

Abstract
Background. Zaire ebolavirus was responsible for 2 outbreaks in Democratic Republic of the Congo (DRC), in 1976 and 1995. The virus reemerged in DRC 12 years later, causing 2 successive outbreaks in the Luebo region, Kasai Occidental province, in 2007 and 2008.
Methods. Viruses of each outbreak were isolated and the full-length genomes were characterized. Phylogenetic analysis was then undertaken to characterize the relationships with previously described viruses.
Results. The 2 Luebo viruses are nearly identical but are not related to lineage A viruses known in DRC or to descendants of the lineage B viruses encountered in the Gabon–Republic of the Congo area, with which they do, however, share a common ancestor.
Conclusions. Our findings strongly suggest that the Luebo 2007 outbreak did not result from viral spread from previously identified foci but from an independent viral emergence. The previously identified epidemiological link with migratory bat species known to carry Zaire ebolavirus RNA support the hypothesis of viral spillover from this widely dispersed reservoir. The high level of similarity between the Luebo2007 and Luebo2008 viruses suggests that local wildlife populations (most likely bats) became infected and allowed local viral persistence and reemergence from year to year.
The genus Ebolavirus comprises 4 species: Reston ebolavirus, Côte d’Ivoire ebolavirus (CIEBOV), Sudan ebolavirus (SEBOV), and Zaire ebolavirus (ZEBOV) [1] as well as the recently discovered Bundibugyo Ebola virus (BEBOV) that may be classified as a new species [2]. Genetic diversity among species ranges from 25% to 35%, and Zaire ebolavirus is the most virulent, accounting for 1390 human cases in 13 recorded outbreaks over the preceding 35 years, with a case-fatality rate of ≤90%.
The first recognized ZEBOV epidemic occurred at Yambuku in the Democratic Republic of the Congo (DRC) in 1976 [3]. An isolated case was registered in Tandala, DRC, in 1977 [4], succeeded by a 17-year epidemically silent period before the Mekouka outbreak in Gabon in 1994. Subsequently, Gabon experienced 2 epidemics in 1996 at Mayibout and Booué [5, 6] whereas DRC saw a reappearance of ZEBOV in 1995, at Kikwit [7, 8]. Between 2001 and 2005, the cross-border area between northeast Gabon and northwest Republic of Congo (Gabon-RC) was hit by 5 ZEBOV outbreaks [9, 10]: Mekambo 2001–2002 (Gabon), Mbomo–Kellé 2001–2002 (RC), Kellé 2003 (RC), Mbandza–Mbomo 2003 (RC), and Etoumbi 2005 (RC). Like the 1994–1996 Gabonese epidemics, these cross-border outbreaks were marked by large wildlife epizootics [11–13], inducing mortality rates of ≤80% in great apes [14–16]. Chimpanzees, gorillas, and duikers were susceptible hosts responsible for viral introduction into human populations [11, 12].
The sequential appearance of ZEBOV outbreaks from 1976 to 2003 led to a hypothesis of wavelike viral dispersion into new territories to explain the explosive emergence of ZEBOV in the Gabon-RC area [17]. Indeed, the spatial and temporal pattern of ZEBOV outbreaks correlated with the genetic distance among strains, having apparently evolved continuously in a single lineage, westward from Yambuku (DRC) in 1976 to Mvoula (RC) in 2003. However, subsequent characterization of new GP and NP sequences demonstrated the existence of a second genetic lineage [10]. The previously recognized lineage A included ZEBOV strains from the 1976–1996 outbreaks (DRC and Gabon), whereas the newly described lineage B included animal-derived sequences since 2001 and the human strains from the Mbandza_Mbomo 2003 and Etoumbi 2005 outbreaks. The strains responsible for the previous 3 outbreaks were phylogenetically linked to lineage A in the GP gene, whereas they grouped with lineage B in the NP gene, strongly suggesting a recent recombination event [10]. Although the spatial and temporal pattern of outbreaks since 2001 still suggested progressive spread of the virus, the identification of 2 genetically divergent lineages did not fit the expected scenario in which all outbreaks during this period would be linked by ongoing transmission. Instead, the existence of the 2 lineages implied independent introductions into human populations following multiple viral spillovers from a reservoir host, a scenario referred to as the multi-emergence hypothesis [10, 11]. The data available from outbreaks up to 2005 thus indicated that the history of ZEBOV could not be explained by a single spreading wave of infections across multiple countries. In addition, ZEBOV sequence data collected from outbreaks up to that point revealed another puzzling aspect of ZEBOV biology, in that observed levels of sequence divergence were low (<5%), especially compared with other filoviruses, such as Marburg virus [18]. Molecular clock-based analyses suggested that these low levels of viral diversity might be due to a recent genetic bottleneck and that the estimated most-recent common ancestor of all viruses in the ZEBOV clade dated back only to the early 1970s [10, 17], just prior to the first outbreak at Yambuku in 1976.
In stark contrast to the temporal and geographical clustering of the Gabon-RC outbreaks, ZEBOV unexpectedly reemerged in DRC in 2007 in the Luebo area (Kasaï Occidental province), nearly 1000 km southeast of the last foci registered only 2 years before in RC. The Luebo 2007 outbreak occurred from June to November, affecting 264 persons and causing 187 deaths (70% case-fatality rate [19]). ZEBOV reappeared in the same area 1 year later, from November 2008 to February 2009, in the Luebo 2008 outbreak. No epidemiological survey was performed and official estimates were 32 human cases and 15 deaths (47% case-fatality rate [20]).
Although the first recorded ZEBOV outbreak took place in DRC in 1976, the virus reemerged in this country only once (Kikwit 1995) before the 2 Luebo outbreaks, whereas the neighboring countries were repeatedly affected during this time frame. In addition to the spatial discontinuity with the Gabon-RC outbreaks network, the Luebo outbreaks were in temporal discontinuity with the Kikwit epidemic that took place only 290 km eastward but 12 years before. Making the issue more complex, epidemiological and ecological investigations suggested that susceptible hosts, such as great apes and duikers, sustaining ZEBOV epizootics in Gabon and RC [11], were not involved in the Luebo epidemics [21]. Instead, the source of the Luebo 2007 outbreak appeared to be the hunting of fruit bats. These hunted bats, including Hypsignatus monstrosus and Epomops franqueti, undergo large annual migrations and are suspected ZEBOV reservoirs [21, 22].
Here, we report virological and phylogenetical analyses to determine whether the Luebo viruses (i) are related to viruses previously seen in DRC; (ii) are descendants of the recent Gabon-RC viruses, or (iii) represent newly emerged variants. We also investigated if the 2 Luebo outbreaks in 2007 and 2008 were caused by the same or different viruses and if the new sequence data collected would change the estimated age of the ZEBOV clade. Our phylogenetic analyses show that the Luebo viruses are not direct descendants of viruses characterized so far but share a common ancestor with lineage B viruses, despite the fact that the 2 groups are found nearly 1000 km apart. The Luebo 2007 and Luebo 2008 strains were nearly identical, despite being sampled 1 year apart. Given the limited divergence from other ZEBOV viruses, the estimated age of the ZEBOV clade overall is unaffected by these new sequence data.
MATERIALS AND METHODS
Ethical Considerations
Blood samples were collected from patients during the acute phase of illness by healthcare workers from the Health Ministries of DRC, Médecins sans Frontières (MSF), and the World Health Organization (WHO), all of whom were participating in the international response. Blood samples were collected at the patient’s home or in hospital isolation wards with verbal consent, and either were sent to international laboratory partners for diagnostic evaluation or were immediately processed for ZEBOV detection in the field laboratories.
Outbreaks
The Luebo 2007 outbreak occurred between May and November 2007 in the Kasaï Occidental province of DRC (Figure 1) as previously described [21]. A diagnosis of ZEBOV hemorrhagic fever was made on September 2007, both by Centre International de Recherches Médicales de Franceville, Gabon (CIRMF) and Centers for Disease Control and Prevention (CDC), Atlanta, Georgia. Outbreak control and investigations were supported by (i) an international WHO team including DRC Ministry of Health (MoH); Institut National de Recherche Biomédicale (INRB), Kinshasa, DRC; MSF; CIRMF; and National Microbiology Laboratory (NML), Winnipeg, Canada and (ii) a CDC team. Field laboratories and an isolation ward were set up for case monitoring. Because of its remote location, the outbreak’s initial cases could not be observed and the international response was considerably delayed. Most investigations were done retrospectively and were impaired by the concomitant occurrence of Salmonella typhi and Shigella outbreaks. The Luebo 2008 outbreak occurred from November 2008 to February 2009. The first identified case was an 18-year-old woman who died from hemorrhagic complications during premature delivery on 27 November. Subsequently, 13 contacts became ill and 9 died. MSF set up an isolation ward, and on 23 December, 3 suspect samples were taken to CIRMF and National Institute for Communicable Disease (NICD), South Africa. On 24 December, laboratory confirmation of 2 ZEBOV infections was obtained, and an alert was declared on 25 December 2008. Further diagnostics were performed at INRB, with laboratory and training support provided by CIRMF and NICD teams involved in the WHO international response. No epidemiological investigation was undertaken, so the source of the outbreak is unknown and the number of cases and deaths may be underestimated.
Figure 1.

Zaire ebolavirus (ZEBOV) outbreak locations since 1976. The Minkebe National Park (Gabon) and Odzala National Park (Republic of the Congo) are in green.
Diagnostic.
Samples received at CIRMF were chemically or heat-inactivated in biosafety level-4 (BSL-4) facilities before reverse-transcriptase polymerase chain reaction (RT-PCR) and antigen-capture assays. RNA was extracted from 140 μL of patients’ sera with the QIAamp viral RNA extraction kit (Qiagen) according to the manufacturer’s instructions. Partial L-gene sequences were amplified with degenerate primer pairs PanFilo-L1/Panfilo-L2 (5′-ATMGRAAYTTTTCYTTYTCWYT-3′/5′-TGWG–GHGGRYTATAAWARTCACTDACAT-3′) and PanFilo-L3/Panfilo-L4 (5′-GCNAARGCMTTYCCHAGYAAYATGATGG-3′/5′-ATAAWARTCACTDACATGCATRTARCA-3′).
A sandwich enzyme-linked immunosorbent assay (ELISA) for ZEBOV antigen detection was applied to 4-fold dilutions (1:4 to 1:256) of heat-inactivated sera (56°C, 30 minutes), as previously described [23]. Investigators coated 96-well plates with a cocktail of monoclonal antibodies against ZEBOV and coated control wells with normal mouse ascetic fluid. After sera incubation, ZEBOV antigens were detected by adding hyperimmune rabbit polyvalent Ebola antiserum [24] followed by peroxidase-conjugated goat antibodies. Reaction development was performed with the tetramethylbenzidine (TMB) detection system (Dynex Technologies).
Virus Isolation.
Tissue culture was performed in a biosafety level 4 laboratory. Vero-cell monolayers in 25 cm2 flasks were incubated for 1 hour at 37°C with 200 μL of serum at 1:10 dilution in Dulbecco’s modified eagle medium (DMEM). Medium containing 2.5% fetal bovine serum and antibiotics was added and the cells were incubated at 37°C with 5% CO2 for 6 days. Supernatants were harvested and stored at −80°C until their use for genomic characterization.
Genomic Characterization.
RNA was extracted from Vero-cell supernatants with the QIAamp viral RNA extraction kit (Qiagen). A set of 20 overlapping primer pairs (available on request) was designed from ZEBOV sequence alignments for RT-PCR (SuperScript III One-Step RT-PCR System with Platinum Taq High Fidelity, Invitrogen). Polymerase chain reaction product sizes averaged 1500 nucleotides and melting temperatures ranged from 55°C to 65°C. Contiguous sequences were assembled using ChromasPro 1.5 software (Technelysium Pty Ltd).
Phylogenetic Analysis
The new sequence data were aligned with existing full-length GP and NP sequences using the MUSCLE algorithm [25] implemented in the Geneious program [26]. Phylogenetic trees without a molecular-clock assumption were estimated for the GP and NP data set using MrBayes version 3.1.2 [27] in 2 independent runs of 2 million states with a sampling frequency of 1000 generations and a burn-in of 1 million samples. Phylogenies with a molecular-clock assumption and specific divergence dates were estimated using BEAST version 1.5 [28]. Analyses were calibrated using the year and month of sampling and applied a relaxed clock model with uncorrelated log–normally distributed rates [29] and a constant population size prior (alternative priors had a negligible effect on node height estimates). 2 independent runs of 10 million states were performed with a sampling frequency of 10000 and a burn-in period of 1 million states. Both Bayesian inference methods applied a SDR06 model of molecular evolution [30] and used sequences from CIEBOV and BEBOV as outgroups for rooting the ZEBOV clade. Runs were examined visually in program Tracer version 1.4.1 [31] to confirm convergence among runs and that chains had reached a stationary phase. Genbank accession numbers for the nucleotide sequences used in this study are available online (see Supplemental content S1).
RESULTS
Genome Description and Genetic Distances
The final sequence contigs cover 99% of the whole genomic sequences, with 18808 and 18775 nucleotides for the Luebo 2007 and Luebo 2008 strains, respectively, with partial leader and trailer sequences. The 2 Luebo strains are almost identical, differing at 11 nucleotide positions, of which 3 lie in noncoding region (NCRs; including intergenic regions and partial leader and trailer sequences), 5 are silent mutations (NP, VP40, GP, VP24, and L proteins), and 3 are nonsynonymous mutations, 1 in VP35 and 2 in the L protein. This resulted in a p-distance of <0.1%, maximum divergences being observed in VP35, VP40, and VP24 coding sequences (CDS) and not in the NP and GP variable genes. Gene sizes, transcriptional signals, GP editing site, cleavage sites, and immunosuppressive motifs were identical to those of other available ZEBOV sequences. The ectopic stop codon identified in the group R (recombinant) viruses [10] was absent from the GP Luebo sequences.
Compared with the fully characterized ZEBOV strains (Figure 2), the Luebo viruses appeared slightly more similar to the Yambuku 1976 strain (1.9% mean p-distance) than to the more recent Kikwit 1995 strain (2.15% mean p-distance). The maximum p-distances were recorded in the NCRs (1.9%–3.5%), NP (1.3%–2.1 %) CDS, and VP40 (1.4%–2.1%) CDS, whereas the smallest distances were observed in VP30 (<0.7%) CDS. Based on the inclusion of partially characterized strains, NP sequences from group B and R viruses are the sequences most closely related to the Luebo strains (p-distance < 2.1%), whereas higher p-distances are observed with lineage A viruses, namely the Booué 1996, Mayibout 1996, and Mekouka 1994 Gabonese strains (2.4%–2.8% p-distance). Conversely, in GP CDS, group A and B viruses appeared equally distant from the Luebo strains (∼2.4%), whereas group R viruses were slightly more divergent, with 2.9%–3.1% p-distance.
Figure 2.

Zaire ebolavirus (ZEBOV) p-distances with respect to Luebo 2007 and Luebo 2008 sequences. Available complete sequences were first aligned for comparison with Luebo viruses. The p-distances between Yambuku 1976 and Kikwit 1995 are reported for reference (in blue). Complete GP coding sequences and partial NP coding sequences were then added to compare distances between Luebo viruses and other group A, B, and R partially characterized strains. NCR:, noncoding regions.
Phylogenetic Analysis
Phylogenetic trees were derived from NP (1448 nucleotides, Figure 3) and GP (2031 nucleotides, Figure 4) CDS. In addition to the Bayesian method, molecular clock–based analysis was used to take into account the expectation that viruses sampled >30 years apart have evolved, and to estimate the lineage divergence dates (Figures 3B and 4B). The CIEBOV and BEBOV sequences were included as outgroups, because these are the viruses most closely related to ZEBOV.
Figure 3.
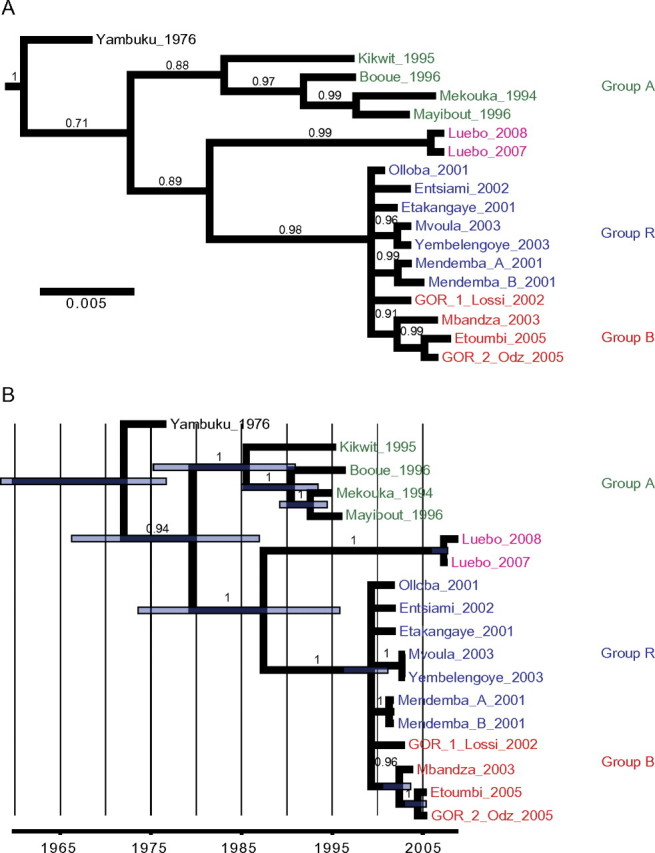
Bayesian phylogenies inferred from NP sequences, representing a consensus tree without molecular clock assumption (A) and a maximum clade credibility tree inferred under a ’dated tips’ molecular clock model (B ). Côte d’Ivoire ebolavirus (CIEBOV) and Bundibugyo Ebola virus (BEBOV) sequences were included as outgroups. Green: group A strains; red: group B strains; blue: group R strains. Branch support in form of Bayesian posterior probabilities is indicated above branches. In panel B, purple bars indicate the upper and lower limits of the 95% highest posterior density interval for estimated node ages.
Figure 4.
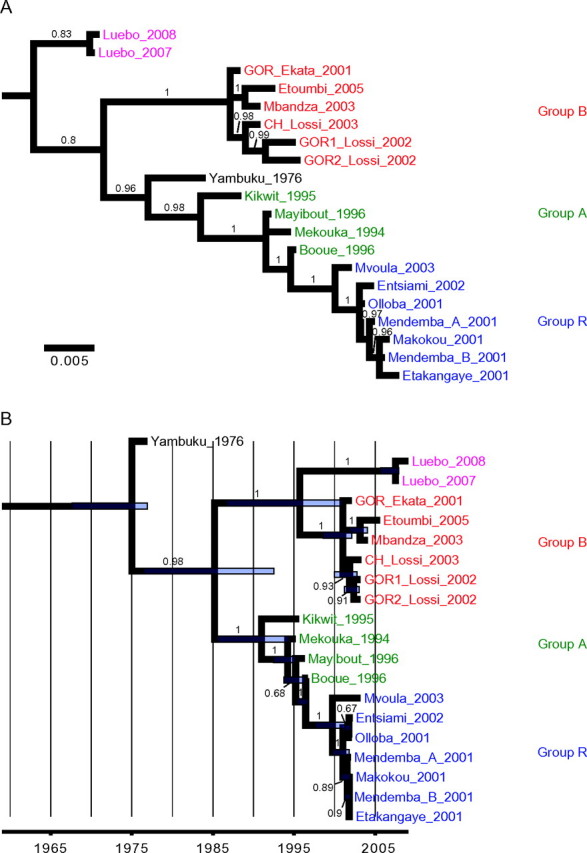
Bayesian phylogenenies inferred from GP sequences, representing a consensus tree without molecular-clock assumption (A) and a maximum clade credibility tree inferred under a ’dated tips’ molecular-clock model (B ). Côte d’Ivoire ebolavirus (CIEBOV) and Bundibugyo Ebola virus (BEBOV) sequences were included as outgroups. Green: group A strains; red: group B strains; blue: group R strains. Bayesian posterior probabilities are indicated above branches. In panel B, purple bars indicate the upper and lower limits of the 95% highest posterior density interval for estimated node ages.
The 4 phylogenetic trees display 2 genetic lineages, with higher support values observed with molecular-clock constraints. Lineage A included ZEBOV strains from 1994–1996 whereas lineage B included animal-derived sequences from 2001 and human isolates from Mbandza 2003 and Etoumbi 2005 outbreaks. With both methods and with high support values, the group R viruses display a phylogenetic incongruence, being associated with lineage B in the NP gene (Figure 3) and with lineage A in the GP gene (Figure 4). Except for the Bayesian analysis of GP sequences without the molecular-clock assumption, the Yambuku 1976 strain fell at the most basal position of the ZEBOV tree, just before the split into lineages A and B.
The Luebo 2007 and Luebo 2008 viruses grouped together tightly in all 4 trees. In most cases, they formed a strongly supported sister group to lineage B viruses, with the exception of the 1 GP analysis, which placed the Luebo viruses at the most basal position of the tree (Figure 4A).
According to the relaxed molecular-clock model applied to the NP (Figure 3B) and GP data (Figure 4B), the most-recent common ancestor (MRCA) of all known ZEBOV isolates was estimated to date back to 1972 and 1975, respectively. Lineage A and B would have split off sometime between 1979 and 1985, whereas the Luebo viruses would have separated from group B viruses between 1987 and 1996. The group R viruses shared an MRCA between 1998 and 2000, respectively.
DISCUSSION
The last 2 ZEBOV outbreaks occurred in DRC, within the same area, 1 year apart, in spatial and temporal discontinuities with the locations and time of occurrence of the previous sequential outbreaks. Following viral isolation, we characterized the full coding sequences of the Luebo 2007 and Luebo 2008 strains and performed phylogenetic analyses to investigate their relationships with previously characterized viruses.
The phylogenetic trees inferred from the GP and NP data sets, with and without a molecular-clock constraint, confirmed previous findings [10] including (i) the split of the ZEBOV clade into distinct lineages, (ii) the clustering of animal-derived sequences in the recently discovered B lineage, and (iii) the phylogenetic incongruence between the GP and NP phylogenies for the group R viruses. As in previous studies [10, 17], some uncertainty remained regarding the inferred position of the ZEBOV tree root. Most of our current analyses support a root-near position of the Yambuku 1976 virus and placed this virus outside groups A and B. The only exception to this was the GP analysis with no molecular clock assumption, which characterized the newly identified viruses from Luebo as a sister clade to all remaining ZEBOV isolates. However, the deeper tree nodes were not well resolved. More-definitive resolution of the early evolutionary history of the ZEBOV clade will likely not be possible until more full-length genomes of both lineage A and B viruses are available.
Surprisingly, from the p-distances observed all along the genome, the Luebo viruses shared greater homology with the Yambuku 1976 strain than with the more recent and geographically closer Kikwit 1995 strain. Consistent with this, our phylogenetic analysis did not identify the Luebo viruses as belonging to the same genetic lineage A as Kikwit 1995. Instead, the Luebo viruses formed a well-supported sister group to the lineage B viruses, previously encountered only in Gabon-RC. The analysis clearly demonstrates that the Luebo viruses are not direct descendants of viruses sampled during previous outbreaks, which strongly suggests that the Luebo outbreaks did not arise through viral spread from any of the recently recognized foci. By contrast, the Luebo viruses are phylogenetically distinct from the strains characterized so far, suggesting that these outbreaks were epidemiologically independent of the 1994–2005 network. The most plausible scenario for such independent emergences is multiple viral spillover from a reservoir host widely dispersed throughout Central Africa.
Despite representing a ZEBOV lineage that is clearly divergent from any viruses previously described, the new DRC sequence data, like those collected during the 2001–2005 outbreaks [10], did not increase the estimated age of diversification of the ZEBOV clade as a whole. Rather, our date estimates confirm earlier results, placing the most recent common ancestor of the entire group at a time just prior to the virus’ first emergence at Yambuku in 1976. Although this recent origin of the ZEBOV clade could result from a recent bottleneck and subsequent dispersion of ZEBOV across much of central Africa, the nature of the event that gave rise to this pattern is unknown.
Whereas the Luebo 2007 outbreak appeared to be linked to migratory fruit bats [21], the source of the 2008 outbreak could not be identified. The scenario of a second spillover from the migratory bat population appears unlikely, given that the 2 viruses were genetically almost identical. Such similarity would seem improbable for a virus originating from a large reservoir population that would be expected to simultaneously maintain multiple, and thus genetically diverse, viral lineages. Interestingly, the latter pattern was observed for Marburg viruses sampled from cavernous bats and humans in Uganda in 2007–2008 [32] and during the Durba outbreak (DRC, 1999–2000) [33], with different introductions into the human population, usually involving genetically divergent viruses. Despite the limited viral sampling, the high similarity between the 2 Luebo viruses is consistent with a direct epidemiological link between the 2 epidemics. Based on our evolutionary rate estimates for the full-length virus and the GP and NP genes (∼1.5–7.5 × 10−4 substitutions per site per year) we would expect between 3 and 15 substitutions to accumulate across the entire ZEBOV genome over the course of a year. The observed divergence of 11 substitutions between the 2007 and 2008 viruses fits this well, raising the possibility that the 2008 virus is a direct descendent of the 2007 virus and that the latter continued to be transmitted in the area around Luebo. However, great apes were not present, signs of wildlife mortality were noticeably absent [21], and it is unlikely that human cases in the area would have been missed over a period of several months, posing the question of how the virus could have persisted so inconspicuously during this period. Following viral introduction into the area in 2007, subsequent transmission among local, nonmigrating bats could provide a possible explanation, especially as local bats species include Hypsignatus monstrosus and Epomops franqueti (E. Leroy, personal observations), both of which are suspected of being natural ZEBOV reservoir species [22].
More generally, an increasing number of epidemiological events due to Ebola viruses have occurred over the last 20 years, contrasting with the prior apparent silent period [1978–1993] (Figure 5). Since 1994, 15 Ebola virus outbreaks have been recorded, of which 11 were due to ZEBOV, also responsible for massive local mortality among great apes [11, 12, 14, 16]. The absence of great apes and of noticeable mortality among other wildlife species in Luebo indicates that transmission of ZEBOV can occur in the absence of any indicators. Whereas the mechanisms underlying such cryptic circulation are elusive, these observations may help to explain the prolonged periods of apparent epidemiological silence, during which ZEBOV genetic diversity increases (Figure 5). However, the possibility remains that isolated cases, limited outbreaks, or epizootics may have been missed.
Figure 5.

Zaire ebolavirus (ZEBOV) epizootics and human outbreaks (red line) are reported, along with outbreaks due to other Ebolavirus species (grey line). Time of occurrence of the main genetic events in Zaire ebolavirus (ZEBOV) evolution are reported in red; MRCA: most recent common ancestor. SEBOV: Sudan ebolavirus; CIEBOV: Côte d’Ivoire ebolavirus; BEBOV: Bundibugyo Ebola virus.
In conclusion, this study highlights that the Luebo outbreaks were phylogenetically, temporally and spatially distinct from all previous outbreaks. In contrast to the continuous ladder-like evolution of ZEBOV observed until 1996, the most recent trees display a more bushlike cladogenetic picture, with coexistence of distinct lineages. The emergence of a novel strain of ZEBOV, with the recent discovery of BEBOV, point to the depth of viral genetic diversity in the underlying natural animal hosts and suggests the possibility of future outbreaks with additional novel viruses or strains. Spillovers and the potential for recombination in ZEBOV raise the possibility—hypothetical at this stage—that viruses with increased transmissibility from the natural host to other animals or humans may emerge. Identifying the environmental drivers that trigger emergence of Ebola viruses from their reservoirs, which may differ from one viral species to another, after they circulate silently for years or decades, remains one of the key research challenges.
Supplementary Data
Supplementary Data are available at The Journal of Infectious Diseases online.
Funding
The Centre International de Recherches Médicales de Franceville (CIRMF) is supported by the government of Gabon, Total-Fina- Elf Gabon, and the Ministére des Affaires Etrangères et Européennes de la France. This work was also partially financed by Global Viral Forecasting, graciously supported by the U.S. Department of Defense Global Emerging Infections, Surveillance and Response Systems (DoD GEIS) and the Defense Threat Reduction Agency (DTRA), Google.org and the Skoll Foundation, and the U.S. Agency for International Development Emerging & Pandemic Threats PREDICT Cooperative Agreement. R.B. was supported by the Research and Policy for Infectious Disease Dynamics (RAPIDD) program of the Science and Technology Directorate, U.S. Department of Homeland Security, and the Fogarty International Center, NIH.
Supplementary Material
Acknowledgments
The authors thank the national and international teams involved in the control of the Ebola outbreaks. The national teams were members of the DRC Health Ministry and Croix Rouge. The international teams were mainly scientific and medical experts of the World Health Organization and Médecins Sans Frontières. We thank T. Ksiazek, S. Nichol, T. Geisbert, P. Rollin, and J. Towner from the Viral Special Pathogens Branch, Division of High-Consequence Pathogens and Pathology of the Centers for Disease Control and Prevention, who generously provided reagents to the Centre International de Recherches Médicales de Franceville.
References
- 1.Fauquet CM, Mayo MA, Maniloff J, Desselberger U, Ball LA, eds. Virus taxonomy, Eighth report of the International Committee on Taxonomy of Viruses. San Diego, California: Elsevier Academic Press; 2005. [Google Scholar]
- 2.Towner JS, Sealy TK, Khristova ML, et al. Newly discovered Ebola virus associated with hemorrhagic fever outbreak in Uganda. PLoS Pathog. 2008;4:e1000212. doi: 10.1371/journal.ppat.1000212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Johnson KM. Ebola haemorrhagic fever in Zaire, 1976. Bull World Health Organ. 1978;56:271–93. [PMC free article] [PubMed] [Google Scholar]
- 4.Heymann DL, Weisfeld JS, Webb PA, Johnson KM, Cairns T, Berquist H. Ebola hemorrhagic fever: Tandala, Zaire, 1977–1978. J Infect Dis. 1980;142:372–6. doi: 10.1093/infdis/142.3.372. [DOI] [PubMed] [Google Scholar]
- 5.Georges-Courbot MC, Sanchez A, Lu CY, et al. Isolation and phylogenetic characterization of Ebola viruses causing different outbreaks in Gabon. Emerg Infect Dis. 1997;3:59–62. doi: 10.3201/eid0301.970107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Georges AJ, Leroy EM, Renaut AA, et al. Ebola hemorrhagic fever outbreaks in Gabon, 1994–1997: Epidemiologic and health control issues. J Infect Dis. 1999;179:S65–75. doi: 10.1086/514290. [DOI] [PubMed] [Google Scholar]
- 7.Khan AS, Tshioko FK, Heymann DL, et al. The reemergence of Ebola hemorrhagic fever, Democratic Republic of the Congo, 1995. Commission de Lutte contre les Epidémies à Kikwit. J Infect Dis. 1999;179:S76–86. doi: 10.1086/514306. [DOI] [PubMed] [Google Scholar]
- 8.Muyembe T, Kipasa M. Ebola haemorrhagic fever in Kikwit, Zaire. International Scientific and Technical Committee and WHO Collaborating Centre for Haemorrhagic Fevers. Lancet. 1995;345:1448. doi: 10.1016/s0140-6736(95)92640-2. [DOI] [PubMed] [Google Scholar]
- 9.Pourrut X, Kumulungui B, Wittmann T, et al. The natural history of Ebola virus in Africa. Microbes Infect. 2005;7:1005–14. doi: 10.1016/j.micinf.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 10.Wittmann TJ, Biek R, Hassanin A, et al. Isolates of Zaire ebolavirus from wild apes reveal genetic lineage and recombinants. Proc Natl Acad Sci U S A. 2007;104:17123–7. doi: 10.1073/pnas.0704076104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leroy EM, Rouquet P, Formenty P, et al. Multiple Ebola virus transmission events and rapid decline of central African wildlife. Science. 2004;303:387–90. doi: 10.1126/science.1092528. [DOI] [PubMed] [Google Scholar]
- 12.Rouquet P, Froment JM, Bermejo M, et al. Wild animal mortality monitoring and human Ebola outbreaks, Gabon and Republic of Congo, 2001–2003. Emerg Infect Dis. 2005;11:283–90. doi: 10.3201/eid1102.040533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lahm SA, Kombila M, Swanepoel R, Barnes RF. Morbidity and mortality of wild animals in relation to outbreaks of Ebola haemorrhagic fever in Gabon, 1994–2003. Trans R Soc Trop Med Hyg. 2007;101:64–78. doi: 10.1016/j.trstmh.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 14.Huijbregts B, Wachter PD, Ndong Obiang LS, Akou ME. Ebola and the decline of gorilla Gorilla gorilla and chimpanzee Pan troglodytes populations in Minkebe Forest, north-eastern Gabon. Oryx. 2003;37:437–43. [Google Scholar]
- 15.Walsh PD, Abernethy KA, Bermejo M, et al. Catastrophic ape decline in western equatorial Africa. Nature. 2003;422:611–4. doi: 10.1038/nature01566. [DOI] [PubMed] [Google Scholar]
- 16.Bermejo M, Rodriguez-Teijeiro JD, Illera G, Barroso A, Vilà C, Walsh PD. Ebola outbreak killed 5000 gorillas. Science. 2006;314:1564. doi: 10.1126/science.1133105. [DOI] [PubMed] [Google Scholar]
- 17.Walsh PD, Biek R, Real LA. Wave-like spread of Ebola Zaire. PLoS Biol. 2005;3:e371. doi: 10.1371/journal.pbio.0030371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Towner JS, Khristova ML, Sealy TK, et al. Marburgvirus genomics and association with a large hemorrhagic fever outbreak in Angola. J Virol. 2006;80:6497–516. doi: 10.1128/JVI.00069-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.ProMED-mail. Ebola hemorrhagic fever—Congo DR. Archive number 20071121.3758. 2007. http://www.promedmail.org/pls/apex/f?p=2400:1000. Accessed 20 October 2010. [Google Scholar]
- 20.World Health Organization. End of Ebola outbreak in the Democratic Republic of the Congo. 2009. http://www.who.int/csr/don/2009_02_17/en/index.html. Accessed 20 October 2010. [Google Scholar]
- 21.Leroy EM, Epelboin A, Mondonge V, et al. Human Ebola outbreak resulting from direct exposure to fruit bats in Luebo, Democratic Republic of Congo, 2007. Vector Borne Zoonotic Dis. 2009;9:723–8. doi: 10.1089/vbz.2008.0167. [DOI] [PubMed] [Google Scholar]
- 22.Leroy EM, Kumulungui B, Pourrut X, et al. Fruit bats as reservoirs of Ebola virus. Nature. 2005;438:575–6. doi: 10.1038/438575a. [DOI] [PubMed] [Google Scholar]
- 23.Ksiazek TG, Rollin PE, Jahrling PB, Johnson E, Dalgard DW, Peters CJ. Enzyme immunosorbent assay for Ebola virus antigens in tissues of infected primates. J Clin Microbiol. 1992;30:947–50. doi: 10.1128/jcm.30.4.947-950.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ksiazek TG, Rollin PE, Williams AJ, et al. Clinical virology of Ebola hemorrhagic fever (EHF): Virus, virus antigen, and IgG and IgM antibody findings among EHF patients in Kikwit, Democratic Republic of the Congo, 1995. J Infect Dis. 1999;179:S177–87. doi: 10.1086/514321. [DOI] [PubMed] [Google Scholar]
- 25.Edgar RC. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Drummond AJ, Ashton B, Buxton S, et al. Geneious v5.1. 2010. http://www.geneious.com. Accessed 20 October 2010. [Google Scholar]
- 27.Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–4. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- 28.Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Drummond AJ, Ho SY, Phillips MJ, Rambaut A. Relaxed phylogenetics and dating with confidence. PLoS Biol. 2006;4:e88. doi: 10.1371/journal.pbio.0040088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shapiro B, Rambaut A, Drummond AJ. Choosing appropriate substitution models for the phylogenetic analysis of protein-coding sequences. Mol Biol Evol. 2006;23:7–9. doi: 10.1093/molbev/msj021. [DOI] [PubMed] [Google Scholar]
- 31.Rambaut A. Tracer v1.3. 2004. http://beast.bio.ed.ac.uk/Tracer. Accessed 20 October 2010. [Google Scholar]
- 32.Towner JS, Amman BR, Sealy TK, et al. Isolation of genetically diverse Marburg viruses from Egyptian fruit bats. PLoS Pathog. 2009;5:e1000536. doi: 10.1371/journal.ppat.1000536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bausch DG, Nichol ST, Muyembe-Tamfum JJ, et al. International Scientific and Technical Committee for Marburg Hemorrhagic Fever Control in the Democratic Republic of the Congo. Marburg hemorrhagic fever associated with multiple genetic lineages of virus. N Engl J Med. 2006;355:909–19. doi: 10.1056/NEJMoa051465. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.