre(−) cell in the IML column was TrkB(+) (Fig. 9D, lane 4). These observations support the view that pre-ganglionic sympathetic neurons of the IML column are depleted of TrkB in our compound mice. To the extent that the hypermetabolic phenotype of mutant SOD1 mice contributes to motor neuron degeneration, it is possible that the beneficial action of eliminating TrkB from cholinergic neurons is mediated, in part, by the modulatory actions of sympathetic neurons on REE.
DISCUSSION
In this work, we show that reduction of TrkB expression by motor neurons and a subpopulation of segmental spinal cord interneurons has a beneficial effect on a mutant SOD1 mouse model of motor neuron disease. Our genetic manipulation slows the course of the disease (particularly the early phase) and maintains motor function. These effects are likely to be subserved by preservation of motor neurons and NMJs and a reduced astrocytic/inflammatory response within the spinal cord. These observations grew out of prior in vitro work implicating TrkB activation in rendering neurons vulnerable to certain insults (18). To our knowledge, this is the first and only in vivo demonstration that reducing TrkB function can have beneficial effects in any neurodegenerative disease.
The effects we see of reducing motor neuron TrkB expression on mutant SOD1 disease are reasonably comparable with many studies in the literature. Extending survival for 28 days is >13 days offered by anti-CD40L treatment (39) or by inactivation of NADPH oxidase (40), 16 days by transplantation-based astrocyte replacement (41), 11 days by retrograde viral delivery of GDNF (42), but <60 days offered by diminishing mutant SOD1 expression in astrocytes (28), 37 days by IGF-1 treatment (42) or 64 days by ablation mutant SOD1 in progenitors of motor and dorsal root ganglion neurons (33). Similar trends are seen when one frames the results as difference between the experimental group and control as a percent of maximum lifespan. Although there is a clear beneficial effect of eliminating TrkB from motor neurons in the mutant SOD1 mice, why is the effect relatively modest? First, we are at the mercy of the expression of Cre from the VAChT promoter, thus the timing of the recombination event is uncontrolled. This is significant because several studies demonstrate morphological and molecular alterations in motor neurons are present early in the disease process-month(s) before the first overt behavioral abnormality can be found (43,44). It is conceivable that TrkB elimination earlier in the disease process would reap a greater benefit. Second, only a subpopulation of motor neurons both expresses Cre and has deletion of the TrkBLoxP allele. One could reasonably postulate that if more motor neurons lacked TrkB, the beneficial effects would be greater. Third, we do not know whether elimination of TrkB function from other cells besides motor neurons might be important for the neuroprotective activity. It is generally agreed that motor neuron death in the SOD1 model is non-cell autonomous; mutant SOD1 expression in astrocytes and microglia contributes to motor neuron death (33,45). Although there is no evidence that astrocytes/microglia express TrkB (46,47), TrkB is expressed by interneurons in the adult spinal cord (46,48). The importance of antagonizing interneuronal TrkB is unknown. Future studies need to address these important issues.
By what mechanism(s) does elimination of TrkB benefit motor neurons in the mutant SOD1 animals? Although several recent publications expand our traditional view of Trk receptor biology, none provides a compelling answer to the question. For example, Harel et al. (49) show that activation of TrkA can cause cell death through its interaction with the protein product of the cerebral cavernous malformation 2 (CCM2) gene. Although CCM2 binds the juxtamembrane region of TrkA, B and C, ligand-induced cell death is only evoked by activation of TrkA. A different form of Trk receptor-mediated cell death was reported by Nikoletopoulou et al. (50). These investigators show that when TrkA and TrkC are expressed at physiological levels from the tau (Mapt) locus, they can trigger neuron death in a manner that does not rely on activation of the tyrosine kinase domain. Part, but not all, of the neuron death can be ascribed to p75. Despite the high degree of homology among Trk family members, expressing TrkB from the tau locus did not induce neuron death. Thus, although these studies expand our view of the cellular consequences of Trk expression by neurons, they fail to explain why elimination of TrkB might promote motor neuron survival under certain circumstances.
Our in vitro studies of motor neurons grown in compartment cultures indicate that susceptibility to mutant SOD1 toxicity depends on activation of TrkB on axons or dendrites. We also found that the baleful actions of TrkB required de novo protein synthesis (51). BDNF/TrkB signaling is known to induce translation of hundreds of mRNAs (52,53). One set of genes of particular interest (whose expression is induced by BDNF) is the NADPH oxidase subunits, gp91phox, p22phox, p47phox and p67phox (54). NADPH oxidase catalyzes the production of reactive oxygen species, and genetic deletion of NADPH oxidase subunits in the mutant SOD1 animal is strongly neuroprotective (40,55). We propose that BDNF induces the expression of a substantial panel of proteins that together is responsible for the ‘susceptibility-to-insult’ effect.
Mutant SOD1 mice are hypermetabolic as assessed by indirect calorimetry (35) and the same has been observed in a subpopulation of sporadic ALS patients (56,57). Weight loss and low body fat in the mutant SOD1 mice cannot be accounted for on the basis of fever, hyperthyroidism or hyperactivity. These animals eat more than controls, yet lose body weight and are ketotic late in the disease. Although the origin of the hypermetabolic phenotype is not well understood, it is likely to make an important contribution to the disease. One method for re-establishing balance in the energy-intake versus energy-utilization equilibrium is by providing more fuel, and in fact, provision of a high-calorie diet extends the life of the mutant SOD1 mice (35). An alternative approach is to reduce REE, and here MC4Rs are key. MC4Rs are expressed by subpopulations of neurons in the hypothalamus, brain stem and IML of the thoracic spinal cord (58). MC4R−/− mice are severely obese (59) and genetic replacement studies implicate MC4R-expressing pre-ganglionic sympathetic neurons in controlling REE (37). In the hypothalamus, BDNF/TrkB signaling has been shown to be a downstream effector of MC4R's control of metabolism (38). If BDNF/TrkB signaling was an important downstream effector of MC4Rs in the pre-ganglionic sympathetic neurons, then loss of TrkB (as we found) might inhibit REE. The only data we have on this point is that G85R-SOD1+/−; VAChT-Cre+/− mice are not statistically significantly heavier than G85R-SOD1+/−; VAChT-Cre−/− mice at any point in their lives. A more in-depth metabolic analysis of G85R-SOD1 mice with BDNF/TrkB signaling ablated in the pre-ganglionic sympathetic neurons is clearly warranted.
If the current observations are relevant to sporadic ALS, then development of compounds that specifically inhibit TrkB and can cross the blood–brain barrier might have therapeutic utility. This is, admittedly, a counter intuitive notion. Nonetheless, Le Corre et al. (60) found that administration of a derivative of K252a (a broad spectrum tyrosine kinase inhibitor that can antagonize TrkB) called SRN-003-556 had potent therapeutic efficacy in a mouse model of tauopathy. On the other hand, BDNF/TrkB signaling is important for normal brain function and so antagonizing this pathway might adversely affect cognition and mood; proper dosing or local administration to the spinal cord might obviate these potential drawbacks.
BDNF/TrkB signaling plays a critical role in the acquisition and maturation of the most advanced features of neuronal operation. Its effects on activity-dependent modification of synapses and dendritic structure underlie the nuanced interaction of an organism with its environment. We suggest that BDNF/TrkB-mediated events also create a fertile substrate upon which proteotoxic events play out in neurodegeneration.
MATERIALS AND METHODS
Antibodies and western blot
Antibodies used in immunostaining and immunoblot were anti-human SOD1 (ab52950, Abcam), anti-glial fibrillary acidic protein (GFAP, AB5804, Millipore), anti-Iba1 (019-19741, Wako), mouse synaptic vesicle antibody SV2 (Developmental Study Hybridoma Center, University of Iowa), anti-neurofilament (clone 2H3, DSHC, University of Iowa), anti-ubiquitin (Z0458, Dako) and anti-actin (A2066, Sigma). For western blot analysis, spinal cords were homogenized in lysis buffer containing 1% Triton X-100. After spinning at 14 000 r.p.m. for 15 min at 4°C, supernatants were saved for western blotting. Pellets were washed with lysis buffer and solubilized in Laemmli sampling buffer. Protein extracts were resolved by SDS–PAGE, transferred onto nitrocellulose membrane (162-0115, Bio-Rad) and analyzed by standard western blot.
Generation of G85R mutant SOD1;VAChT-Cre+ mice
A five-step breeding strategy was used to generate G85R SOD1; VAChT-Cre+ mice. First, heterozygous mice expressing Cre recombinase under the control of the VAChT promoter (29) were crossed with heterozygous mice containing one floxed TrkB allele (8). The progeny of this cross that were used going forward have the genotype VAChT-Cre+/−; TrkB+/LoxP and they are noted in bold in the F1 generation of ‘step 1’. VAChT-Cre+/−; TrkB+/LoxP mice were identified and intercrossed to produce F1 littermates with nine genotypes. The key progeny of this cross that were used going forward have the genotype VAChT-Cre+/−; TrkBLoxP/LoxP and they are noted in bold in the F1 generation of ‘step 2’. Next, G85R SOD1 transgenic mice were crossed with TrkB+/LoxP mice. The key progeny of this cross that were used going forward have the genotype G85R-SOD1+/−; TrkB+/LoxP and they are noted in bold in the F1 generation of ‘step 3’. G85R-SOD1+/; TrkB+/LoxP mice were identified and intercrossed to produce F1 littermates with nine different genotypes. The key progeny of this cross that were used going forward have the genotype G85R-SOD1+/−; TrkBLoxP/LoxP and they are noted in bold in the F1 generation of ‘step 4’. Lastly, in step 5, male G85R-SOD1+/−; TrkBLoxP/LoxP mice were crossed with female VAChT-Cre+/−; TrkBLoxP/LoxP to generate mice with four different genotypes: (i) G85R-SOD1+/−; VAChT-Cre+/−, (ii) G85R-SOD1+/−; VAChT-Cre−/− , (iii) G85R-SOD1−/−;VAChT-Cre+/− and (iv) G85R-SOD1−/−; VAChT-Cre−/−. All animal experiments were performed in accordance with The Children Hospital of Philadelphia Animal Care and Use Committee guidelines.
 .
.
Genotyping
Genomic DNA was extracted from biopsies of mice tails and analyzed by PCR. The primer set for VAChT-Cre was 5′-ACC TGA TGG ACA TGT TCA GG-3′ and 5′-CGA GTT GAT AGC TGG CTG G-3′. The primer set for detecting mouse and human SOD1 was 5′-CCA TCT TTC TTC CCA GAG CA-3′ (specific for human SOD1), 5′-gct aac tca gga gct ggc ata-3′ (specific for mouse SOD1) and 5′-CAG CAG TCA CAT TGC CCA GGT CTC CA-3′ (for human and mouse SOD1). The primer set for TrkB was 5′-CCA AGG TGA TCA ACA GCC CAA GTC-3′ and 5′-CCT TGA TGA TGG TTG CAC CCA CAC-3′.
Analysis of disease onset and survival
All animals we used were age- and sex-matched. Beginning at 30 weeks, all animals were assessed weekly with a set of behavioral tests (body weight, rotarod test, grip strength) in randomized order. Disease onset was determined as the time when mice reached their peak of bodyweight before denervation-induced muscle atrophy and weight loss. The onset of paralysis was defined as irreversible 30% drop of grip strength. End-stage was defined as the time at which mouse could not right itself within 30 s when placed on its side. The survival data were analyzed by the Kaplan–Meyer lifespan test using MedCalc software.
Rotarod
Each mouse was trained on the rotating beam (diameter 3.5 cm) of a rotarod apparatus (Columbus Instruments, OH, USA) for 2 min before experiment. Then the time for which each mouse could remain on the rotating beam set to accelerate at 0.2 r.p.m./s was measured. Each animal was given three tries and the longest latency to fall was recorded.
Grip strength
The digital grip strength meter (Columbus Instruments, OH, USA) was used to measure the strength of fore- and hindlimbs of mice. Each animal was tested five times at a single age and the three maximum values obtained were averaged.
Immunohistochemistry and evaluation of gliosis
Mice were sacrificed at the indicated ages, perfused with 4% paraformaldehyde and spinal cords were dissected. After postfixation in 4% paraformaldehyde for 2 h, spinal cords were sectioned at 50 μm. For immunohistochemistry, sections were incubated with 0.3% Triton X-100 for 1 h and blocked in buffers containing 5% goat or horse serum and 1% BSA. Then sections were incubated with primary antibodies overnight at cold room. After washes, sections were incubated with fluorescence-labeled secondary antibody for 1.5 h. For the ABC method, sections were treated with 1% H2O2 for 30 min to inactivate endogenous peroxidase prior primary antibody incubation. After primary antibody incubation, sections were incubated with biotinylated secondary antibody (Vector Laboratories, Burlingame, CA, USA). Staining was developed by incubation in ABC complex (Vectastain ABC Kit; Vector Laboratories) followed by incubation in DAB solution.
Laser capture microscopy
Single-cell isolation from frozen spinal cord tissue was performed with a Positioning Ablation Laser Micromanipulation (PALM) system (Carl Zeiss MicroImaging GmbH). Frozen lumbar spinal cord was first sectioned at 7 μm thickness and transfer to MembraneSlide NF 1.0 PEN (Carl Zeiss). Then slides were treated with 95, 75 and 50% of ethanol for 30 s, respectively. After stained with Neutral Red or hematoxylin and eosin Y solution for 40 s, slides were dehydrated in 50, 75, 95, 2× 100% ethanol (30 s each step) and two times in xylene (5 min). Large motor neurons in the anterior horn were dissected from each slide. Each neuron was collected into single 0.5 ml of Eppendorf tube containing 50 μl of Buffer RLT (Qiagen). All samples were kept at –80°C until use.
Single-cell RT-PCR and nested PCR
Total RNA was extracted from microdissected spinal cord neurons using RNeasy Micro Kit (Qiagen) and subjected to RT-PCR analysis using OneStep RT-PCR Kit (Qiagen). Primer pairs used were 5′-GGA GCA TCT CTC GGT CTA TG-3′ and 5′-GAT AGT AGT CGG TGC TGT AC-3′ for TrkB; 5′-cat ttg ggc cag cta aac at-3′ and 5′-tgc atg atc tcc ggt att ga-3′ for Cre recombinase; 5′-TAT GAT GAC AGG CAA CAA GA-3′ and 5′-GGT TGC TCA TCA GGT ATG TT-3′ for ChAT. The cycling conditions for the RT-PCR reaction were 30 min at 50°C, 15 min at 95°C, 40 cycles of 30 s at 94°C, 1 min at 55°C and 2 min at 72°C, followed by 10 min at 72°C. Nested PCR was conducted on 5 μl of the RT-PCR product under the same conditions with the exception of the annealing temperature of 58°C for TrkB and ChAT and 62°C for Cre. The nested PCR was performed individually for each primer pair in 50 µl. Primer sets used for nested PCR were 5′-AGT TGG CGA GAC ATT CCA AG-3′ and 5′-CCC TAA GGA ACT TGT TGA GG-3′ for TrkB; 5′-ctg att tcg acc agg ttc gt-3′ and 5′-ccc ggc aaa aca ggt agt ta-3′ for Cre; and 5′-GGA AAT GTT CCC CAG AAA CT-3′ and 5′-CTC AGT GCC AGA AGA TGG TT-3′ for ChAT. The PCR products were separated and visualized in an ethidium bromide-stained 2% agarose gel.
NMJ analysis
To study the NMJ, gastrocnemius, EDL and soleus muscles were quickly dissected, pinned in Sylgard-lined dish at resting length and fixed with 4% paraformaldehyde in PBS (pH 7.4) for 15 min at room temperature. Motor endplates were visualized by incubation with Alexa 594-conjugated alpha-bungarotoxin (1:1000, Invitrogen) for 15 min at room temperature. After three washes in PBS, muscles were treated with −20°C methanol for 5 min. Distal axons and their terminals were visualized by incubation with anti-neurofilament (clone 2H3, 1:300) and anti-SV2 (1:300) overnight at room temperature followed by Alexa 488-conjugated secondary antibody (1:1000) incubation. Muscles were filleted and mounted superficial side-up on slides and analyzed using confocal microscopy. For quantification of endplate denervation under fluorescence microscopy, junctions were scored as ‘innervated’ if there was complete overlap between the endplate and the axon terminal, or ‘denervated’ if the endplate was not associated with an axon. Some NMJs showed partial overlap between endplate and terminal. These were also labeled as ‘denervated’. Five mice from each experimental group (G85R-SOD1+/−; VAChT-Cre−/− and G85R-SOD1+/−;VAChT-Cre+/−) were quantified.
Quantification of motor axon degeneration
After perfusion with 2% paraformaldehyde/2% glutaraldehyde, L4 and L5 roots were dissected and post-fixed in the same fixatives for 4 h. Subsequently, tissues were incubated in 1% osmium tetroxide in 0.1 m phosphate buffer for 1 h, washed with PB and dehydrated in series of ethanol. Ventral and dorsal roots were then treated twice with propylene oxide for 5 min, incubated sequentially with Embed 812:propylene oxide mixtures (1:1 and 2:1) and pure Embed 812 mixture (Electron Microscopy Sciences) overnight. Tissues were then embedded in fresh Embed 812 mixture at 60°C for 48 h. Cross-sections of the roots were cut at a thickness of 1 μm, stained with toludine blue and examined by light microscopy. Measurements of large (>4 μm) and small (<4 μm) axon numbers and calibers were made with Image-Pro Plus software (Media Cybernetics, Silver Spring, MD, USA) in cross-sections of L5 ventral root. Multiple overlapping images were captured so that all axons were counted.
FUNDING
This work was supported by the US Public Health Service (NS 052325 and NS 064232) and the ALS Association.
ACKNOWLEDGEMENTS
We thank Don Cleveland for the G85R mutant SOD1 mice and Daniel Martinez from Pathology Core Laboratories of Children's Hospital of Philadelphia for technical support of laser capture microdissection.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Marsh H.N., Scholz W.K., Lamballe F., Klein R., Nanduri V., Barbacid M., Palfrey H.C. Signal transduction events mediated by the BDNF receptor gp 145trkB in primary hippocampal pyramidal cell culture. J. Neurosci. 1993;13:4281–4292. doi: 10.1523/JNEUROSCI.13-10-04281.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tao X., West A.E., Chen W.G., Corfas G., Greenberg M.E. A calcium-responsive transcription factor, CaRF, that regulates neuronal activity-dependent expression of BDNF. Neuron. 2002;33:383–395. doi: 10.1016/s0896-6273(01)00561-x. doi:10.1016/S0896-6273(01)00561-X. [DOI] [PubMed] [Google Scholar]
- 3.Seil F.J., Drake-Baumann R. TrkB receptor ligands promote activity-dependent inhibitory synaptogenesis. J. Neurosci. 2000;20:5367–5373. doi: 10.1523/JNEUROSCI.20-14-05367.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McAllister A.K., Katz L.C., Lo D.C. Neurotrophin regulation of cortical dendritic growth requires activity. Neuron. 1996;17:1057–1064. doi: 10.1016/s0896-6273(00)80239-1. doi:10.1016/S0896-6273(00)80239-1. [DOI] [PubMed] [Google Scholar]
- 5.McAllister A.K., Katz L.C., Lo D.C. Opposing roles for endogenous BDNF and NT-3 in regulating cortical dendritic growth. Neuron. 1997;18:767–778. doi: 10.1016/s0896-6273(00)80316-5. doi:10.1016/S0896-6273(00)80316-5. [DOI] [PubMed] [Google Scholar]
- 6.Akaneya Y., Tsumoto T., Kinoshita S., Hatanaka H. Brain-derived neurotrophic factor enhances long-term potentiation in rat visual cortex. J. Neurosci. 1997;17:6707–6716. doi: 10.1523/JNEUROSCI.17-17-06707.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boulanger L., Poo M.M. Gating of BDNF-induced synaptic potentiation by cAMP. Science. 1999;284:1982–1984. doi: 10.1126/science.284.5422.1982. doi:10.1126/science.284.5422.1982. [DOI] [PubMed] [Google Scholar]
- 8.Minichiello L., Korte M., Wolfer D., Kuhn R., Unsicker K., Cestari V., Rossi-Arnaud C., Lipp H.P., Bonhoeffer T., Klein R. Essential role for TrkB receptors in hippocampus-mediated learning. Neuron. 1999;24:401–414. doi: 10.1016/s0896-6273(00)80853-3. doi:10.1016/S0896-6273(00)80853-3. [DOI] [PubMed] [Google Scholar]
- 9.Balkowiec A., Kunze D.L., Katz D.M. Brain-derived neurotrophic factor acutely inhibits AMPA-mediated currents in developing sensory relay neurons. J. Neurosci. 2000;20:1904–1911. doi: 10.1523/JNEUROSCI.20-05-01904.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jovanovic J.N., Czernik A.J., Fienberg A.A., Greengard P., Sihra T.S. Synapsins as mediators of BDNF-enhanced neurotransmitter release. Nat. Neurosci. 2000;3:323–329. doi: 10.1038/73888. doi:10.1038/73888. [DOI] [PubMed] [Google Scholar]
- 11.Han B.H., Holtzman D.M. BDNF protects the neonatal brain from hypoxic-ischemic injury in vivo via the ERK pathway. J. Neurosci. 2000;20:5775–5781. doi: 10.1523/JNEUROSCI.20-15-05775.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kobayashi N.R., Fan D.P., Giehl K.M., Bedard A.M., Wiegand S.J., Tetzlaff W. BDNF and NT-4/5 prevent atrophy of rat rubrospinal neurons after cervical axotomy, stimulate GAP-43 and Talpha1-tubulin mRNA expression, and promote axonal regeneration. J. Neurosci. 1997;17:9583–9595. doi: 10.1523/JNEUROSCI.17-24-09583.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yan Q., Elliott J., Snider W.D. Brain-derived neurotrophic factor rescues spinal motor neurons from axotomy-induced cell death. Nature. 1992;360:753–755. doi: 10.1038/360753a0. doi:10.1038/360753a0. [DOI] [PubMed] [Google Scholar]
- 14.Sendtner M., Holtmann B., Kolbeck R., Thoenen H., Barde Y.A. Brain-derived neurotrophic factor prevents the death of motoneurons in newborn rats after nerve section. Nature. 1992;360:757–759. doi: 10.1038/360757a0. doi:10.1038/360757a0. [DOI] [PubMed] [Google Scholar]
- 15.Koh J.Y., Gwag B.J., Lobner D., Choi D.W. Potentiated necrosis of cultured cortical neurons by neurotrophins. Science. 1995;268:573–575. doi: 10.1126/science.7725105. doi:10.1126/science.7725105. [DOI] [PubMed] [Google Scholar]
- 16.Ishikawa Y., Ikeuchi T., Hatanaka H. Brain-derived neurotrophic factor accelerates nitric oxide donor-induced apoptosis of cultured cortical neurons. J. Neurochem. 2000;75:494–502. doi: 10.1046/j.1471-4159.2000.0750494.x. doi:10.1046/j.1471-4159.2000.0750494.x. [DOI] [PubMed] [Google Scholar]
- 17.Mojsilovic-Petrovic J., Jeong G.B., Crocker A., Arneja A., David S., Russell D.S., Kalb R.G. Protecting motor neurons from toxic insult by antagonism of adenosine A2a and Trk receptors. J. Neurosci. 2006;26:9250–9263. doi: 10.1523/JNEUROSCI.1856-06.2006. doi:10.1523/JNEUROSCI.1856-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hu P., Kalb R.G. BDNF heightens the sensitivity of motor neurons to excitotoxic insults through activation of TrkB. J. Neurochem. 2003;84:1421–1430. doi: 10.1046/j.1471-4159.2003.01599.x. doi:10.1046/j.1471-4159.2003.01599.x. [DOI] [PubMed] [Google Scholar]
- 19.Fryer H.J., Wolf D.H., Knox R.J., Strittmatter S.M., Pennica D., O'Leary R.M., Russell D.S., Kalb R.G. Brain-derived neurotrophic factor induces excitotoxic sensitivity in cultured embryonic rat spinal motor neurons through activation of the phosphatidylinositol 3-kinase pathway. J. Neurochem. 2000;74:582–595. doi: 10.1046/j.1471-4159.2000.740582.x. doi:10.1046/j.1471-4159.2000.740582.x. [DOI] [PubMed] [Google Scholar]
- 20.Van der Zee C.E., Ross G.M., Riopelle R.J., Hagg T. Survival of cholinergic forebrain neurons in developing p75NGFR-deficient mice. Science. 1996;274:1729–1732. doi: 10.1126/science.274.5293.1729. doi:10.1126/science.274.5293.1729. [DOI] [PubMed] [Google Scholar]
- 21.Yoon S.O., Casaccia-Bonnefil P., Carter B., Chao M.V. Competitive signaling between TrkA and p75 nerve growth factor receptors determines cell survival. J. Neurosci. 1998;18:3273–3281. doi: 10.1523/JNEUROSCI.18-09-03273.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dupuis L., Pehar M., Cassina P., Rene F., Castellanos R., Rouaux C., Gandelman M., Dimou L., Schwab M.E., Loeffler J.P., et al. Nogo receptor antagonizes p75NTR-dependent motor neuron death. Proc. Natl Acad. Sci. USA. 2008;105:740–745. doi: 10.1073/pnas.0703842105. doi:10.1073/pnas.0703842105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vargas M.R., Pehar M., Cassina P., Beckman J.S., Barbeito L. Increased glutathione biosynthesis by Nrf2 activation in astrocytes prevents p75NTR-dependent motor neuron apoptosis. J. Neurochem. 2006;97:687–696. doi: 10.1111/j.1471-4159.2006.03742.x. doi:10.1111/j.1471-4159.2006.03742.x. [DOI] [PubMed] [Google Scholar]
- 24.Pehar M., Cassina P., Vargas M.R., Xie Y., Beckman J.S., Massa S.M., Longo F.M., Barbeito L. Modulation of p75-dependent motor neuron death by a small non-peptidyl mimetic of the neurotrophin loop 1 domain. Eur. J. Neurosci. 2006;24:1575–1580. doi: 10.1111/j.1460-9568.2006.05040.x. doi:10.1111/j.1460-9568.2006.05040.x. [DOI] [PubMed] [Google Scholar]
- 25.Kust B.M., Brouwer N., Mantingh I.J., Boddeke H.W., Copray J.C. Reduced p75NTR expression delays disease onset only in female mice of a transgenic model of familial amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 2003;4:100–105. doi:10.1080/14660820310012745. [PubMed] [Google Scholar]
- 26.Turner B.J., Cheah I.K., Macfarlane K.J., Lopes E.C., Petratos S., Langford S.J., Cheema S.S. Antisense peptide nucleic acid-mediated knockdown of the p75 neurotrophin receptor delays motor neuron disease in mutant SOD1 transgenic mice. J. Neurochem. 2003;87:752–763. doi: 10.1046/j.1471-4159.2003.02053.x. doi:10.1046/j.1471-4159.2003.02053.x. [DOI] [PubMed] [Google Scholar]
- 27.Kim H.J., Hwang J.J., Behrens M.M., Snider B.J., Choi D.W., Koh J.Y. TrkB mediates BDNF-induced potentiation of neuronal necrosis in cortical culture. Neurobiol. Dis. 2003;14:110–119. doi: 10.1016/s0969-9961(03)00103-7. doi:10.1016/S0969-9961(03)00103-7. [DOI] [PubMed] [Google Scholar]
- 28.Yamanaka K., Chun S.J., Boillee S., Fujimori-Tonou N., Yamashita H., Gutmann D.H., Takahashi R., Misawa H., Cleveland D.W. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat. Neurosci. 2008;11:251–253. doi: 10.1038/nn2047. doi:10.1038/nn2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Misawa H., Nakata K., Toda K., Matsuura J., Oda Y., Inoue H., Tateno M., Takahashi R. VAChT-Cre. Fast and VAChT-Cre.Slow: postnatal expression of Cre recombinase in somatomotor neurons with different onset. Genesis. 2003;37:44–50. doi: 10.1002/gene.10224. doi:10.1002/gene.10224. [DOI] [PubMed] [Google Scholar]
- 30.Kramer E.R., Aron L., Ramakers G.M., Seitz S., Zhuang X., Beyer K., Smidt M.P., Klein R. Absence of Ret signaling in mice causes progressive and late degeneration of the nigrostriatal system. PLoS. Biol. 2007;5:e39. doi: 10.1371/journal.pbio.0050039. doi:10.1371/journal.pbio.0050039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bruijn L.I., Becher M.W., Lee M.K., Anderson K.L., Jenkins N.A., Copeland N.G., Sisodia S.S., Rothstein J.D., Borchelt D.R., Price D.L., et al. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron. 1997;18:327–338. doi: 10.1016/s0896-6273(00)80272-x. doi:10.1016/S0896-6273(00)80272-X. [DOI] [PubMed] [Google Scholar]
- 32.Liu J., Shinobu L.A., Ward C.M., Young D., Cleveland D.W. Elevation of the Hsp70 chaperone does not effect toxicity in mouse models of familial amyotrophic lateral sclerosis. J. Neurochem. 2005;93:875–882. doi: 10.1111/j.1471-4159.2005.03054.x. doi:10.1111/j.1471-4159.2005.03054.x. [DOI] [PubMed] [Google Scholar]
- 33.Boillee S., Yamanaka K., Lobsiger C.S., Copeland N.G., Jenkins N.A., Kassiotis G., Kollias G., Cleveland D.W. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006;312:1389–1392. doi: 10.1126/science.1123511. doi:10.1126/science.1123511. [DOI] [PubMed] [Google Scholar]
- 34.Leigh P.N., Whitwell H., Garofalo O., Buller J., Swash M., Martin J.E., Gallo J.M., Weller R.O., Anderton B.H. Ubiquitin-immunoreactive intraneuronal inclusions in amyotrophic lateral sclerosis. Morphology, distribution, and specificity. Brain. 1991;114:775–788. doi: 10.1093/brain/114.2.775. doi:10.1093/brain/114.2.775. [DOI] [PubMed] [Google Scholar]
- 35.Dupuis L., Oudart H., Rene F., Gonzalez de Aguilar J.L., Loeffler J.P. Evidence for defective energy homeostasis in amyotrophic lateral sclerosis: benefit of a high-energy diet in a transgenic mouse model. Proc. Natl Acad. Sci. USA. 2004;101:11159–11164. doi: 10.1073/pnas.0402026101. doi:10.1073/pnas.0402026101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bamshad M., Song C.K., Bartness T.J. CNS origins of the sympathetic nervous system outflow to brown adipose tissue. Am. J. Physiol. 1999;276:R1569–R1578. doi: 10.1152/ajpregu.1999.276.6.R1569. [DOI] [PubMed] [Google Scholar]
- 37.Rossi J., Balthasar N., Olson D., Scott M., Berglund E., Lee C.E., Choi M.J., Lauzon D., Lowell B.B., Elmquist J.K. Melanocortin-4 receptors expressed by cholinergic neurons regulate energy balance and glucose homeostasis. Cell Metab. 2011;13:195–204. doi: 10.1016/j.cmet.2011.01.010. doi:10.1016/j.cmet.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu B., Goulding E.H., Zang K., Cepoi D., Cone R.D., Jones K.R., Tecott L.H., Reichardt L.F. Brain-derived neurotrophic factor regulates energy balance downstream of melanocortin-4 receptor. Nat. Neurosci. 2003;6:736–742. doi: 10.1038/nn1073. doi:10.1038/nn1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lincecum J.M., Vieira F.G., Wang M.Z., Thompson K., De Zutter G.S., Kidd J., Moreno A., Sanchez R., Carrion I.J., Levine B.A., et al. From transcriptome analysis to therapeutic anti-CD40L treatment in the SOD1 model of amyotrophic lateral sclerosis. Nat. Genet. 2010;42:392–399. doi: 10.1038/ng.557. doi:10.1038/ng.557. [DOI] [PubMed] [Google Scholar]
- 40.Wu D.C., Re D.B., Nagai M., Ischiropoulos H., Przedborski S. The inflammatory NADPH oxidase enzyme modulates motor neuron degeneration in amyotrophic lateral sclerosis mice. Proc. Natl Acad. Sci. USA. 2006;103:12132–12137. doi: 10.1073/pnas.0603670103. doi:10.1073/pnas.0603670103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lepore A.C., Rauck B., Dejea C., Pardo A.C., Rao M.S., Rothstein J.D., Maragakis N.J. Focal transplantation-based astrocyte replacement is neuroprotective in a model of motor neuron disease. Nat. Neurosci. 2008;11:1294–1301. doi: 10.1038/nn.2210. doi:10.1038/nn.2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kaspar B.K., Llado J., Sherkat N., Rothstein J.D., Gage F.H. Retrograde viral delivery of IGF-1 prolongs survival in a mouse ALS model. Science. 2003;301:839–842. doi: 10.1126/science.1086137. doi:10.1126/science.1086137. [DOI] [PubMed] [Google Scholar]
- 43.Saxena S., Cabuy E., Caroni P. A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat. Neurosci. 2009;12:627–636. doi: 10.1038/nn.2297. doi:10.1038/nn.2297. [DOI] [PubMed] [Google Scholar]
- 44.Amendola J., Durand J. Morphological differences between wild-type and transgenic superoxide dismutase 1 lumbar motoneurons in postnatal mice. J. Comp. Neurol. 2008;511:329–341. doi: 10.1002/cne.21818. doi:10.1002/cne.21818. [DOI] [PubMed] [Google Scholar]
- 45.Lobsiger C.S., Cleveland D.W. Glial cells as intrinsic components of non-cell-autonomous neurodegenerative disease. Nat. Neurosci. 2007;10:1355–1360. doi: 10.1038/nn1988. doi:10.1038/nn1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Copray S., Kernell D. Neurotrophins and trk-receptors in adult rat spinal motoneurons: differences related to cell size but not to ‘slow/fast’ specialization. Neurosci. Lett. 2000;289:217–220. doi: 10.1016/s0304-3940(00)01305-7. doi:10.1016/S0304-3940(00)01305-7. [DOI] [PubMed] [Google Scholar]
- 47.Zhang J., Huang E.J. Dynamic expression of neurotrophic factor receptors in postnatal spinal motoneurons and in mouse model of ALS. J. Neurol. 2006;66:882–895. doi: 10.1002/neu.20269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang J., Chen D., Gong X., Ling H., Zhang G., Wood A., Heinrich J., Cho S. Cyclic-AMP response element-based signaling assays for characterization of Trk family tyrosine kinases modulators. Neurosignals. 2006;15:26–39. doi: 10.1159/000094385. doi:10.1159/000094385. [DOI] [PubMed] [Google Scholar]
- 49.Harel L., Costa B., Fainzilber M. On the death Trk. Dev. Neurobiol. 2010;70:298–303. doi: 10.1002/dneu.20769. [DOI] [PubMed] [Google Scholar]
- 50.Nikoletopoulou V., Lickert H., Frade J.M., Rencurel C., Giallonardo P., Zhang L., Bibel M., Barde Y.A. Neurotrophin receptors TrkA and TrkC cause neuronal death whereas TrkB does not. Nature. 2010;467:59–63. doi: 10.1038/nature09336. doi:10.1038/nature09336. [DOI] [PubMed] [Google Scholar]
- 51.Jeong G.B., Mojsilovic-Petrovic J., Boccitto M., Kalb R. Signaling events in axons and/or dendrites render motor neurons vulnerable to mutant superoxide dismutase toxicity. J. Neurosci. 2011;31:295–299. doi: 10.1523/JNEUROSCI.4824-10.2011. doi:10.1523/JNEUROSCI.4824-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schratt G.M., Nigh E.A., Chen W.G., Hu L., Greenberg M.E. BDNF regulates the translation of a select group of mRNAs by a mammalian target of rapamycin-phosphatidylinositol 3-kinase-dependent pathway during neuronal development. J. Neurosci. 2004;24:7366–7377. doi: 10.1523/JNEUROSCI.1739-04.2004. doi:10.1523/JNEUROSCI.1739-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Takei N., Kawamura M., Ishizuka Y., Kakiya N., Inamura N., Namba H., Nawa H. Brain-derived neurotrophic factor enhances the basal rate of protein synthesis by increasing active eukaryotic elongation factor 2 levels and promoting translation elongation in cortical neurons. J. Biol. Chem. 2009;284:26340–26348. doi: 10.1074/jbc.M109.023010. doi:10.1074/jbc.M109.023010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kim S.H., Won S.J., Sohn S., Kwon H.J., Lee J.Y., Park J.H., Gwag B.J. Brain-derived neurotrophic factor can act as a pronecrotic factor through transcriptional and translational activation of NADPH oxidase. J. Cell Biol. 2002;159:821–831. doi: 10.1083/jcb.200112131. doi:10.1083/jcb.200112131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Marden J.J., Harraz M.M., Williams A.J., Nelson K., Luo M., Paulson H., Engelhardt J.F. Redox modifier genes in amyotrophic lateral sclerosis in mice. J. Clin. Invest. 2007;117:2913–2919. doi: 10.1172/JCI31265. doi:10.1172/JCI31265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Desport J.C., Preux P.M., Magy L., Boirie Y., Vallat J.M., Beaufrere B., Couratier P. Factors correlated with hypermetabolism in patients with amyotrophic lateral sclerosis. Am. J. Clin. Nutr. 2001;74:328–334. doi: 10.1093/ajcn/74.3.328. [DOI] [PubMed] [Google Scholar]
- 57.Desport J.C., Torny F., Lacoste M., Preux P.M., Couratier P. Hypermetabolism in ALS: correlations with clinical and paraclinical parameters. Neurodegener. Dis. 2005;2:202–207. doi: 10.1159/000089626. doi:10.1159/000089626. [DOI] [PubMed] [Google Scholar]
- 58.Mountjoy K.G., Mortrud M.T., Low M.J., Simerly R.B., Cone R.D. Localization of the melanocortin-4 receptor (MC4-R) in neuroendocrine and autonomic control circuits in the brain. Mol. Endocrinol. 1994;8:1298–1308. doi: 10.1210/mend.8.10.7854347. doi:10.1210/me.8.10.1298. [DOI] [PubMed] [Google Scholar]
- 59.Huszar D., Lynch C.A., Fairchild-Huntress V., Dunmore J.H., Fang Q., Berkemeier L.R., Gu W., Kesterson R.A., Boston B.A., Cone R.D., et al. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell. 1997;88:131–141. doi: 10.1016/s0092-8674(00)81865-6. doi:10.1016/S0092-8674(00)81865-6. [DOI] [PubMed] [Google Scholar]
- 60.Le Corre S., Klafki H.W., Plesnila N., Hubinger G., Obermeier A., Sahagun H., Monse B., Seneci P., Lewis J., Eriksen J., et al. An inhibitor of tau hyperphosphorylation prevents severe motor impairments in tau transgenic mice. Proc. Natl Acad. Sci. USA. 2006;103:9673–9678. doi: 10.1073/pnas.0602913103. doi:10.1073/pnas.0602913103. [DOI] [PMC free article] [PubMed] [Google Scholar]