

Abstract
Background:
Identification of orthologous relationships between genes from widely divergent taxa allows partial reconstruction of the gene complement of ancestral genomes. C2H2 zinc-finger genes are one of the largest and most complex gene superfamilies in metazoan genomes, with hundreds of members in the human genome. Here we analyze C2H2 zinc-finger genes from three taxa - Drosophila, Caenorhabditis elegans and human - from which near-complete genome sequence data are available.
Results:
Our analyses conclusively identify 39 families of genes, of which 38 can be defined as orthology groups in that they are descended from single ancestral genes in the common ancestor of Drosophila, C. elegans and humans.
Conclusions:
On the basis of current metazoan phylogeny, these 39 groups represent the minimum complement of C2H2 zinc-finger genes present in the genome of the bilaterian common ancestor.
Background
Model organisms such as the fruit fly Drosophila melanogaster and the nematode Caenorhabditis elegans are commonly used to investigate gene function. Frequently, genes with similar sequence can be identified in the human genome, allowing prediction of human gene function by extrapolation from Drosophila and/or C. elegans. Implicit in such extrapolations is that the genes being compared are orthologous, that is, they derive from the same ancestral gene in the common ancestor of the model organism and humans [1]. Correct identification of such relationships is therefore essential if extrapolation of function is to be fully exploited. In one form, such identifications typically utilize database comparisons with algorithms such as BLAST, with the highest-scoring sequences inferred to be orthologs [2, 3]. Additional criteria can then be applied to confirm orthologous relationships, including checking that orthologs have similar domain structures, and ensuring that no sequence from a more distantly related taxon is more closely related to one proposed ortholog than to another. In more complex analyses, molecular phylogenetic reconstruction of gene family history is employed. Such reconstructions help distinguish speciation from gene duplication, thereby revealing orthologous and paralogous relationships.
With the near-completion of the human, C. elegans and Drosophila genome sequences, it is becoming possible to extend the identification of such relationships to analyses of large, complex gene superfamilies in the Metazoa. Such an exercise essentially reconstructs the minimum gene complement, for a particular superfamily, that would have been present in the last common ancestor of these three taxa and, given their phylogenetic relationship [4], gives insight into the genome complexity of the bilaterian common ancestor. Here we present an analysis of the C2H2 zinc finger (C2H2 ZNF) genes: a superfamily that, with over 600 members in humans, contains 1-2% of all human genes. C2H2 ZNF genes primarily encode DNA- and chromatin-binding transcription factors, and include familiar and well-studied developmental genes such as Krox-20, snail, Gli, Krüppel and hunchback, as well as numerous genes whose function is yet to be established. By defining orthologous relationships within this superfamily, we aim to reconstruct the minimum complement of C2H2 ZNFs present in the bilaterian common ancestor.
Results
The organization of a typical C2H2 ZNF includes two features that make inference of evolutionary history complicated (Figure 1). The first is the conservation in almost all C2H2 ZNFs of a number of key residues critical for the structure of the domain. This means all C2H2 ZNFs have a high baseline of identity. The second is repetition of the C2H2 ZNF motif in individual genes. This makes BLAST scores unreliable indicators of evolutionary relationships, as the score depends on the length of matching sequence and will be misleadingly high for genes that have independently evolved multiple contiguous fingers. Finger repetition also means that molecular phylogenetics can only be employed where the relationships of individual fingers between genes can be determined. This is only possible for subgroups where a robust phylogenetic framework has already been established, and is consequently of little use in defining such subgroups.
Figure 1.
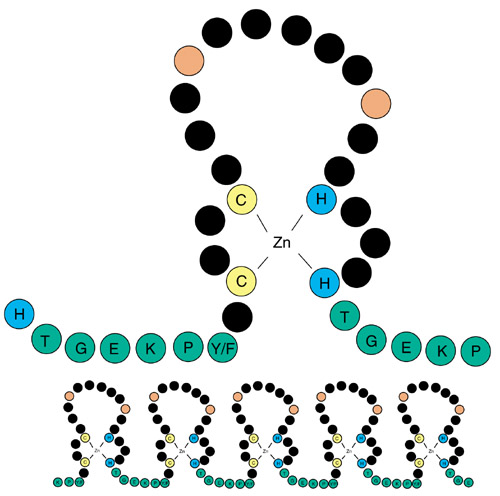
Schematic diagram of a C2H2 zinc-finger motif. The paired cysteines (C) and histidines (H) that bind the zinc ion are shown in yellow and blue, respectively. The linker sequence, shown in green with its consensus sequence in the single-letter amino acid code, frequently joins adjacent fingers. This is apparent in the lower panel, which shows the typical arrangement of fingers in a C2H2 ZNF protein. The two large hydrophobic residues, which are also structurally important, are shown in red. The black residues are not structurally important and include those responsible for contacting DNA during sequence-specific binding [16]. The precise number of 'black' residues between the cysteines, histidines and on the loop may vary [10].
The limitations of BLAST and molecular phylogenetics lead us to seek alternative criteria for defining orthology of C2H2 ZNF genes. We used percentage amino-acid sequence identity over the ZNF region, as determined by FASTA [5], as a preliminary indicator of relationships. First, we compiled datasets of all Drosophila, C. elegans and human proteins that contained C2H2 ZNFs. For a preliminary view of the levels of identity between species, we used FASTA to compare the Drosophila and C. elegans datasets to the human dataset and recorded the highest identity match in the human dataset for each Drosophila and C. elegans gene. To visualize the results, we combined identity scores (which potentially range from 0 to 100%) into 5% intervals and plotted the proportion of each dataset that had its highest match in each interval (Figure 2). The results were essentially the same for Drosophila and C. elegans, with a peak of highest identity centered at about 40% and a tail of genes with matches higher than 50%. A large majority of invertebrate genes had their highest identity matches to human genes within the peak in the 25-50% range.
Figure 2.
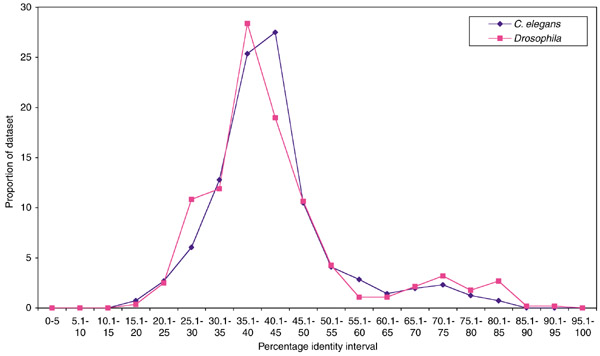
Highest percentage-identity match in 5% intervals for the E < 10 datasets of Drosophila and C. elegans compared to the human dataset. Baseline identity between typical C2H2 ZNF domains is between 20 and 44%, and this is where most genes show their highest identity. Values higher than this range are strongly suggestive of orthology. We also examined the difference between this analysis and an analysis of more stringent datasets (E < 1). All but one of the sequences detected at E < 10 but excluded from E < 1 had maximum identity matches below 40%.
In a typical C2H2 ZNF motif, between 20 and 44% of the amino acids are structurally important and highly conserved, with variation within this range mostly arising from the presence or absence of a six-residue linker sequence that frequently joins adjacent fingers (Figure 1). Therefore the peak centered at 40% in Figure 2 can be largely explained by the baseline of identity that occurs between most C2H2 ZNF sequences. A similarity score of 45% and above indicates a closer relationship and therefore possible orthology. These values, however, cannot be used either to definitively exclude or conclude orthology without further evaluation because of the limited but significant variation in baseline identity. We therefore examined highest matches by eye to judge whether they indicated orthology. We used the presence of conserved amino acids in the zinc fingers other than those important for structure as a criterion to assess this. Specifically, we did not include the paired cysteines and histidines that bind the zinc ion (Figure 1). The consensus linker, where present, was also excluded. We also compared all Drosophila and C. elegans C2H2 ZNF sequences to available human genome and expressed sequence tag (EST) sequences to detect potential orthologs absent from our human C2H2 ZNF dataset. This step was essential as, because of the incomplete cataloguing of human protein data, our human C2H2 ZNF protein dataset is certain to be incomplete. With these analyses we defined a total of 39 families of genes (Table 1) which we propose represent 'orthology groups', as we infer that each group is descended from a single ancestral gene in the most recent common ancestor of Drosophila, C. elegans and humans. Multiple genes from one species within a group are therefore paralogs. To our knowledge, 17 of these groups have not previously been defined. As an additional check of orthology we also compared our C2H2 ZNF datasets to a yeast C2H2 ZNF dataset [6]. No yeast sequences were more closely related to single orthology group members than to all group members, which supports our group definitions. Each orthology group typically contains genes with the same number and arrangement of fingers. This fulfilled another standard prediction of orthology (similar domain structure), and allowed us to use molecular phylogenetics to examine, where relevant, the pattern of evolution within a group and to determine whether our assumption of descent from a single gene in the most recent common ancestor was supported (Figure 3). In all but one case, molecular phylogenetics either produced trees that were too poorly resolved to confirm or disprove our inference of orthology or produced trees that supported our inference of orthology. The exception was the KLF family (Table 1), which tree topology suggested might include more than one orthology group; data from additional taxa will be necessary to further resolve this family. All sequences that showed an identity score >55% were in orthology groups. Conversely, we consider some sequences with scores of <44% to be in orthology groups.
Table 1.
The 39 groups of orthologous C2H2 ZNF genes defined by our analyses
| Gene family | Human | Drosophila | C. elegans |
| 1 Sp | Sp1 (SP:P08047) | Btd (CT35305; SPTR:Q24266) | T22C8.5 (SPTR:Q22678) |
| Sp2 (SP:Q02086) | DSp1 (CT2914; SPTR:Q9U1K4) | Y40B1A.4 (SPTR:Q9XW26) | |
| Sp3 (SP:Q02447) | |||
| Sp4 (SP:Q02446) | |||
| 2 Zic | Zic1 (SP:Q15915) | Opa (CT1819; SPTR:P39768) | C47C12.3 (SPTR:Q94178) |
| Zic2 (SP:O95409) | |||
| Zic3 (SP:O60481) | |||
| 3 Ovo | Ovol1 (SP:O14753) | Ovo (CT21113, CT36311; SPTR:P51521) | F34D10.5 (SPTR:Q19996) |
| SPTR:O00110 | |||
| SPTR:Q9Y4M0 | |||
| 4 Snail | Slug (SP:O43623) | Snail (CT13146; SPTR:P08044) | C55C2.1 (SPTR:O01830) |
| Snai1 (SP:O95863) | Escargot (CT12561; SPTR:P25932) | F43G9.11 (SPTR:Q93721) | |
| SnaiP1 (Snai1 pseudogene) | Worniu (CT13175; SPTR:Q9NK88) | ||
| Scratch (CT1817; SPTR:Q24140) | |||
| CT33426 (SPTR:Q9W0P9) | |||
| CT34835 (SPTR:Q9VZK3) | |||
| 5 Gli | Gli/Gli1 (SP:P08151) | Ci (CT6641; SPTR:P19538) | Tra-1 (Y47D3A.6; SPTR:Q9U2C0) |
| Gli2 (SP:P10070) | |||
| Gli3 (SP:P10071) | |||
| 6 Egr/Krox | Egr1/Krox-24 (SP:P18146) | Stripe (CT23724; SPTR:Q24163) | C27C12.2 (SPTR:Q18250) |
| Egr2/Krox-20 (SP:P11161) | Y55F3AM.7 (SPTR:Q9N374) | ||
| Egr3 (SP:Q06889) | |||
| Egr4 (SP:Q05215) | |||
| 7 KLF | EZF/GKLF (SPTR:Q9UNP3) | CT2144 (SPTR:Q9VZN4) | F56F11.3 (SPTR:Q9TZ64) |
| LKLF (SPTR:Q9UKR6) | CT27882 (SPTR:Q9W1W2) | F53F8.1 (SPTR:O62259) | |
| UKLF (SP:O75840) | CT14096 (SPTR:Q9VPQ5) | mua1/F54H5.4 (SPTR: P91329) | |
| BKLF (SP:P57682) | CT9920 (SPTR:O77251) | ||
| EKLF (SP:Q13351) | |||
| KKLF (SPTR:Q9UIH9) | |||
| ZNF741 (SPTR:O95600) | |||
| NSLP1 (SPTR:Q9Y356) | |||
| BTEB1 (SP:Q13886) | |||
| ZF9/CPBP (SP:Q99612) | |||
| BTEB2/CKLF (SP:Q13887) | |||
| AP-2REP (SPTR:Q9UHZ0) | |||
| TIEG1 (SP:Q13118) | |||
| TIEG2 (SP:O14901) | |||
| 8 Zfh-1 | SPTR:O60315 | Zfh-1 (CT2773; SPTR:P28166) | F28F9.1 (SPTR:Q94196) |
| NIL-2-A (SP:P37275) | |||
| 9 Zfh-2 | ATBF1 (SPTR:Q13719) | Zfh-2 (CT3397; SPTR:P28167) | ZC123.3 (SPTR:O45019) |
| 10 Odd-like | EM:AI126171 | Odd (CT12867; SPTR:P23803) | YKC4 (B0280.4; SPTR:P41995) |
| Sob (CT10899; SPTR:Q24571) | C34H3.2 (SPTR:Q9N5X6) | ||
| Bowl (CT9648, CT37221; | |||
| CT40018, SPTR: Q9VQU9) | |||
| 11 Spalt | HSAL1 (SPTR:Q99881) | Spalt-major (CT20082; SPTR:P39770) | SEM-4 (SPTR:Q17396) |
| HSAL2 (SPTR:Q9Y467) | Spalt-related (CT15643; SPTR:Q24163) | ||
| SALL3 (SPTR:Q9UGH1) | |||
| 12 Disco | Basonuclin (SPTR:Q01954) | Disco (CT27904; SPTR:P23792) | F55C5 (SPTR: Q20815) |
| SPTR:Q9NXV0 | CT26340 (SPTR:Q9VXJ5) | ||
| 13 GFI | GFI-1, GFI-1B (SPTR:Q99684) | CT31381 (SPTR:Q9VM77) | F45B8.4 (SPTR:O02265) |
| 14 YY1 | TYY1 (SPTR:P25490) | Pho (CT39329; SPTR:O76247) | |
| SPTR:O15391 | CT11601 (SPTR:Q9VSZ3) | ||
| 15 BLIMP-1 | BLIMP-1 (SPTR:O95914) | CT16759 (SPTR:Q9VRN4) | F25D7.3 (SPTR:Q93560) |
| 16 Zep | Zep1 (SP:P15822) | Schnurri (CT23537; PIR:A56922) | T05A10.1 (SPTR:Q22190) |
| Zep2 (SP:P31629) | |||
| KBP-1 (SPTR:Q99302) | |||
| 17 IA-1* | IA-1 (SP:Q01101) | CT31935 (SPTR:Q9VH29) | K11G9.4 (SPTR:Q23011) |
| Nerfin-1 (CT33443; SPTR:Q9V3B8) | |||
| 18 Evi-1* | Evi-1 SP:Q03112) | CT29074 (SPTR:Q9VJ55) | R53.3 (A and B) (SPTR:Q22024) |
| CT29650 (SPTR:Q9VJ52) | |||
| 19 SAP61 | SAP 61 (SPTR:Q12874) | Noisette (CT7078; SPTR:O46106) | T13H5.4 (SPTR:Q22469) |
| 20 SP62* | SP62 (SP:Q15428) | CT30142 (SPTR:Q9VU15) | F11A10.2 (SPTR:Q19335) |
| 21 Kin-17 | KIN-17 (SPTR: O60870) | Kin-17 (CT17834; SPTR:O76926) | Y52B11A.9 (SPTR:Q9XWF2) |
| 22 Hindsight | FinB (SPTR:Q9Y474) | Hindsight | |
| RREB-1 (SP:Q92766) | (CT11247; PIR:T13594) | ||
| 23 MTF | MTF-1 (SPTR:Q14872) | CT12477 (SPTR:Q9NFS1) | |
| 24 ZNF207* | ZNF207 (SP:O43670) | CT39886 (SPTR:Q9VJI6) | B0035.1 (SPTR:Q93156) |
| 25 ZNF277* | ZNF277 (SP:Q9NRM2) | CT27874 (SPTR:Q9W1V7) | F46B6.7 (SPTR:Q20448) |
| 26 Fez | SPTR:Q9NWB9 | CT22557 (SPTR:Q9VQ56) | Y38H8A.5 (SPTR:O62425) |
| 27 OAZ* | OAZ (SPTR:Q9NZ13) | CT33481 (SPTR:Q9V724) | |
| 28 Zfam 1* | HSPC038 (SPTR:Q9Y5V0) | CT35941 (SPTR:Q9VUU8) | C01F6.9 (SPTR:O62023) |
| CT40578 (SPTR:Q9VUU7) | |||
| 29 Zfam 2* | SPTR:Q9NWA7 | CT27270 (SPTR:Q9W3S1) | F13H6.1 (SPTR:O16350) |
| 30 Zfam 3* | SPTR:Q9NTN4 | CT15069 (SPTR:Q9VCS3) | |
| 31 Zfam 4* | EM:AI907237 | CT17352 (SPTR:Q9U9A8) | Lin29 (SPTR:Q9N6B5) |
| 32 Zfam 5* | EM:Z64553 | CT21013 (SPTR:Q9VX08) | C16A3.4 (SPTR:Q18036) |
| 33 Zfam 6* | Ptg-12 (EM:X97303) | CT36542 (SPTR:Q9VZF0) | ZK686.4 (SP:P34670) |
| 34 Zfam 7* | EM:AC005606 | CT31867 (SPTR:Q9W149) | |
| 35 Zfam 8* | EM:AK000711 | CT4004 (SPTR:Q9V9Z6) | |
| 36 Zfam 9* | EM:HS626B19 | CT32584 (SPTR:Q9VRV0) | |
| 37 Zfam 10* | Br140/BRPF1 (SP:P55201) | CT5659 (SPTR:Q9V4J4) | |
| Br140-like (SP:O95696) | |||
| 38 Zfam 11* | EM:AI077328 | CT32574 (SPTR:Q9VRQ6) | |
| 39 Zfam 12* | HPCMF (SPTR:Q9P0J7) | CT32121 (SPTR:Q9VHI5) |
*Families we believe not to have been defined previously. Human genes are identified by gene name and, where names have not yet been given, by database accession number. Drosophila sequences are identified by gene name and corresponding Gadfly protein symbol in brackets [14], or just by symbol where no name has been ascribed. C. elegans sequences are identified by name where possible and by coding sequence identifier [15]. All sequences are also identified by accession number: where possible these are SWISSPROT TREMBL accession numbers (designated SPTR). In a few cases only SWISSPROT accession numbers (designated SP) could be identified. For a minority of human genes no protein database entries have been made. These derive from EST or genomic sequences and the corresponding EMBL nucleotide database accession number (designated EM) is given.
Figure 3.
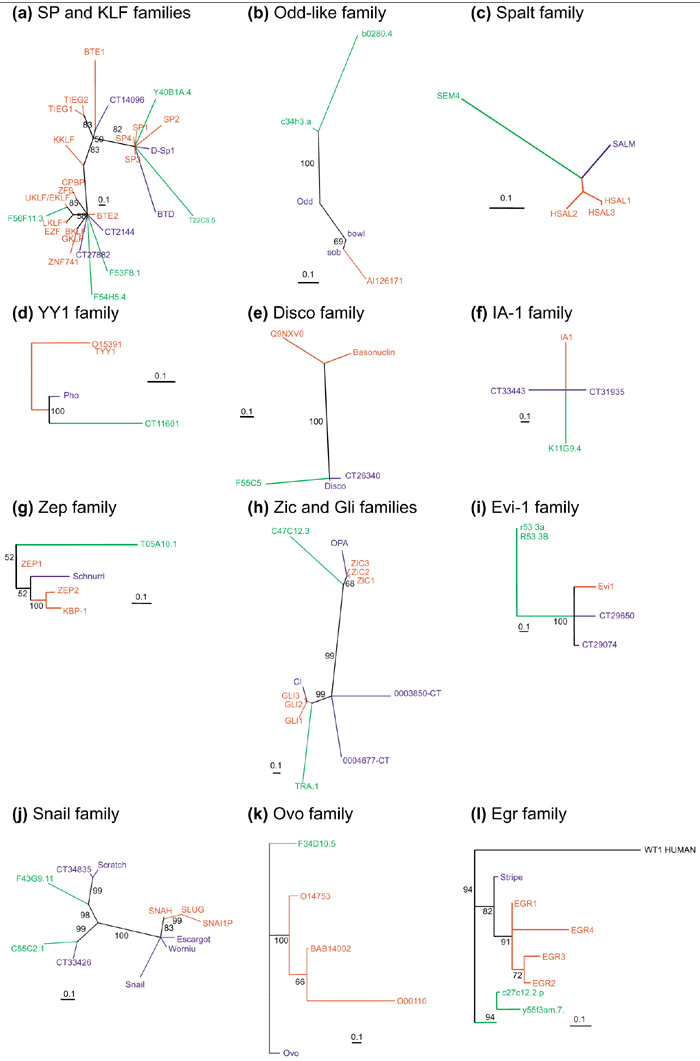
Phylogenies of the gene families identified in our analysis for which more than three family members were present. (a) SP and KLF families; (b) Odd-like family; (c) Spalt family; (d) YY1 family; (e) Disco family; (f) IA-1 family; (g) Zep family; (h) Zic and Gli families; (i) Evi-1 family; (j) Snail family; (k) Ovo family; (l) Egr family. In each tree, the scale bar indicates a maximum likelihood branch length of 0.1 inferred substitutions per site and the numbers next to relevant branches are percentage quartet-puzzling support values. Genes and branches are color coded according to species: human genes are red, Drosophila genes are blue and C. elegans genes are green. Most trees are unrooted and built with members of only a single orthology group, as in only two cases could sequences from separate groups be confidently aligned. One of these exceptions is the SP and KLF families (a), which were analyzed together as their similar ZNF number and structure suggest relatively recent common ancestry. The other is the Zic and Gli families (h), which have a similar number and arrangement of C2H2 fingers. This tree also includes two 'orphan' Drosophila genes that have a similar finger arrangement. The phylogenetic analyses, with the exception of the KLF group, either failed to resolve relationships sufficiently to confirm or disprove orthology or showed that each group was descended from a single gene present in the common ancestor of humans, C. elegans and Drosophila. We therefore call these families 'orthology groups', implying that genes from different species within each family are orthologs. Consequently, genes from one species within a family are paralogs. For the KLF and SP genes, the tree topology shows monophyly of the SP genes and suggests that multiple KLF orthology groups may be present, although the poor resolution does not allow definition of these.
Discussion
The 39 families identified above represent the conservative minimum of C2H2 ZNF genes present in the common ancestor of Drosophila, C. elegans and humans. They have, however, essentially been defined on one criterion - sequence identity at defined sites. It is possible that other features of zinc-finger genes could indicate orthology in the absence of sequence conservation, including similarities in the spacing between the paired histidines and cysteines, finger number, finger organization, intron/exon structure, the presence of other conserved domains and similarity of function. An example of this is the invertebrate hunchback and vertebrate Ikaros-related genes (Ikaros, Helios, Eos and Aiolos), which have low levels of sequence identity but a similar unusual arrangement of zinc fingers. Such examples may also represent orthology groups; their definition is, however, more subjective and we have not included them in our 39 groups.
Even including speculative orthology groups such as hunchback/Ikaros, genes for which orthology can be determined represent less than 25% of the C2H2 ZNF gene complement of each genome. This suggests that many orthologous relationships may not have been identified using our criteria. Whereas lineage-specific gene loss may account for our inability to identify orthologs for a proportion of the remaining 'nonassignable' genes, for most genes orthology is presumably cryptic to the point that it can no longer be recognized. This is presumably a result of high rates of sequence divergence. A key question, then, is how many orthology groups are hidden in this remaining approximately 75% of genes? Direct extrapolation from our finding that 39 orthology groups contain about 25% of genes would suggest that another 117 orthology groups remain undetected. Evidence from human and Xenopus genomes, however, suggests that the number may be much less, as in both taxa a considerable number of C2H2 ZNF genes (KRAB C2H2 ZNF genes in humans and FAX and FAR C2H2 ZNF genes in Xenopus) have been reported to have evolved by separate mass gene duplications [7, 8, 9]. Such lineage-restricted gene duplication suggests that a considerable proportion of the nonassignable genes may have evolved from a comparatively small number of ancestral genes. We therefore suggest that our 39 orthology groups represent a much larger proportion of the total existing groups than the 25% of genes they contain would suggest. Identifying precisely how many other groups there are, however, is a major bioinformatic challenge that will require data from other, phylogenetically well placed, taxa.
Conclusions
We have conclusively identified 39 families of C2H2 ZNF genes by comparing Drosophila and C. elegans sequences with human sequences. Of these, 17 have not been previously defined, and we propose that 38 represent definitive groups of orthologous genes, each deriving from a single gene in the common ancestor of these three organisms. Therefore, on the basis of current metazoan phylogeny [4], a member of each of these groups was primitively present in all triploblast bilaterian taxa, and they represent the minimum C2H2 ZNF complement in the bilaterian common ancestor.
Materials and methods
Drosophila and C. elegans sequences were identified by searching the complete predicted protein sets (gadfly and wormpep 24, respectively) with a Hidden Markov Model profile generated from the PFAM C2H2 ZNF seed alignment [10]. We searched at two stringencies, E < 10 and E < 1, identifying 394 and 332 Drosophila and 220 and 156 C. elegans sequences, respectively. Examination of the datasets showed the E < 10 datasets to contain some other types of zinc fingers (for example ring fingers), and that the E < 1 dataset excluded some genuine C2H2 ZNFs. We used this method rather than relying on previous identifications of C2H2 ZNF genes (see for example [11]) as we wanted to be confident we had identified all members of this superfamily. Such stringent criteria could not be applied to identification of human sequences, where many genes are currently represented only by short or fragmented sequences in genomic or EST databases. Inclusion of such sequences in the dataset could potentially have biased our preliminary analyses because of their short length. Instead, human sequences were identified using the listing provided by the SMART database [12], and edited to remove short sequences (<100 amino acids). This provided a sufficiently large and diverse dataset of long sequences for our preliminary analyses, but raised the possibility that human orthologs of Drosophila and C. elegans sequences might be missed because of their exclusion from our human dataset. We circumvented this by using FASTA comparisons of all Drosophila and C. elegans C2H2 ZNF protein sequences against all available human genomic DNA and EST sequences to identify orthologs absent from our human dataset. Molecular phylogenetic analyses were performed using the maximum likelihood method with one fixed and eight gamma-distributed rates, implemented by Puzzle [13].
Additional data files
The following additional data files are available: alignments for all orthology groups and datasets.
Supplementary Material
Alignments for all orthology groups
Datasets
References
- Fitch WM. Homology, a personal view on some of the problems. Trends Genet. 2000;16:227–231. doi: 10.1016/s0168-9525(00)02005-9. [DOI] [PubMed] [Google Scholar]
- Tatusov RL, Koonin EV, Lipman DJ. A genomic approach to protein families. Science. 1997;278:631–637. doi: 10.1126/science.278.5338.631. [DOI] [PubMed] [Google Scholar]
- Tatusov RL, Galperin M, Natale D, Koonin EV. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000;28:33–36. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adoutte A, Balavoine G, Lartillot N, Lespinet O, Prud'homme B, de Rosa R. The new animal phylogeny: Reliability and implications. Proc Natl Acad Sci USA. 2000;97:4453–4456. doi: 10.1073/pnas.97.9.4453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson WR, Lipmann DJ. Improved tools for biological sequence comparison. Proc Natl Acad Sci USA. 1988;85:2444–2448. doi: 10.1073/pnas.85.8.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohm S, Frishman D, Mewes HW. Variations of the C2H2 zinc finger motif in the yeast genome and classification of yeast zinc finger proteins. Nucleic Acids Res. 1997;25:2464–2469. doi: 10.1093/nar/25.12.2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellefroid E, Poncelet D, Lecocq P, Revelant PO, Martial J. The evolutionarily conserved Krüppel-associated box domain defines a subfamily of eukaryotic multifingered proteins. Proc Natl Acad Sci USA. 1991;88:3608–3612. doi: 10.1073/pnas.88.9.3608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knochel W, Poting A, Koster M, el Baradi T, Nietfeld W, Bouwmeester T, Piele T. Evolutionary conserved modules associated with zinc fingers in Xenopus laevis. Proc Natl Acad Sci USA. 1989;86:6097–6100. doi: 10.1073/pnas.86.16.6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klocke B, Koster M, Hille S, Bouwmeester T, Bohm S, Pieler T, Knochel W. The FAR domain defines a new Xenopus laevis zinc finger protein subfamily with specific RNA homopolymer binding activity. Biochim Biophys Acta. 1994;1217:81–89. [PubMed] [Google Scholar]
- Bateman A, Birney E, Durbin R, Eddy S, Howe K, Sonnhammer E. The Pfam Protein Families Database. Nucleic Acids Res. 2000;28:263–266. doi: 10.1093/nar/28.1.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke N, Berg J. Zinc fingers in Caenorhabditis elegans: finding families and probing pathways. Science. 1998;282:2018–2022. doi: 10.1126/science.282.5396.2018. [DOI] [PubMed] [Google Scholar]
- Schultz J, Copley RR, Doerks T, Ponting CP, Bork P. SMART: A Web-based tool for the study of genetically mobile domains. Nucleic Acids Res. 2000;28:231–134. doi: 10.1093/nar/28.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strimmer K, von Hassler S. Quartet puzzling: a quartet maximum likelihood method for reconstructing tree topologies. Mol Biol Evol. 1996;13:964–969. [Google Scholar]
- The Berkeley Drosophila Genome Project http://www.fruitfly.org/
- The C. elegans Protein Database Wormpep http://www.sanger.ac.uk/Projects/C_elegans/wormpep/
- Pavletich N, Pavo C. Crystal structure of a five-finger GLI-DNA complex: new perspectives on zinc fingers. Science. 1993;261:1701–1707. doi: 10.1126/science.8378770. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Alignments for all orthology groups
Datasets