

Abstract
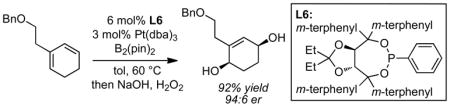
The enantioselective 1,4-diboration of cyclic dienes with a new taddol-derived phosphonite ligand occurs with excellent enantioselectivity. Oxidation delivers the derived 1,4 diol whereas homologation can be used to deliver a chiral 1,6 diol.
The enantioselective 1,4-diboration of 1,3-dienes is a reaction that provides 2-alkene-1,4-diols upon oxidative work-up.1,2,3 This overall reaction does not have a generally effective complement in contemporary catalytic enantioselective methodology and it is important to gain a greater understanding of the substrate scope.4 Previous examples from our laboratory suggested that this transformation can be realized with Pt(0) catalysis in the presence of a chiral TADDOL-derived phosphonite ligands. These studies included an example of an enantioselective 1,4-diboration of a prochiral cyclic diene (Scheme 1), a reaction that delivers a new chiral carbocyclic product with good efficiency. Using a chiral phosphonite ligand, this reaction was found to be highly selective with n-alkyl-substituted substrates such as 1a. However, further study of the scope of this reaction revealed that with aryl or branched alkyl substituents on the substrate enantioselection was severely diminished (vide infra). In an effort to render this transformation more generally useful for asymmetric synthesis, we have undertaken a systematic survey of substituted TADDOL-derived phosphonites5 and report herein, that with an appropriately modified ligand structure, significantly improved generality is observed.
Scheme 1.

To identify a chiral phosphonite ligand structure that is effective with a range of substrates, 2-butyl-1,3-cyclohexadiene (1a) and 2-cyclohexyl-1,3-cyclohexadiene (2a) were chosen as representative substrates and their diboration reactions were surveyed with a range of taddol-derived phosphonite ligands. As depicted in Table 1, with the ligand described in our preliminary report (L2, entries 3 and 4), the diboration of n-butyl substituted substrate 1a occurred with excellent enantioselectivity, however, the level of selectivity with cyclohexyl substituted substrate 2a was markedly inferior. In an effort to ameliorate this problem, the size of the substituents on the aryl rings of the ligand backbone was modified. Previous experience showed that the meta positions of the aryl rings are most consequential1,6 and, indeed, dramatic changes in enantioselection were noticed when the methyl groups of the ligand (R) were replaced with either smaller (ligand L1) or larger substituents (ligands L3 and L4). As observed in Table 1, while the use of alkyl substituents on the ligand backbone did not lead to a generally effective ligand structure, use of aryl-substituted ligands was highly rewarding and offered excellent levels of enantioselection for both substrates (ligands L5 and L6, entries 9–12). Comparison of ligands L5 and L6 showed that the more encumbered dioxolane protecting group provided a subtle enhancement in stereoselection and this ligand was chosen for further study. It is also important to note that with only 1.2 equivalents of ligand relative to platinum, reduced selectivity was observed in the diboration of 1a (77:23 er). We attribute this outcome to incomplete precomplexation of the catalyst; precomplexation at 80 °C with 1.2 equivalents of ligand returned selectivity to higher levels.
Table 1.
Evaluation of Ligands in the Pt-Catalyzed Enantioselective Diboration of 1 and 2.a
 | ||||||
|---|---|---|---|---|---|---|
| entry | diene | ligand | R | R1 | % yieldb | erc |
| 1 | 1a | L1 | H | Me | 31 | 55:45 |
| 2 | 2a | L1 | H | Me | 69 | 55:45 |
| 3 | 1a | L2 | Me | Me | 87 | 94:6 |
| 4 | 2a | L2 | Me | Me | 89 | 75:25 |
| 5 | 1a | L3 | Et | Me | 89 | 82:18 |
| 6 | 2a | L3 | Et | Me | 79 | 56:44 |
| 7 | 1a | L4 | t-Bu | Me | 60 | 61:39 |
| 8 | 2a | L4 | t-Bu | Me | 19 | 52:48 |
| 9 | 1a | L5 | Ph | Me | 92 | 94:6 |
| 10 | 2a | L5 | Ph | Me | 87 | 95:5 |
| 11 | 1a | L6 | Ph | Et | 97 | 96:4 |
| 12 | 2a | L6 | Ph | Et | 87 | 96:4 |
Reaction conditions: 1.05 equiv. B2(pin)2, precomplexation for 1 h at room temp, reaction time = 12 h, oxidation at room temp for 4 h.
Yield of purified material.
Enantiomer ratio was determined by GC, or HPLC analysis on a chiral stationary phase.
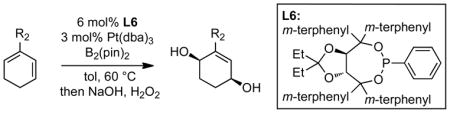
With a more generally effective ligand in hand, the catalytic enantioselective diboration of a range of substituted cyclic dienes was examined (Table 2).7,8 The standard set of reaction conditions employed 6 mol% ligand L6, 3 mol% Pt(dba)3, and 1.05 equivalents of B2(pin)2 in toluene solvent for 12 hours at 60 °C. While these conditions were effective for many substrates, it was found that for the phenyl-substituted diene in entry 3, selectivity was improved by executing the reaction at room temperature after pre-activating the catalyst at 60 °C for 20 minutes. Additionally, substrates in entries 8 and 11 reacted sluggishly under the standard conditions and required an increased catalyst loading to achieve the yields depicted in Table 2. Notable features of the substrate scope are that the reaction accommodates both aliphatic and aromatic substitution and that both small and large substituents are tolerated. The example in entry 11 suggests that seven-membered rings are processed selectively, even though these substrates require added catalyst and, even then, the cycloheptadiene provided moderated yields of reaction product. Another notable point that this substrate survey revealed is that protected alcohol and protected aldehyde functionality is well tolerated.
Table 2.
Catalytic Enantioselective Diboration of Cyclic Dienes.a
 | ||||
|---|---|---|---|---|
| entry | substrate | productb | % yieldc | erd |
| 1 |
 1a |
 1b |
97 | 96:4 |
| 2 |
 2a |
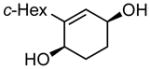 2b |
87 | 96:4 |
| 3 |
 3a |
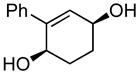 3b |
72 63e |
85:15 93:7 |
| 4 |
 4a |
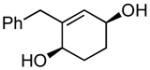 4b |
94 | 94:6 |
| 5 |
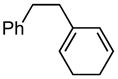 5a |
 5b |
91 | 95:5 |
| 6 |
 6a |
 6b |
92 | 96:4 |
| 7 |
 7a |
 7b |
75 | 96:4 |
| 8f |
 8a |
 8b |
94 | 96:4 |
| 9 |
 9a |
 9b |
92 | 94:6 |
| 10 |
 10a |
 10b |
84 | 95:5 |
| 11g |
 11a |
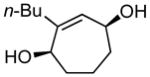 11b |
32 | 96:4 |
Conditions: 1.05 equiv. B2(pin)2, precomplexation for 1 h at room temp, reaction time = 12 h, oxidation at room temp for 4 h.
Absolute configuration determined by x-ray crystallography for 2b and 6b; by comparison to the literature for 1b (ref. 1), and assumed by analogy for others.
Yield of purified material. Value is an average of two experiments.
Enantiomer ratio was determined by GC, or HPLC, analysis on a chiral stationary phase.
Reaction at 60 °C for 20 min, then ambient for 48 hours.
Reaction employed 6 mol% Pt(dba)3, 12 mol% ligand L6 for 24 hours.
Reaction with 10 mol% Pt(dba)3, 20 mol% ligand L6 for 48 hours
One attractive feature of the diboration products is that the organoboron group may be readily converted to a range of useful functional groups or used in strategic C-C bond-forming processes. While oxidation cleanly delivers hydroxylated end products as depicted in Table 1, homologation can provide other derivatives.9 While Matteson homologation reactions10 have previously been applied to 1,2-bis(boronates) that arise from alkene diboration,11 this transformation has not been applied to 2-alkene-1,4-bis(boronates) that would arise from diene diboration. To examine the efficacy of this process, the sequence in Scheme 2 was examined. Thus, diene 6a was subjected to catalytic diboration in the presence of the chiral catalyst and after 12 hours of reaction, the solvent was removed in vacuo, tetrahydrofuran was added, the mixture cooled to −78 °C and treated with chloromethyllithium (2.2 equiv). Upon oxidative work-up, this sequence delivered the 1,6-diol reaction product 12 in excellent yield and stereoselectivity.
Scheme 2.

It was also considered that the 2-alkene-1,4-bis(boronates) might engage in allylation reactions but the mode of regioselection was uncertain.12 To learn more about this process and about the types of synthesis targets that might be accessed with these structures, the sequence in Scheme 3 was carried out. Thus, after diboration of diene 2a in toluene at 60 °C for 12 h, isovaleraldehyde (3 equiv) was added to the reaction mixture and the concoction heated to 80 °C for 30 h. After oxidation, this reaction furnished diol 14 in excellent yield, diastereo- and enantiomeric purity.8 An exciting outcome of this sequence is that C-C bond formation appeared to occur by association of the aldehyde to putative intermediate allylboronate 13, followed by progression to the product through ts-1.13 Importantly, the chair-like arrangement of the reacting array of atoms was maintained with good fidelity and furnished the product in a predictable manner. That the intermediate allylation product, prior to oxidation, maps well onto cladiellin diterpene natural products portends the use of this overall synthesis transform in complex molecule synthesis.14
Scheme 3.

In conclusion, we have described the development of a Pt-catalyzed diboration that bears general applicability to prochiral substituted cyclohexadiene substrates.
Experimental
((4R,5R)-2,2-dimethyl-1,3-dioxolane-4,5-diyl)bis(di([1,1′:3′,1″-terphenyl]-5′-yl)methanol) (15)
Prepared from 1-bromo-3,5-diphenylbenzene15 following literature procedures (495 mg, 62% yield).5c 1H NMR (500 MHz, CDCl3): δ 1.14 (6H, s), 5.11 (2H, s), 7.32–7.40 (16H, m), 7.41–7.44 (8H, m), 7.52–7.54 (8H, m), 7.68–7.70 (10H, m), 7.76 (4H, d, J = 1.7 Hz), 7.83 (2H, t, J = 1.7 Hz), 7.97 (4H, d, J = 1.7 Hz); 13C NMR (100 MHz, CDCl3): δ 146.9, 143.9, 141.8, 141.4, 141.3, 141.1, 129.01, 128.96, 127.59, 127.57, 127.5, 126.6, 125.9, 125.7, 125.6, 110.3, 81.9, 78.8, 27.6; IR (neat): 3324.0 (br), 3034.5 (w), 1595.2 (m), 1497.5 (w), 1427.2 (w), 1239.8 (w), 1165.5 (w), 1049.1 (w), 879.7 (m), 759.2 (s), 742.1 (s), 696.3 (s), 614.2 (w) cm−1; HRMS-(ESI+) for C79H62O4Na [M+Na]: calculated: 1097.4546, found: 1097.4557; [α]D20: +37.80 (c = 0.84, CHCl3, l = 50 mm).
((4R,5R)-2,2-diethyl-1,3-dioxolane-4,5-diyl)bis(di([1,1′:3′,1″-terphenyl]-5′-yl)methanol). (16)
Prepared from 1-bromo-3,5-diphenylbenzene14 following literature procedures (3.75 g, 62% yield).5c 1H NMR (500 MHz, CDCl3): δ 0.63 (6H, t, J = 7.4 Hz), 1.35–1.43 (4H, m), 4.60 (2H, br), 4.88 (2H, s), 7.29–7.33 (8H, m), 7.35–7.39 (16H, m), 7.51–7.53 (8H, m), 7.62–7.64 (8H, m), 7.68–7.69 (2H, m), 7.77–7.78 (6H, m), 7.91 (4H, d, J = 1.7 Hz); 13C NMR (125 MHz, CDCl3): δ 147.2, 143.8, 141.8, 141.4, 141.3, 141.0, 128.93, 128.92, 127.53, 127.51, 127.48, 127.47, 126.6, 125.8, 125.7, 125.6, 113.1, 81.4, 79.0, 30.1, 8.4; IR (neat): 3203.1 (br), 3035.4 (w), 2970.8 (w), 1595.5 (m), 1497.8 (w), 1426.7 (w), 1174.6 (w), 1031.1 (w), 897.7 (w), 759.2 (s), 740.8 (s), 727.5 (m), 696.5 (s), 614.1 (w) cm−1; HRMS-(MALDI+) for C81H66O4Na [M+Na]: calculated: 1125.4853, found: 1125.4852; [α]D20: −22.80 (c = 1.01, CHCl3, l = 50 mm).
(3aR,8aR)-4,4,8,8-tetra([1,1′:3′,1″-terphenyl]-5′-yl)-2,2-dimethyl-6-phenyltetrahydro-[1,3]dioxolo[4,5-e][1,3,2]dioxaphosphepine (L5)
Prepared from 15 in direct analogy to the literature (384 mg, 70% yield).5c 1H NMR (500 MHz, CDCl3): δ 0.34 (3H, s), 1.59 (3H, s), 5.15 (1H, d, J = 8.6 Hz), 6.24 (1H, dd, J = 8.6 Hz, 4.6 Hz), 7.30–7.31 (6H, m), 7.34–7.47 (18H, m), 7.54–7.61 (15H, m), 7.67–7.70 (5H, m), 7.72–7.74 (2H, m), 7.81–7.85 (5H, m), 8.00 (4H, s), 8.24 (2H, m); 13C NMR (100 MHz, CDCl3): δ 147.9, 147.0, 146.9, 143.23, 143.22, 142.33, 142.31, 141.88, 141.82, 141.6, 141.5, 141.41, 141.40, 141.33, 141.31, 141.2, 140.9, 131.3, 130.3, 130.0, 129.1, 129.03, 128.97, 128.90, 128.83, 127.7, 127.63, 127.59, 127.54, 127.51, 127.4, 126.8, 126.2, 126.1, 125.8, 125.7, 125.6, 112.2, 84.63, 84.59, 84.0, 83.9, 83.3, 83.1, 82.90, 82.85, 28.3, 25.1; 31P NMR (162 MHz, CDCl3): δ 156.7; IR (neat): 3034.2 (w), 1594.8 (m), 1497.2 (w), 1427.2 (w), 1160.3 (w), 1031.8 (m), 996.8 (m), 758.4 (s), 735.4 (s), 695.1 (s), 613.8 (w) cm−1; HRMS (ESI+) for C85H65O4NaP [M+Na]: calculated: 1203.4518, found: 1203.4495; [α]D20: −103.26 (c = 1.12, CHCl3, l = 50 mm).
(3aR,8aR)-4,4,8,8-tetra([1,1′:3′,1″-terphenyl]-5′-yl)-2,2-diethyl-6-phenyltetrahydro-[1,3]dioxolo[4,5-e][1,3,2]dioxaphosphepine (L6)
Prepared from 16 in direct analogy to the literature (494 mg, 70% yield).5c 1H NMR (400 MHz, CDCl3): δ 0.28 (3H, t, J = 7.4 Hz), 0.72 (2H, q, J = 7.5 Hz), 0.82 (3H, t, J = 7.5 Hz), 1.71 (2H, q, J = 7.4 Hz), 5.26 (1H, d, J = 8.7 Hz), 6.02 (1H, dd, J = 8.7 Hz, 3.9 Hz), 7.28–7.43 (23H, m), 7.44–7.54 (8H, m), 7.58–7.63 (12H, m), 7.68–7.80 (6H, m), 7.89 (2H, d, J = 1.6 Hz), 7.93–7.98 (2H, m), 8.01 (2H, d, J = 1.6 Hz), 8.15 (2H, d, J = 1.6 Hz); 13C NMR (100 MHz, CDCl3): δ 147.8, 147.22, 147.19, 143.32, 143.30, 142.59, 142.57, 141.9, 141.6, 141.5, 141.49, 141.47, 141.4, 141.3, 141.2, 140.9, 131.3, 130.3, 130.0, 129.05, 129.04, 129.01, 128.95, 128.8, 128.7, 127.6, 127.56, 127.54, 127.50, 127.4, 127.01, 126.97, 126.05, 125.96, 125.7, 125.6, 125.4, 116.1, 84.3, 84.2, 83.79, 83.77, 83.4, 83.3, 82.6, 82.4, 30.2, 27.9, 8.7, 8.6; 31P NMR (162 MHz, CDCl3): δ 157.6; IR (neat): 3034.2 (w), 2970.8 (w), 1594.3 (w), 1426.6 (w), 1263.9 (w), 1170.4 (w), 1031.4 (w), 930.6 (m), 758.3 (m), 733.0 (s), 693.4 (s), 613.1 (m), 489.8 (w) cm−1; HRMS (ESI+) for C87H69O4NaP [M+Na]: calculated: 1231.4831, found: 1231.4832; [α]D20: −87.30 (c = 0.51, CHCl3, l = 50 mm).
Preparation of cyclic dienes by Kumada coupling.7
Magnesium (292 mg, 12.0 mmol) and a single crystal of I2 were diluted with diethyl ether (20 mL). Bromocyclohexane (1.23 mL, 10.0 mmol) was slowly added. The stirred reaction was heated to 40 °C for 3 h and cooled to room temperature. In a separate flask, NiCl2(dppe) (16 mg, 30 mmol), cyclohexa-1,5-dien-1-yl diphenyl phosphate (1.00 g, 3.05 mmol), and Et2O (10 mL) were cooled to 0 °C under N2. The Grignard reagent was then added via syringe. Upon complete reaction (TLC analysis; 1–2 h) saturated NH4Cl solution (10 mL) was added. Extraction with diethyl ether (2×20 mL), drying, concentration, and purification by silica gel chromatography (100% hexanes) afforded a colorless oil (331 mg, 68% yield). A gravity column was required to separate the desired product from 1,1′-bi(cyclohexane), which can only be detected by NMR.
[1,1′-bi(cyclohexane)]-1,5-diene (2a)
(331 mg, 68% yield). 1H NMR (500 MHz, CDCl3): δ 1.09–1.19 (3H, m), 1.23–1.32 (2H, m), 1.66–1.77 (5H, m), 1.85 (1H, t, J = 11.6 Hz), 2.05–2.13 (4H, m), 5.44–5.46 (1H, m), 5.80–5.83 (1H, m), 5.88 (1H, dd, J = 11.2 Hz, 1.5 Hz); 13C NMR (125 MHz, CDCl3): δ 141.3, 126.9, 126.6, 118.1, 43.7, 32.2, 26.9, 26.6, 22.8, 22.6; IR (neat): 3033.1 (w), 2921.6 (s), 2850.2 (s), 2822.3 (w), 1447.7 (m), 1425.5 (w), 1165.1 (w), 998.9 (w), 948.2 (w), 891.6 (w), 806.6 (m), 736.8 (m), 687.7 (w), 592.2 (m), 570.2 (w), 518.7 (w) cm−1; HRMS (ESI+) for C12H19 [M+H]: calculated: 163.1487, found:163.1488; Rf = 0.78 (100% hexanes, stain in KMnO4).
(cyclohexa-1,5-dien-1-ylmethyl)benzene (4a)
(2.41 g, 77% yield) 1H NMR (500 MHz, CDCl3): δ 2.09–2.21 (4H, m), 3.37 (2H, s), 5.51–5.54 (1H, m), 5.76–5.84 (2H, m), 7.19–7.22 (3H, m), 7.27–7.31 (2H, m); 13C NMR (100 MHz, CDCl3): δ 140.2, 135.0, 129.1, 128.5, 127.19, 127.15, 126.2, 122.1, 42.0, 22.65, 22.58; IR (neat): 3027.6 (w), 2930.4 (w), 2872.8 (w), 2821.6 (w), 1493.7 (w), 1452.6 (w), 1425.8 (w), 1164.5 (w), 1078.5 (w), 953.9 (m), 787.9 (w), 755.2 (w), 726.8 (s), 697.0 (s), 668.0 (m), 620.0 (m), 601.4 (m), 530.8 (w), 487.5 (w), 445.7 (w) cm−1; HRMS (ESI+) for C13H15 [M+H]: calculated: 171.1174, found: 171.1168.
(2-(cyclohexa-1,5-dien-1-yl)ethyl)benzene (5a)
(234 mg, 85% yield) 1H NMR (400 MHz, CDCl3): δ 2.12 (4H, br), 2.34 (2H, t, J = 8.0 Hz), 2.70–2.74 (2H, m), 5.49 (1H, br), 5.86–5.92 (2H, m), 7.19–7.21 (3H, m), 7.27–7.31 (2H, m); 13C NMR (MHz, CDCl3): δ 142.4, 135.4, 128.7, 128.4, 127.4, 127.1, 125.9, 120.8, 37.8, 35.2, 22.62, 22.57; IR (neat): 3026.9 (m), 2930.4 (m), 2871.5 (w), 2821.2 (m), 1603.2 (w), 1495.6 (m), 1453.1 (m), 1437.7 (w), 1425.9 (w), 1164.7 (w), 1030.1 (w), 939.1 (w), 805.2 (m), 745.8 (s), 696.5 (s), 593.3 (m), 566.4 (m), 544.4 (m), 491.7 (m), 462.2 (w) cm−1; HRMS (ESI+) for C14H17 [M+H]: calculated: 185.1330, found: 185.1339.
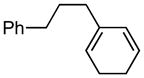
(3-(cyclohexa-1,5-dien-1-yl)propyl)benzene (6a)
(1.06 g, 58% yield) 1H NMR (400 MHz, CDCl3): δ 1.71–1.79 (2H, m), 2.06–2.13 (6H, m), 2.61 (2H, t, J = 7.7 Hz), 5.50 (1H, br), 5.84 (2H, br), 7.16–7.20 (3H, m), 7.26–7.30 (2H, m); 13C NMR (100 MHz, CDCl3): δ 142.8, 135.7, 128.7, 128.4, 127.4, 127.0, 125.8, 120.6, 35.6, 35.3, 30.3, 22.7, 22.6; IR (neat): 3026.8 (w), 2930.2 (m), 2856.4 (w), 2821.7 (w), 1495.5 (w), 1453.1 (w), 1029.7 (w), 941.3 (w), 805.7 (w), 740.8 (s), 696.4 (s), 589.0 (w), 544.4 (w), 494.2 (w), 467.7 (w) cm−1; HRMS (ESI+) for C15H19 [M+H]: calculated: 199.1487, found: 199.1496.
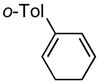
2′-methyl-3,4-dihydro-1,1′-biphenyl (7a)
(225 mg, 88% yield) 1H NMR (500 MHz, CDCl3): δ 2.20–2.25 (2H, m), 2.29–2.35 (5H, m), 5.71–5.73 (1H, m), 5.89–5.93 (1H, m), 5.96–5.99 (1H, m), 7.12–7.18 (4H, m); 13C NMR (125 MHz, CDCl3): δ 142.2, 137.4, 135.6, 130.3, 128.8, 128.0, 127.1, 126.2, 125.9, 124.6, 22.8, 22.2, 20.3; IR (neat): 3035.2 (w), 2930.6 (w), 2822.3 (w), 1484.2 (w), 1453.7 (w), 997.5 (w), 823.7 (w), 756.3 (s), 734.4 (s), 611.0 (m), 501.2 (w), 453.5 (m) cm−1; HRMS (ESI+) for C13H15 [M+H]: calculated: 171.1174, found: 171.1174.
1-(cyclohexa-1,5-dien-1-yl)naphthalene (8a)
(243 mg, 79% yield) 1H NMR (500 MHz, CDCl3): δ 2.31–2.37 (2H, m), 2.41–2.46 (2H, m), 5.95 (1H, d, J = 4.3 Hz), 5.98 (1H, dt, J = 9.5 Hz, 4.4 Hz), 6.15 (1H, d, J = 9.5 Hz), 7.34 (1H, d, J = 6.8 Hz), 7.43–7.49 (3H, m), 7.78 (1H, d, J = 8.3 Hz), 7.84–7.88 (1H, m), 8.02–8.05 (1H, m); 13C NMR (125 MHz, CDCl3): δ 140.6, 136.5, 134.0, 131.6, 128.6, 128.5, 127.5, 126.3, 126.2, 125.9, 125.85, 125.81, 125.7, 125.6, 23.0, 22.2; IR (neat): 3035.4 (w), 2931.7 (w), 2819.5 (w), 1389.3 (w), 961.5 (w), 797.0 (m), 773.1 (s), 726.1 (m), 646.8 (m), 424.1 (w) cm−1; HRMS (ESI+) for C16H15 [M+H]: calculated: 207.1174, found: 207.1172.
2-(2-(cyclohexa-1,5-dien-1-yl)ethyl)-1,3-dioxolane (10a)
(867 mg, 55% yield) 1H NMR (500 MHz, CDCl3): δ 1.74–1.78 (2H, m), 2.05–2.11 (4H, m), 2.13–2.17 (2H, m), 3.82–3.89 (2H, m), 3.93–4.00 (2H, m), 4.87 (1H, t, J = 4.5 Hz), 5.50–5.52 (1H, m), 5.81–5.85 (2H, m); 13C NMR (125 MHz, CDCl3): δ 135.1, 127.2, 127.1, 120.5, 104.4, 65.1, 32.9, 30.1, 22.6, 22.5; IR (neat): 2929.4 (m), 2874.6 (m), 2822.5 (w), 1403.7 (m), 1131.9 (s), 1032.9 (s), 942.8 (s), 887.1 (m), 820.7 (w), 736.9(m), 591.7 (m), 551.3 (w) cm−1; HRMS (ESI+) for C11H17O2 [M+H]: calculated: 181.1229, found: 181.1220.
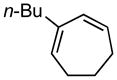
2-butylcyclohepta-1,3-diene (11a)
(325 mg, 73% yield) 1H NMR (500 MHz, CDCl3): δ 0.89 (3H, t, J = 7.3 Hz), 1.26–1.33 (2H, m), 1.35–1.41 (2H, m), 1.81–1.86 (2H, m), 2.01 (2H, t, J = 7.1 Hz), 2.22 (2H, dt, J = 5.6 Hz, 5.6 Hz), 2.28 (2H, dt, J = 5.9 Hz, 5.4 Hz), 5.62 (1H, t, J = 5.9 Hz), 5.69 (1H, d, J = 11.7 Hz), 5.83 (1H, dt, J = 11.7 Hz, 5.1 Hz); 13C NMR (100 MHz, CDCl3): δ 137.3, 133.5, 129.2, 128.6, 39.4, 31.88, 31.81, 30.1, 27.9, 22.5, 14.2; IR (neat): 2955.3 (m), 2924.5 (s), 2856.9 (m), 1437.1 (m), 1044.8 (w), 851.9 (w), 775.9 (w), 735.0 (m), 554.6 (m) cm−1; HRMS (ESI+) for C11H19 [M+H]: calculated: 151.1487, found: 151.1494.
Representative Procedure for Diene Diboration/Oxidation
In the glove box, Pt(dba)3 (13.5 mg, 15.0 mmol), ligand L6 (36.3 mg, 30.0 mmol), toluene (5.0 mL, 0.1 M) were mixed. After stirring for 1 h, B2(pin)2 (133.3 mg, 0.525 mmol) and [1,1′-bi(cyclohexane)]-1,5-diene (2a, 81.1 mg, 0.50 mmol) were added. The vessel was removed from the glovebox, and stirred at 60 °C for 12 h. The mixture was then cooled to 0 °C and slowly charged with tetrahydrofuran (3 mL), 3 M NaOH (3 mL) and 30 % H2O2 (1.5 mL). After stirring for 4 h at ambient temperature, the mixture was cooled to 0 °C and treated with aqueous sodium thiosulfate (3 mL, dropwise slowly). Extraction with ethyl acetate (3×20 mL), drying, concentration and purification (silica gel; hexane: ethyl acetate = 1:1 to 1:2) afforded a white solid (85.4 mg, 87% yield).
(2S,5R)-[1,1′-bi(cyclohexan)]-6-ene-2,5-diol (2b)
(85.4 mg, 87% yield) Rf = 0.17 (hexane:ethyl acetate = 1:1, stain in phosphomolybdic acid); 1H NMR (500 MHz, CDCl3): δ 1.01 (1H, dtd, J = 13.6 Hz, 11.9 Hz, 3.6 Hz), 1.11–1.20 (1H, m), 1.23–1.35 (3H, m), 1.61–1.82 (8H, m), 1.83–1.89 (3H, m), 2.08 (1H, tt, J = 11.5 Hz, 3.1 Hz), 4.10 (1H, br), 4.16 (1H, br), 5.53 (1H, d, J = 2.7 Hz); 13C NMR (125 MHz, CDCl3): δ 147.8, 126.4, 67.4, 65.8, 41.3, 33.7, 31.9, 29.9, 27.7, 27.1, 26.8, 26.5; IR (neat): 3300.3 (br), 2921.8 (s), 2850.4 (s), 1446.8 (m), 1285.9 (w), 1072.4 (m), 979.9 (m), 956.6 (w) cm−1; HRMS (ESI+) for C12H24N1O2 [M+NH4]: calculated: 214.1807, found: 214.1818; [α]D: +9.78 (c = 0.92, CHCl3, l = 50 mm); mp: 115.1–117.0 °C. Enantiomer ratio determined by GLC (diacetate derivative, Supelco β-dex, 180 °C, 20 psi), major enantiomer 33.27 min, minor enantiomer 33.64 min, 96:4 e.r. Absolute stereochemistry was determined by crystallography using anomalous dispersion (Flack parameter = 0.11).
(2S,5R)-2,3,4,5-tetrahydro-[1,1′-biphenyl]-2,5-diol (3b)
(59.9 mg, 63% yield) Rf = 0.21 (hexanes: ethyl acetate = 1:2, stain in PMA); 1H NMR (500 MHz, CDCl3): δ 1.77–1.85 (1H, m), 1.87–1.94 (1H, m), 1.98–2.07 (2H, m), 4.35(1H, br), 4.67 (1H, br), 6.13 (1H, dd, J = 2.9 Hz, 0.7 Hz), 7.29–7.32 (1H, m), 7.35–7.39 (2H, m), 7.49–7.52 (2H, m); 13C NMR (125 MHz, CDCl3): δ 141.2, 138.9, 130.9, 128.9, 128.1, 126.6, 67.5, 65.4, 29.3, 27.4; IR (neat): 3222.8 (br), 2938.0 (w), 1490.1 (w), 1441.9 (w), 1307.2 (w), 1047.1 (s), 969.6 (s), 950.3 (m), 760.1 (m), 697.3 (s), 508.4 (m), 485.0 (m) cm−1; HRMS (ESI+) for C12H13O1 [M+H-H2O]: calculated: 173.0966, found: 173.0966; [α]D20: +64.49 (c = 0.83, CHCl3, l = 50 mm); mp: 137.0–139.0 °C. Enantiomer ratio determined by GLC (diacetate derivative, Supelco β-dex, 180 °C, 20 psi), major enantiomer 48.65 min, minor enantiomer 49.65 min, 93:7 e.r.
(1S,4R)-2-benzylcyclohex-2-ene-1,4-diol (4b)
(96.0 mg, 94% yield) Rf = 0.23 (hexanes: ethyl acetate = 1:2, stain in PMA); 1H NMR (500 MHz, CDCl3): δ 1.65–1.76 (4H, m), 1.79–1.88 (2H, m), 3.43 (1H, d, J = 15.1 Hz), 3.52 (1H, d, J = 15.1 Hz), 3.95 (1H, br), 4.18 (1H, br), 5.58 (1H, br), 7.20–7.23 (3H, m), 7.28–7.31 (2H, m); 13C NMR (100 MHz, CDCl3): δ 142.3, 139.2, 129.7, 129.3, 128.7, 126.5, 66.9, 66.2, 40.5, 29.3, 28.0; IR (neat): 3313.5 (br), 2939.6 (m), 2867.3 (w), 1493.6 (w), 1453.1 (m), 1278.4 (w), 1070.6 (m), 1030.2 (m), 980.3 (m), 962.0 (m), 755.7 (w), 700.7 (s) cm−1; HRMS (ESI+) for C13H20O2N1 [M+NH4]: calculated: 222.1494, found: 222.1489; [α]D20: +100.80 (c = 0.92, CHCl3, l = 50 mm); mp: 114.8–116.6 °C. Enantiomer ratio determined by HPLC (Chiraldex OD-R, 3% iPrOH/hexanes, 1 mL/min, 220 nm), major enantiomer 45.91 min, minor enantiomer 63.95 min, 94:6 e.r.
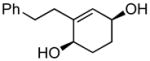
(1R,4S)-2-phenethylcyclohex-2-ene-1,4-diol (5b)
(50.0 mg, 91% yield) Rf = 0.18 (hexanes: ethyl acetate = 1:2, stain in PMA); 1H NMR (500 MHz, CDCl3): δ 1.64–1.71 (1H, m), 1.74–1.80 (1H, m), 1.81–1.89 (2H, m), 2.41 (1H, ddd, J = 14.9 Hz, 10.1 Hz, 6.4 Hz), 2.48–2.54 (1H, m), 2.74 (1H, ddd, J = 13.7 Hz, 9.8 z, 6.4 Hz), 2.83 (1H, dd, J = 13.7 Hz, 10.1 Hz, 5.7 Hz), 4.04 (1H, br), 4.15 (1H, br), 7.17–7.20 (3H, m), 7.26–7.30 (2H, m); 13C NMR (125 MHz, CDCl3): δ 142.3, 142.0, 128.62, 128.57, 128.4, 126.2, 67.1, 66.8, 35.5, 34.6, 29.4, 28.1; IR (neat): 3300.4 (br), 2935.0 (s), 2858.3 (m), 1495.8 (w), 1453.6 (m), 1276.4 (w), 1042.3 (m), 979.1 (m), 954.8 (m), 873.4 (w), 749.0 (m), 699.3 (s) cm−1; HRMS (ESI+) for C14H22O2N1 [M+NH4]: calculated: 236.1651, found: 236.1657; [α]D20: +9.82 (c = 0.62, CHCl3, l = 50 mm); mp: 114.5–116.0 °C. Enantiomer ratio determined by HPLC (Chiraldex OD-R, 3% iPrOH/hexanes, 1 mL/min, 220 nm), major enantiomer 67.78 min, minor enantiomer 84.01 min, 95:5 e.r.
(1R,4S)-2-(3-phenylpropyl)cyclohex-2-ene-1,4-diol (6b)
(94.0 mg, 92% yield) Rf = 0.28 (hexanes: ethyl acetate = 1:2, stain in PMA); 1H NMR (500 MHz, CDCl3): δ 1.64–1.72 (1H, m), 1.73–1.81 (2H, m), 1.82–1.89 (3H, m), 2.10–2.16 (1H, m), 2.21–2.27 (1H, m), 2.58–2.68 (2H, m), 4.00–4.03 (1H, m), 4.15(1H, br), 5.57 (1H, br), 7.17–7.19 (3H, m), 7,26–7.29 (2H, m); 13C NMR (125 MHz, CDCl3): δ 142.6, 142.5, 128.6, 128.5, 128.0, 126.0, 66.86, 66.82, 35.9, 33.4, 29.6, 29.4, 28.1; IR (neat): 3299.1 (br), 3025.2 (w), 2937.0 (s), 2859.7 (m), 1495.4 (m), 1453.1 (m), 1356.3 (w), 1287.8 (w), 1045.6 (m), 979.5 (m), 954.8 (m), 747.5 (m), 699.1 (s) cm−1; HRMS (ESI+) for C15H19O1 [M+H-H2O]: calculated: 215.1436, found: 215.1443; [α]D20: +31.37 (c = 0.91, CHCl3, l = 50 mm); mp: 114.2–116.1 °C. Enantiomer ratio determined by HPLC (diacetate derivative, Chiraldex OD-R, 1% iPrOH/hexanes, 220 nm), major enantiomer 10.96 min, minor enantiomer 9.71 min, 96:4 e.r. Absolute stereochemistry was determined by crystallography using anomalous dispersion (Flack parameter = −0.01).
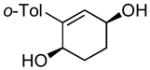
(2R,5S)-2′-methyl-2,3,4,5-tetrahydro-[1,1′-biphenyl]-2,5-diol (7b)
(76.6 mg, 75% yield) Rf = 0.26 (hexanes: ethyl acetate = 1:2, stain in PMA); 1H NMR (500 MHz, CDCl3): δ 1.83–1.92 (1H, m), 1.93–2.00 (3H, m), 2.30 (3H, s), 4.28–4.33 (1H, m), 4.34–4.36 (1H, m), 5.75 (1H, d, J = 3.2 Hz), 7.12–7.22 (4H, m); 13C NMR (100 MHz, CDCl3): δ 143.3, 139.4, 136.0, 131.6, 130.6, 129.2, 127.8, 125.9, 67.5, 66.5, 28.3, 28.0, 20.1; IR (neat): 3277.4 (br), 2944.8 (m), 2899.3 (m), 1483.5 (m), 1439.7 (m), 1290.4 (w), 1269.6 (w), 1042.6 (s), 980.8 (s), 954.0 (s), 842.1 (w), 764.3 (m), 656.8 (s), 606.0 (s), 445.5 (m) cm−1; HRMS (ESI+) for C13H15O1 [M+H-H2O]: calculated: 187.1123, found: 187.1122; [α]D20: +90.55 (c = 0.98, CHCl3, l = 50 mm); mp: 116.2–119.0 °C. Enantiomer ratio determined by GLC (diacetate derivative, Supelco β-dex, 160 °C, 20 psi), major enantiomer 98.20 min, minor enantiomer 99.65 min, 96:4 e.r.
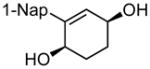
(1R,4S)-2-(naphthalen-1-yl)cyclohex-2-ene-1,4-diol (8b)
(33.9 mg, 94% yield) Rf = 0.21 (hexanes: ethyl acetate = 1:2, stain in PMA); 1H NMR (500 MHz, CDCl3): δ 1.94–2.02 (1H, m), 2.04–2.11 (3H, m), 4.42 (1H, br), 4.52 (1H, br), 5.92 (1H, d, J = 2.9 Hz), 7.36 (1H, dd, J = 7.1 Hz, 1.0 Hz), 7.45–7.51 (3H, m), 7.82 (1H, d, J = 8.3 Hz), 7.86–7.89 (1H, m), 7.93–7.95 (1H, m); 13C NMR (125 MHz, CDCl3): δ 142.0, 137.7, 134.0, 133.4, 131.9, 128.7, 128.3, 126.4, 126.3, 126.2, 125.53, 125.51, 68.0, 66.9, 28.5, 27.9; IR (neat): 3273.1 (br), 2898.9 (m), 1440.8 (w), 1272.2 (w), 1051.3 (m), 981.8 (m), 801.8 (s), 778.3 (s), 667.5 (m), 597.4 (m) cm−1; HRMS (ESI+) for C16H15O1 [M+H-H2O]: calculated: 223.1123, found: 223.1134; [α]D20: +26.13 (c = 0.15, CHCl3, l = 50 mm); mp: 176.5–179.2 °C. Enantiomer ratio determined by HPLC (dibenzoate derivative, Chiraldex OD-R, 1% iPrOH/hexanes, 1.0 mL/min, 220 nm), major enantiomer 18.86 min, minor enantiomer 22.62 min, 96:4 e.r.
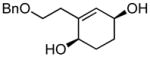
(1R,4S)-2-(2-(benzyloxy)ethyl)cyclohex-2-ene-1,4-diol (9b)
(34.3 mg, 92% yield) Rf = 0.17 (hexanes: ethyl acetate = 1:2, stain in PMA); 1H NMR (500 MHz, CDCl3): δ 1.69–1.76 (2H, m), 1.80–1.85 (2H, m), 2.30–2.36 (1H, m), 2.47–2.52 (1H, m), 3.56–3.60 (1H, m), 3.62–3.66 (1H, m), 3.99 (1H, br), 4.13 (1H, br), 4.52 (1H, d, J = 12.0 Hz), 4.55 (1H, d, J = 12.0 Hz), 5.59 (1H, d, J= 2.8 Hz), 7.28–7.37 (5H, m); 13C NMR (125 MHz, CDCl3): δ 141.5, 137.7, 129.8, 128.7, 128.11, 128.09, 73.5, 70.4, 66.95, 66.90, 35.3, 28.6, 28.1; IR (neat): 3343.2 (br), 2927.7 (s), 2857.1 (s), 1453.9 (m), 1360.3 (m), 1275.2 (w), 1074.6 (s), 980.3 (s), 737.7 (s), 698.2 (s), 608.1 (w); HRMS (ESI+) for C15H19O2 [M+H-H2O]: calculated: 231.1385, found: 231.1379; [α]D20: −5.41 (c = 0.53, CHCl3, l = 50 mm). Enantiomer ratio determined by HPLC (Chiraldex OD-R, 3% iPrOH/hexanes, 1.0 mL/min, 220 nm), major enantiomer 74.35 min, minor enantiomer 67.69 min, 94:6 e.r.
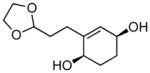
(1R,4S)-2-(2-(1,3-dioxolan-2-yl)ethyl)cyclohex-2-ene-1,4-diol (10b)
(90.0 mg, 84% yield) Rf = 0.23 (100% ethyl acetate, stain in PMA); 1H NMR (500 MHz, CDCl3): δ 1.63–1.75 (2H, m), 1.76–1.89 (4H, m), 2.18–2.31 (2H, m), 3.81–3.87 (2H, m), 3.92–3.97 (2H, m), 3.98 (1H, br), 4.10 (1H, br), 4.87 (1H, t, J = 4.7 Hz), 5.55 (1H, br); 13C NMR (125 MHz, CDCl3): δ 141.9, 128.1, 104.4, 66.69, 66.66, 65.07, 65.04, 32.0, 29.3, 28.1, 27.9; IR (neat): 3353.7 (br), 2937.5 (w), 2878.1 (s), 1443.1 (w), 1408.9 (m), 1265.6 (w), 1135.1 (s), 1044.3 (s), 977.8 (s), 955.6 (s), 895.6 (m), 644.4 (w); HRMS (ESI+) for C11H17O3 [M+H-H2O]: calculated: 197.1178, found: 197.1169; [α]D20: +15.91 (c = 0.95, CHCl3, l = 50 mm). Enantiomer ratio determined by HPLC (dibenzoate derivative, Chiraldex OD-R, 3% iPrOH/hexanes, 1.0 mL/min, 220 nm), major enantiomer 24.53 min, minor enantiomer 14.52 min, 95:5 e.r.
(1R,4S)-2-butylcyclohept-2-ene-1,4-diol (11b)
(14.7 mg, 32% yield) Rf = 0.26 (hexanes: ethyl acetate = 3:2, stain in PMA); 1H NMR (500 MHz, CDCl3): δ 0.91 (3H, t, J = 7.2 Hz), 1.29–1.36 (2H, m), 1.37–1.43 (2H, m), 1.55–1.69 (3H, m), 1.84–1.93 (2H, m), 2.08 (2H, td, J = 7.6 Hz, 1.2 Hz), 2.28–2.37 (1H, m), 4.12 (1H, d, J = 6.6 Hz), 4.26 (1H, ddd, J = 6.9 Hz, 6.8 Hz, 1.5 Hz), 5.71 (1H, d, J = 6.8 Hz); 13C NMR (100 MHz, CDCl3): δ 149.7, 129.5, 72.3, 68.2, 37.9, 34.5, 33.8, 30.6, 22.6, 19.5, 14.2; IR (neat): 3315.0 (br), 2924.3 (s), 2856.1 (m), 1647.7 (w), 1453.7 (m), 1272.8 (w), 1059.3 (s), 1028.1 (s), 937.1 (m), 654.6 (w); HRMS (ESI+) for C11H19O1 [M+H-H2O]: calculated: 167.1436, found: 167.1430; [α]D20: +6.28 (c = 0.53, CHCl3, l = 50 mm). Enantiomer ratio determined by HPLC (dibenzoate derivative, Chiraldex OD-R, 1% iPrOH/hexanes, 0.25 mL/min, 220 nm), major enantiomer 28.34 min, minor enantiomer 31.49 min, 96:4 e.r.
((1R,4S)-2-(3-phenylpropyl)cyclohex-2-ene-1,4-diyl)dimethanol (12)
Pt(dba)3 (6.7 mg, 7.5 mmol) and phosphonite ligand (L6) (18.1 mg, 15.0 mmol), and toluene (2.5 mL, 0.1 M) were mixed under Ar and stirred for 1 h. Then, B2(pin)2 (66.7 mg, 262.5 mmol) and (3-(cyclohexa-1,5-dien-1-yl)propyl)benzene (49.6 mg, 0.25 mmol) were added. After stirring at 60 °C for 12 h, the volatiles were removed. THF (2.5 mL) was added, the flask cooled to −78 °C, and bromochloromethane (35.7 mL, 0.55 mmol) and n-BuLi (0.19 mL, 0.55 mmol) added sequentially. After 10 min, the cooling bath was removed and the contents stirred for 12 h. Then, THF (2 mL), 3 M NaOH (2 mL) and 30 % H2O2 (1 mL) were added, slowly, at 0 °C and allowed to stir for 4 h at ambient. After cooling to 0 °C, saturated aqueous sodium thiosulfate was added (2 mL, dropwise slowly). Extraction with ethyl acetate (3×20 mL), drying, concentration, and purification by silica gel chromatography (hexanes: ethyl acetate = 1:1) afforded a colorless oil (57.3 mg, 88% yield). Rf = 0.24 (hexanes: ethyl acetate = 1:1, stain in PMA); 1H NMR (500 MHz, CDCl3): δ 1.45–1.53 (1H, m), 1.59–1.76 (3H, m), 1.77–1.88 (1H, m), 1.89–1.95 (1H, m), 2.02–2.13 (2H, m), 2.23 (1H, br), 2.32 (1H, br), 2.55–2.67 (2H, m), 3.50–3.59 (3H, m), 3.66 (1H, dd, J = 10.6 Hz, 2.8 Hz), 5.49 (1H, br), 7.17–7.20 (3H, m), 7.26–7.30 (2H, m); 13C NMR (125 MHz, CDCl3): δ 142.6, 139.7, 128.6, 128.5, 125.9, 125.6, 67.3, 64.5, 39.8, 38.8, 35.9, 35.3, 30.2, 24.5, 22.0; IR (neat): 3314.4 (br), 2935.6 (w), 2862.4 (w), 1451.8 (w), 1028.7 (m), 734.2 (w), 697.4 (s), 488.5 (w) cm−1; HRMS (ESI+) for C17H25O2 [M+H]: calculated: 261.1855, found: 261.1864; [α]D20: +8.09 (c = 0.82, CHCl3, l = 50 mm). Enantiomer ratio determined by HPLC (Chiraldex AD-H, 5% iPrOH/hexanes, 1.0 mL/min, 220 nm), major enantiomer 39.48 min, minor enantiomer 34.56 min, 95:5 e.r.
(2R,3S)-2-((S)-1-hydroxy-3-methylbutyl)-[1,1′-bi(cyclohexan)]-6-en-3-ol (14)
In a glovebox, a stir bar, Pt(dba)3 (13.5 mg, 15.0 mmol), phosphonite ligand (L6) (36.3 mg, 30.0 mmol), and toluene (5.0 mL, 0.1 M) were added to a vial. After stirring for 1 h, B2(pin)2 (133.3 mg, 0.525 mmol) and [1,1′-bi(cyclohexane)]-1,5-diene (81.1 mg, 0.50 mmol) were added. The vial was sealed, removed from the glovebox, and stirred at 60 °C for 12 h. After cooling to room temperature isovaleraldehyde was added (129.2 mg, 1.50 mmol) under nitrogen atmosphere. The vial was sealed again and stirred at 80 °C for 30 h. The mixture was cooled to 0 °C and charged with THF (3 mL), 3 M NaOH (3 mL) and 30 % H2O2 (3 mL). The reaction was stirred for 4 h at ambient, then cooled to 0 °C and saturated aqueous sodium thiosulfate (3 mL, dropwise slowly) added dropwise. Extraction (ethyl acetate, 3×20 mL), drying, concentration, and purification on silica gel (100% dichloromethane, then hexanes : ethyl acetate = 4:1) afforded a white solid (114.6 mg, 86% yield). Rf = 0.18 (hexanes:ethyl acetate = 5:1, stain in PMA); 1H NMR (500 MHz, CDCl3): δ 0.79–0.89 (1H, m), 0.93 (3H, d, J = 6.6 Hz), 0.96 (3H, d, J = 6.6 Hz), 1.14–1.19 (1H, m), 1.23–1.34 (4H, m), 1.60–1.73 (5H, m), 1.79–1.97 (5H, m), 2.01–2.08 (1H, m), 2.25–2.33 (2H, m), 4.12 (1H, ddd, J = 9.0 Hz, 4.4 Hz, 4.4 Hz), 4.31 (1H, ddd, J = 7.3 Hz, 3.2 Hz, 3.2 Hz), 5.51 (1H, br); 13C NMR (100 MHz, CDCl3): δ 141.1, 120.7, 70.4, 69.5, 45.7, 44.6, 41.9, 34.7, 31.0, 28.3, 27.2, 27.0, 26.8, 25.0, 23.9 22.3, 21.7; IR (neat): 3311.0 (br), 2922.7 (s), 2850.4 (m), 1448.1 (m), 1261.2 (w), 1068.9 (m), 1045.3 (m), 845.9 (w), 755.2 (m), 568.4 (w); HRMS (ESI+) for C17H29O1 [M+H-H2O]: calculated: 249.2218, found: 249.2210; [α]D20: +24.83 (c = 0.60, CHCl3, l = 50 mm); mp: 78.0–82.1 °C. Enantiomer ratio determined by GLC (Supelco β-dex, 150 °C, 20 psi), major enantiomer 242.57 min, minor enantiomer 246.66 min, 95:5 e.r. Absolute stereochemistry was determined by crystallography using anomalous dispersion (Flack parameter = 0.0)
Supplementary Material
Acknowledgments
Support by the NIGMS (GM-59417) and the NSF (DBI-06195766, BC Mass Spectrometry Center) is gratefully acknowledged. AllyChem is acknowledged for a generous donation of B2(pin)2.
Footnotes
Supporting Information Available: Compound characterization (spectra). This material is available free of charge via the Internet at http://pubs.acs.org/.
References
- 1.Burks HE, Kliman LT, Morken JP. J Am Chem Soc. 2009;131:9134. doi: 10.1021/ja809610h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Non-enantioselective diene diboration: Ishiyama T, Yamamoto M, Miyaura N. Chem Commun. 1996:2073.Ishiyama T, Yamamoto M, Miyaura N. Chem Commun. 1997:689.Clegg W, Thorsten J, Marder TB, Norman NC, Orpen AG, Peakman TM, Quayle MJ, Rice CR, Scott AJ. J Chem Soc, Dalton Trans. 1998:1431.Morgan JB, Morken JP. Org Lett. 2003;5:2573. doi: 10.1021/ol034936z.
- 3.Recent reviews: Burks HE, Morken JP. Chem Commun. 2007:4717. doi: 10.1039/b707779c.Ramirez J, Lillo V, Segarra AM, Fernandez E. Comp Rend Chim. 2007;10:138.Beletskaya I, Moberg C. Chem Rev. 2006;106:2320. doi: 10.1021/cr050530j.Ishiyama T, Miyaura N. Chem Record. 2004;3:271. doi: 10.1002/tcr.10068.Marder TB, Norman NC. Topics in Catalysis. 1998;5:63.
- 4.1,4-Dihydroxylation of dienes, and related reactions, have been accomplished, in moderate selectivity, under Pd-catalyzed oxidative conditions, see: Thorarensen A, Palmgren A, Itami K, Bäckvall JE. Tetrahedron Lett. 1997;38:8541.Itami K, Palmgren A, Thorarensen A, Bäckvall JE. J Org Chem. 1998;63:6466.Verboom RC, Plietker BJ, Bäckvall JE. J Organomet Chem. 2003;687:508.El-Qisairi AK, Qaseer HA, Fennelly JP, Chiarelli MP, Henry PM. Catalysis Commun. 2008;9:1661.
- 5.For a review of these ligands in asymmetric catalysis, see: Lam HW. Synthesis. 2011:2011.For selected primary references, see: Sakaki J, Schweizer WB, Seebach D. Helv Chem Acta. 1993;76:2654.Seebach D, Hayakawa M, Sakaki J, Schweizer WB. Tetrahedron. 1993;49:1711.Alexakis A, Burton J, Vastra J, Benhaim C, Fournioux X, van den Heuvel A, Leveque JM, Maze F, Rosset S. European Journal of Organic Chemistry. 2000:4011.Bee C, Han SB, Hassan A, Iida H, Krische MJ. J Am Chem Soc. 2008;130:2746. doi: 10.1021/ja710862u.
- 6.(a) Boele MDK, Kamer PCJ, Lutz M, Spek AL, de Vries JG, van Leeuwen PWNM, van Strijdonck GPF. Chem Eur J. 2004;10:6232. doi: 10.1002/chem.200400154. [DOI] [PubMed] [Google Scholar]; (b) Corey EJ, Matsumura Y. Tetrahedron Lett. 1991;32:6289. [Google Scholar]
- 7.Substituted cyclohexadienes are reaily accessed by Kumada coupling of the corresponding cyclohexadienyl phosphate, see: Karlström ASE, Itami K, Bäckvall JE. J Org Chem. 1999;64:1745. doi: 10.1021/jo982060h.
- 8.The absolute configuration of compounds 2b, 6b, and 14 was determined by x-ray crystallography (anomalous dispersion). The coordinates have been deposited with the CCDC (deposition numbers CCDC 825560 – 825562).
- 9.For an overview of homologations that apply to boronic esters, see: Thomas SP, French RM, Jheengut V, Aggarwal VK. Chem Rec. 2009;9:24. doi: 10.1002/tcr.20168.
- 10.(a) Sadhu KM, Matteson DS. Organometallics. 1985;4:1687. [Google Scholar]; (b) Chen AC, Ren Li, Crudden CM. Chem Commun. 1999:611. [Google Scholar]; (c) Chen AC, Ren Li, Crudden CM. J Org Chem. 1999;64:9704. [Google Scholar]; (d) Ren L, Crudden CM. Chem Commun. 2000:721. [Google Scholar]
- 11.Kliman LT, Mlynarski SN, Morken JP. J Am Chem Soc. 2009;131:13210. doi: 10.1021/ja9047762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.For a review of allylboration reactions, see: Hall DG. Pure & Appl Chem. 2008;80:913.Lachance H, Hall DG. Organic Reactions. 2008;73:1.
- 13.For relevant allylations involving cyclic allylic boronates, see: Gao X, Hall DG. J Am Chem Soc. 2005;127:1628. doi: 10.1021/ja042827p.Gao X, Hall DG, Deligny M, Favre A, Carreaux F, Carboni B. Chem Eur J. 2006;12:3132. doi: 10.1002/chem.200501197.Lallemand JY, Six Y, Ricard L. Eur J Org Chem. 2002:502.Vaultier M, Truchet F, Carboni B, Hoffmann RW, Denne I. Tetrahedron Lett. 1987;28:4169.
- 14.For reviews of cladiellins and related products, see: For compilations of related natural products, see: Sung PJ, Chen MC. Heterocycles. 2002;57:1705.Bernardelli P, Paquette LA. Heterocycles. 1998;49:531.
- 15.Du CJF, Hart H, Ng KKD. J Org Chem. 1986;51:3162. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.