

Abstract
Skin cancer is the most common form of cancer in the United States. The main cause of this cancer is DNA damage induced by the UV component of sunlight. In humans and mice, UV damage is removed by the nucleotide excision repair system. Here, we report that a rate-limiting subunit of excision repair, the xeroderma pigmentosum group A (XPA) protein, and the excision repair rate exhibit daily rhythmicity in mouse skin, with a minimum in the morning and a maximum in the afternoon/evening. In parallel with the rhythmicity of repair rate, we find that mice exposed to UV radiation (UVR) at 4:00 AM display a decreased latency and about a fivefold increased multiplicity of skin cancer (invasive squamous cell carcinoma) than mice exposed to UVR at 4:00 PM. We conclude that time of day of exposure to UVR is a contributing factor to its carcinogenicity in mice, and possibly in humans.
Keywords: circadian clock, cryptochrome, sunbathing, tanning salons
Skin cancer is the most common form of cancer in the United States. With over 1.3 million new cases each year, it constitutes nearly 40% of all diagnosed cancers (1). Moreover, because of changes in lifestyle and the environment, the incidence of skin cancer is steadily increasing (2). The main causative agent of skin cancer is the UV component of sunlight. UV radiation (UVR) produces two major lesions in DNA, the cyclobutane pyrimidine dimer (CPD) and the (6-4) photoproduct [(6-4) PP], both of which are mutagenic and carcinogenic in animal model systems and are thought to be the primary cause of skin cancer in humans (3–7).
In mice and humans, nucleotide excision repair is the sole repair system for removing CPDs and (6-4) PPs from DNA. As a consequence, humans with hereditary mutations in excision repair genes suffer from xeroderma pigmentosum, a syndrome characterized by a nearly 5,000-fold increase in skin cancer in sunlight-exposed areas of the afflicted individuals (8). Excision repair involves photoproduct removal by dual incisions bracketing the lesion, removal of the damage in the form of a 24- to 32-nt-long oligomer, filling in the resulting single-stranded gap, and sealing by ligase (9). The dual incision is carried out by six excision repair factors: RPA, xeroderma pigmentosum group A (XPA), XPC, TFIIH, XPG, and XPF-ERCC1 (10). Recently, in a study that analyzed liver and brain tissues from mice, it was found that XPA, a critical protein involved in damage recognition and a rate-limiting factor in excision repair, is controlled by the core molecular circadian clock (11, 12). As a consequence, excision repair activity exhibited circadian rhythmicity in these organs, increasing during the day to reach a maximum at 4–6:00 PM and decreasing during the night to a minimum at 4–6:00 AM.
Here we analyzed the expression pattern of XPA and excision repair activity in mouse skin. We found that protein and repair activity exhibit a circadian rhythm similar to that found in the liver and brain. To determine whether this rhythmicity affected UV-induced skin cancer development we exposed a group of mice to UVB (280–320 nm) when excision repair activity was at its nadir (4:00 AM) and a second group when excision repair was at its zenith (4:00 PM), and observed skin cancer development in the two groups. We found that animals that were exposed to UVR when excision repair activity was low developed skin cancers at a faster rate and at about fivefold higher frequency compared with mice that were exposed to UVR when excision repair was at its zenith. We conclude that time of day of exposure to UVR is an important determinant in the carcinogenicity of UVR. Furthermore, because the human molecular clock is essentially identical to the mouse clock except for the phase of the output (diurnal versus nocturnal), the susceptibility of humans to UVR-induced skin cancers is likely to exhibit a daily rhythm as well. This daily rhythmicity should be taken into account in making public health recommendations to reduce skin cancer risk from occupational, therapeutic, and recreational/cosmetic exposures to sunlight or other sources of UVR.
Results
Daily Rhythmicity of XPA in Mouse Skin.
Previously, we reported that the transcription of the xpa gene and the level of XPA protein exhibited circadian rhythmicity in some mouse tissues such as the brain and the liver but not in others such as testis (11, 12). This rhythmicity or the lack thereof (in clock mutant mice) was associated with an oscillatory or a constant rate of repair activity as a function of time of day, respectively. To find out whether excision repair in mouse skin changes as a function of time of day, we harvested mouse skin every 4 h over a 1-d period and probed it for XPA protein levels by immunoblotting. We also probed the tissue for cryptochrome 1 (CRY1) protein, as it is known that CRY1 is a primary repressor of clock-controlled genes, and the CRY1 protein levels, as a rule, are antiphase with proteins controlled by the circadian clock (13–15). Fig. 1A shows the results of a representative immunoblot. In mice kept under light/dark (LD) 12:12 conditions (12 h light:12 h dark), the XPA level reaches its maximum at zeitgeber (ZT) 10 (ZT 0 is when light is turned on), which corresponds to an early evening hour. The CRY1 repressor is at its minimum when XPA is at its peak, and vice versa, as expected for a relation between a repressor and its target gene product when both the repressor and the target protein have relatively short half-lives because of posttranslational modification and degradation (14–16). Fig. 1B shows quantitative analysis of the immunoblot data. These measurements were done in C57BL/6 mice, and they can be directly compared with the previous studies done in C57BL/6 mice on XPA rhythmicity in other tissues.
Fig. 1.

Circadian oscillation of XPA in mouse skin. (A) Mice kept under an LD 12:12 cycle were killed at the indicated zeitgeber times (ZT 0 = light on) and the levels of the indicated proteins were determined from whole skin by immunoblotting. Light and dark boxes indicate the light-on and light-off phases, respectively. Note the robust and antiphase oscillations of the XPA and CRY1 proteins. The core clock protein, Clock, as expected (14), does not exhibit rhythmicity. Actin was used as a loading control. ST, subjective time, with light on at 7:00 AM and off at 7:00 PM. (B) Quantitative analysis of XPA and CRY1 oscillation in the mouse skin. Error bars represent means ± SD (n = 2 mice at each time point). (C) XPA expression in the epidermis of SKH-1 hairless mice. The mice were exposed to UVR at ZT 21 (subjective 4:00 AM) or ZT 9 (subjective 4:00 PM) and epidermis was harvested at 0, 1, 2, 4, 6, and 12 h postirradiation and analyzed for expression of the indicated proteins by immunoblotting. Note that XPA and CRY1 exhibit antiphase rhythms, whereas the oxidative base damage repair enzyme 8-oxo-guanine DNA glycosylase (OGG-1), implicated in repair of oxidative base damage caused by UVR, shows no rhythmicity. GAPDH is a loading control. A representative experiment is shown only with 0, 1, and 2 h postirradiation for clarity and both AM and PM samples were run on the same gel. This experiment was repeated twice with two mice at each time point. (D) Immunohistochemical (IHC) analysis of XPA expression in SKH-1 epidermis at two circadian time points, ZT 22 (subjective 5:00 AM) and ZT 10 (subjective 5:00 PM). (Left) Representative IHC images (blue, nuclei; brown, XPA). Red dashed lines follow the basement membrane, located between the dermis (D) and the epidermis (E). (Right) Quantitative analysis of IHC data. Error bars indicate means ± SD (n = 2 mice at each time point). XPA positive cells were recorded manually using the Aperio ImageScope software, counting at least 10 fields (each field consists of ∼80 cells) per mouse. P value is based on 3-way ANOVA.
The C57BL/6 mouse strain is commonly used in circadian research but the majority of UV carcinogenesis studies have been carried out with the SKH-1 hairless mouse strain for practical reasons and because these mice develop squamous cell carcinomas (SCCs) similar to humans (17, 18). Hence, we decided to do our UV carcinogenesis study with this strain and first we examined the levels of XPA in epidermal keratinocytes, which are the target cells for UVB-induced DNA damage and precursor cells of SCCs. From SKH-1 mice we isolated epidermis that contains primarily keratinocytes to analyze the effect of the circadian clock on XPA expression and also to determine whether the expression level was affected by the UVB dose that we planned to use in the skin carcinogenesis study. The results shown in Fig. 1C and Fig. S1 A and B reveal that CRY1 and XPA expression in SKH-1 mice show the same phases as in C57BL/6 mice with XPA being high at early evening and low at early morning hours and with CRY1 exhibiting an antiphase pattern to XPA. Importantly, neither the XPA nor the CRY1 levels were significantly affected by the UVB dose we used in our carcinogenesis study, indicating that SKH-1 would be an appropriate mouse model for studying the effect of circadian rhythm on UVR carcinogenesis.
XPA levels in the epidermis of SKH-1 mice were also probed by immunohistochemistry (IHC), as shown in Fig. 1D. The data are in agreement with the immunoblot analysis with a statistically significant difference in XPA levels in the SKH-1 epidermis at 5:00 AM (low) and 5:00 PM (high) (Fig. 1D, Right). The IHC data suggest that not all epidermal cells are XPA positive; presumably this is due in part to the limitation of the IHC technique for quantitative analysis and in part to the presence of cell types with variable XPA expression levels. Taken together, these data indicate that XPA levels are controlled by the circadian clock in the mouse skin and hence the rate of repair of UVR-induced photoproducts may also exhibit a daily rhythmicity.
We also examined whether the circadian clock affected the expression of 8-oxo-guanine DNA glycosylase (OGG-1) because some studies have implicated this oxidative damage in UV carcinogenesis (19). As is apparent from Fig. 1C, the levels of OGG-1 do not vary at the times corresponding to the zenith and nadir values of CRY1, indicating that this enzyme is not controlled by the circadian clock and therefore should 8-oxo-guanine play a role in UV carcinogenesis, OGG-1 would not contribute to a potential circadian sensitivity.
Daily Rhythmicity of Excision Repair Rate in Mouse Epidermis.
To determine the effect of time of day of UV irradiation on the repair rate of UV photoproducts, one group of mice was irradiated at 4:00 AM (XPA nadir) and one group at 4:00 PM (XPA zenith), and the skin was harvested at 0, 1, 2, 4, 6, and 12 h post-UV and epidermal DNA was isolated for measuring photoproduct repair by slot blot (20). The results are shown in Fig. 2 A and B for (6-4) PPs and in Fig. 2 C and D for CPDs. Several interesting points emerge from these data. First, in agreement with previous reports, the (6-4) PPs are repaired at a faster rate than the CPDs (21, 22), in both the 4:00 AM and 4:00 PM groups. Second, and most relevant to the subject of this study, both the (6-4) PPs and the CPDs are repaired at faster rates in the PM group than the AM group, consistent with the level of XPA protein at the time of irradiation in the two groups. Third, the difference between the rate of repair of PM and AM groups is much more striking for the CPDs than the (6-4) PPs, consistent with the notion that a decrease in XPA level has a more profound effect on the rates of repair of the poorly recognized CPDs than the efficiently recognized (6-4) PPs, under conditions in which both types of DNA lesions are competing for the same limiting XPA protein (23). Finally, whereas the repair of the (6-4) PP appears to exhibit parabolic kinetics typical of a second order reaction, the kinetics of CPD repair appears to be biphasic for both the AM and the PM groups. The PM group exhibits a linear rate for up to about 6 h and then levels off with only about 50% of CPDs repaired. The AM group shows essentially no repair for the first 6 h and then proceeds with a linear rate. The unusual kinetics are consistent with the change in XPA levels as a function of circadian time over the course of the experiment. In the PM group, repair in the first 6 h takes place when the XPA level starts at its maximum and then declines slowly (Fig. 1 A and B). This slow decline does not affect the (6-4) PP repair, which is rather rapid but considerably affects the rate of CPD repair 4 h after irradiation when the level of XPA is reduced to a level that cannot sustain CPD repair in the presence of the residual but more efficient (6-4) PP substrate. In the AM group the rate of (6-4) PP repair is predictably slower than the PM group and appears to enter a faster rate after 6 h when the XPA level is significantly elevated. The rate of CPD repair in the AM group is rather interesting, with essentially no repair in the first 6 h after irradiation and then a linear repair rate presumably because of a combination of two factors: the elimination of a substantial amount of the competing (6-4) PP substrate during this period and circadian elevation in XPA level.
Fig. 2.

Circadian regulation of excision repair of UV-induced DNA damage in mouse epidermis. SKH-1 hairless mice kept under an LD 12:12 cycle were irradiated with 300 J/m2 of UVB at the indicated ZT times (either ZT 21 or ZT 09) and skin patches were collected 0–12 h after irradiation to measure UV photoproduct repair by slot blot. (A) Repair of (6-4) PP. A representative slot blot is shown. (Left) Immunoblot analysis of α-(6-4) PP signal. (Right) SYBR-GOLD staining of total DNA. (B) Quantitative analysis of (6-4) PP repair. Error bars indicate means ± SD (n = 2 mice at each time point). (C) Repair of CPDs analyzed by slot blot. (D) Quantitative analysis of CPD repair (n = 2 mice at each time point). (E) Slot blot analysis of (6-4) PP and CPD repair in Cry1−/−Cry2−/− mice. Skin was harvested at 0 and 6 h after irradiation and photoproduct levels were probed by immunoblotting. (F) Quantitative analysis of repair in the skin of Cry1−/−Cry2−/− mice irradiated at ZT 21 (4:00 AM) and ZT 09 (4:00 PM). The varied kinetics of repair in wild-type mice (A–D) presumably reflect the higher inherent rate of repair of the (6-4) PP and the cyclic expression of XPA. Error bars indicate means ± SD (n = 2 mice at each time point).
To ascertain that the effect of time of day of UV irradiation on the overall rate of repair was the consequence of circadian control of the XPA level and not due to some unknown environmental factor, we used a clock mutant mouse strain in the repair assay. Cryptochrome is a key component of the molecular clock, and mice and humans have two cryptochrome isoforms, Cry1 and Cry2. Mice mutated in both genes lack circadian rhythmicity (24, 25), so we used Cry1−/−Cry2−/− mice in the C57BL/6 genetic background. The mice were irradiated with UVB at either 4:00 AM or 4:00 PM and the rates of repair of UV photoproducts in the skin were measured. As seen in Fig. 2 E and F, in this genetic background, where XPA is at a constant and moderately elevated level throughout the day, the repair rates for the AM and the PM groups are similar, whereas C57BL/6 wild-type mice show circadian-dependent excision repair of (6-4) PPs (Fig. S2), further supporting the conclusion that circadian control of the XPA protein causes the daily rhythmicity of the repair rate of UV photoproducts in the mouse skin, independent of their genetic backgrounds.
Circadian Rhythmicity of DNA Replication in Mouse Epidermis.
In addition to DNA damage, cell proliferation is a determining factor in mutagenesis and carcinogenesis (3–7). Skin, in addition to the gastrointestinal epithelium and bone marrow, display relatively high levels of cell proliferation. It has been reported that mouse skin has a robust circadian clock (26). Moreover, cellular proliferation in the skin and gastrointestinal epithelium shows circadian rhythmicity, albeit of low amplitude (27, 28). We wished to analyze DNA replication in the skin of SKH-1 mice as a function of time of day, and to this end, we injected mice with BrdU (29, 30) at either 4:00 AM or 4:00 PM, harvested various organs 90 min later, isolated DNA, and probed for BrdU incorporation during replication by slot blot. The results are shown in Fig. 3 A and B. In agreement with previous studies using other methods to measure cell proliferation (27), there is more DNA synthesis in the AM group than the PM group. Of note, the BrdU incorporation appears to be a reliable assay for measuring DNA replication (Fig. S3): When we analyzed intestine and kidney there was much greater BrdU incorporation into the intestinal DNA, which is consistent with the presence of a highly proliferative intestinal epithelium, than into the kidney DNA, which is derived from terminally differentiated cells. Importantly, Fig. 3 C and D shows that BrdU incorporation into skin and intestinal DNA does not vary with time of day in Cry1−/−Cry2−/− mice, whereas wild-type mice of the same genetic background (C57BL/6) show circadian-dependent DNA replication in these tissues (Fig. S4), which indicates that replication is under the control of the circadian clock. Taken together, the replication and the repair data suggest that there is more DNA replication at a time of the day when DNA repair is not efficient because of the limiting amount of XPA protein. This combination of low repair and high replication is expected to render mice more prone to skin cancer when exposed to UVR in the early morning hours compared with evening hours. We tested this prediction by performing a skin carcinogenesis study.
Fig. 3.

Circadian rhythm of DNA replication in mouse tissues. SKH-1 hairless mice were kept under an LD 12:12 cycle. BrdU or vehicle alone (“mock”) was injected intraperitoneally at either ZT 21 (4:00 AM) or ZT 09 (4:00 PM); 90 min later epidermis, intestine, and kidney tissues were harvested and the level of DNA replication was examined by slot blot. (A and B) Results obtained with wild-type SKH-1 hairless mice. (A) Representative slot blot. (B) Quantitative analysis of slot blots. Error bars indicate means ± SD (n = 3 mice at each time point). (C and D) Results obtained with clock-deficient (Cry1−/−Cry2−/−) mice. (C) Representative slot blot. (D) Quantitative analysis of the slot blots. Error bars indicate means ± SD (n = 3 mice at each time point).
Effect of Time of Day of UV irradiation on Skin Carcinogenesis.
SKH-1 hairless mice maintained under an LD 12:12 lighting schedule were divided into three groups. A control group was not subjected to UVR. The second group (AM group) was irradiated with 353 J/m2 of UVB at 4:00 AM (3 h before lights on) three times a week for 25 wk. The third group was irradiated similarly to the second group except UVR was given at 4:00 PM (3 h before lights off). The mice were inspected three times a week and the time of first tumor appearance, number of tumors per mouse, and tumor size per mouse were recorded over the 25-wk period. The results are shown in Fig. 4. Both the AM and the PM groups developed skin tumors, whereas the control mice did not exhibit overt skin lesions over the course of the experiment (Fig. 4 A and B and Fig. S5). Importantly, when the tumorigenesis data were plotted in the form of a Kaplan–Meier plot, it was seen that the time to the first tumor in the AM group was significantly shorter than the time to the first tumor in the PM group, with a median latency of 19 wk for the AM group and 21 wk for the PM group (Fig. 4B). All 29 mice in the AM group developed tumors by week 22, and 25 of 26 mice in the PM group developed tumors by week 25. More strikingly, when the average number of tumors per mouse were plotted, the values were 24 tumors per mouse in the AM group and 12 tumors per mouse for the PM group at the end of the 25-wk observation period (Fig. 4C). Similarly, the average tumor diameter for the AM group was nearly twice as large as for the PM group (Fig. 4D).
Fig. 4.

Effect of time of day of exposure to UVB on skin carcinogenesis (visual diagnosis) in SKH-1 hairless mice. (A) Physical appearance of representatives of the three experimental groups 23 wk after initiation of irradiation. (B) Kaplan–Meier analysis of tumor-free survival. (C) Average number of tumors per mouse following initiation of UV treatments. (D) Average maximal tumor area per mouse in the AM and PM groups.
The data presented in Fig. 4 were derived from the counting of exophytic macroscopic skin tumors, which include papillomas, keratoacanthomas, and SCCs. Because most SCCs are endophytic lesions, we carried out histopathological examination of all endophytic and exophytic skin lesions in the AM and PM groups to find out whether the time of day of UVB exposure affected skin cancer (SCCs) induced in SKH-1 mice. Representative hemotoxylin/eosin-stained sections of tumor tissues from the two groups are shown in Figs. S6, S7, and S8. Quantitative analyses of invasive carcinomas of the two groups is shown in Fig. 5, and values for all tumors including the “in situ” tumors in the AM and PM groups are summarized in Table S1. Invasive SCCs were further classified as invading through the basement membrane or through the underlying muscle. About fivefold higher number of invasive tumors were observed in the AM-treated population than in the PM-treated population (t = 0.00015), which is presumably a result of more mutations in the AM group leading to more tumors of all types, including rapidly growing and aggressive tumors. These analyses lead to the conclusion that the same UVB dose is more carcinogenic in SKH-1 mice when given in early morning hours than when it is given in the early evening hours.
Fig. 5.

Quantitative summary of histopathological analysis performed on skin harvested 25 wk after initiation of the experiment. Note that there are significantly more invasive SCC tumors in the AM group than in the PM group of mice (t = 0.00015). The t value for SCC basement membrane invasive is 0.0002, and for SCC muscle invasive it is 0.023.
Discussion
Our findings are briefly summarized as follows (Fig. 6): In mouse skin there is more DNA replication and less repair in the morning and less replication and more repair in the evening. Because UV-induced skin cancers arise from mutagenic replication of epidermal keratinocyte DNA, the same UV dose is more carcinogenic in early morning hours than when given in the early evening hours. We wish to discuss this conclusion within the context of other studies on the connection of the circadian clock to various diseases and the implications of our findings to skin cancer in humans.
Fig. 6.
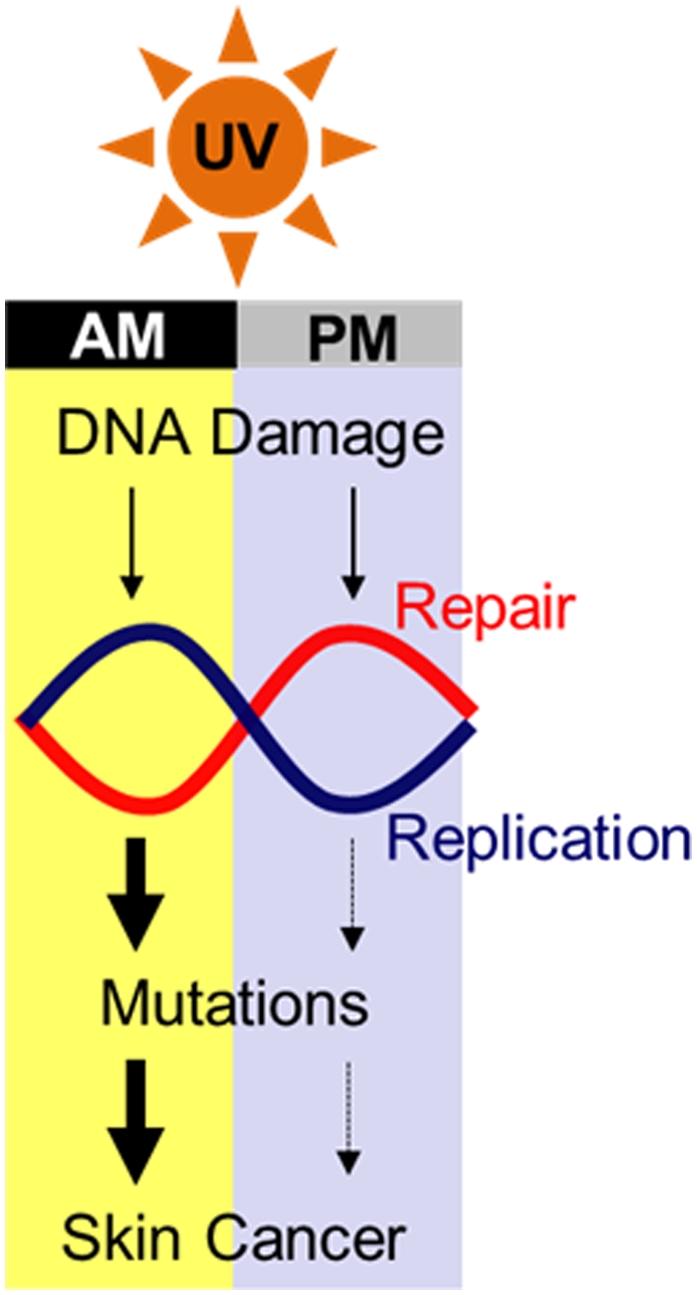
Model for the role of the circadian clock in UV-induced skin carcinogenesis. The higher level of DNA replication combined with the nadir of DNA repair capacity in the early morning hours compared with afternoon/evening renders mice more susceptible to mutation and cancer when exposed to sunlight in the morning.
Circadian Rhythm and Disease.
The circadian clock is known to affect the onset and severity of some common diseases such as asthma and cardiovascular incidents because of the control of some key pathways involved in the pathophysiology of these diseases by the circadian circuitry (15, 31, 32). As a consequence, recommendations have been made for taking the appropriate preventive and therapeutic measures to prevent the occurrence of or alleviate the severity of the disease state. Here, we show that time of day of UV exposure affects its carcinogenic potential in mice. However, in comparing the circadian rhythm of carcinogenicity of UV to the effect of the clock on other diseases three points must be taken into consideration. First, DNA damage and repair are two of the multiple factors that impact cancer development. Many of the other intra- and intercellular pathways and signaling networks that are affected by the circadian clock contribute to carcinogenesis (16) and they may have contributed to the circadian rhythmicity of the UVB carcinogenesis we have observed. Further studies with repair-deficient and clock mutant animals are needed to deconvolute the relative contributions of the various pathways to the rhythmicity of the carcinogenic effect of UVR. Second, in comparing skin cancer to cardiovascular disease and asthma vis-à-vis their circadian clock connection, an important distinction exists. Cardiovascular incidents (heart attack, angina) and asthma attack reflect the organism's pathophysiology at that particular time of the day, whereas cancer is a multistage process and a long-term endpoint of a specific incident (DNA damage and mutation) that occurred at a particular time of the day. This distinction notwithstanding, the most parsimonious interpretation of our data is that mice exposed to UVR in the early morning hours are more likely to develop skin cancer than those receiving the same dose in the evening hours because of a poor repair rate, and therefore, avoiding sunlight exposure in the morning hours will likely reduce cancer risk. Finally, we wish to comment on the effect of the circadian clock on the skin tumor types in SKH-1 mice. It has been reported that repair-proficient SKH-1 mice exposed to UVB develop squamous cell carcinoma, about 50% of which are associated with p53 mutation (6). In contrast, xpa−/− SKH-1 mice exposed to UVB (low dose of equal carcinogenicity) developed papillomas and squamous cell carcinomas that were associated more frequently with ras mutation than p53 mutation (33). Our PM and AM groups could be considered phenocopies of the repair proficient and xpa mutant SKH-1 mice, respectively. However, under our experimental setup we did not detect a differential distribution of the various cancer types in the two groups. Possibly, a more specifically designed radiation protocol may uncover such a difference.
Circadian Rhythm and Human Skin Cancer.
A significant goal in undertaking this study was to find out whether the circadian rhythm could be used to advantage to reduce skin cancer incidence in humans. Mice are nocturnal and humans are diurnal animals and, therefore, the core circadian clock and their outputs exhibit opposite phases (13–15, 31). On the basis of this well-established fact, we predict that humans will have a higher rate of repair in the morning and would be less prone to the carcinogenic effect of UVR in the morning hours and it might be advisable for humans, to the extent possible, to restrict their occupational, therapeutic, recreational, and cosmetic UVR exposure to the morning hours. We note that this is a deductive conclusion based on our findings in mice reported in this paper and the evidence that humans have a robust circadian clock in their skin, which is antiphase to the mouse clock (34, 35) and with the peak xpa transcript around 7:00 AM (35). However, we also note that the phase of the circadian rhythm in humans exhibits interindividual variability (different chronotypes) and that recommendations for best times for UVR exposure may have to be tailored to the various chronotypes. Fortunately, noninvasive methods for assigning chronotypes exist (35), and with rare exceptions the phase differences between various chronotypes are not large enough to affect the general fact that humans are diurnal animals and hence most likely have maximum repair capacity in the morning hours. With these considerations, then, we suspect that by restricting UVR exposure to morning hours would reduce the risk of skin cancer in humans. This recommendation, however, must be considered provisional until actual DNA repair rates are measured in the skin of human volunteers.
Materials and Methods
UVB Treatment of Mice.
All animal procedures were in accordance with the National Institutes of Health guidelines and were approved by the Institutional Animal Care and Use Committee of the University of North Carolina, Chapel Hill. Male outbred SKH-1 (6 wk old) mice were obtained from Charles River Laboratories. The mice were maintained on an LD 12:12 schedule for 3 wk before UVR treatment. The time of day is expressed in two ways: One is the standard chronobiology terminology where ZT (zeitgeber) = 0 is light on and ZT = 12 is light off for animals under 12 h light:12 h dark cycles. To relate ZT time to time expression in common practice, we also indicate time by AM and PM designation whereby ZT 0 (light on) is taken as 7:00 AM and ZT 12 (light off) is expressed as 7:00 PM. Mice were irradiated in a relatively small chamber (18 inches high, 12 inches deep, and 22 inches long), which was purchased from Plastic Design. The UVB lamp, located at the top of the chamber above the mice, was from UVP; it emits 290–350 nm light with a peak emission at 312 nm. The lid of the mouse cage was removed and the cage was placed into the chamber for irradiation. The UVB dose rate was determined by a UVX radiometer from UVP. For in vivo excision repair and protein expression studies, mice were treated with a single dose of 300 J/m2 UVB at a rate of 15 J/m2/sec. Depilation of C57BL/6 wild-type and cry knockout mice was done 24 h before UV irradiation by anesthetizing with isofluorane and then plucking the hair completely from an area (about 2 cm in diameter) of the dorsal aspect. For tumorigenesis studies, 9- to 10-wk-old SKH-1 mice were treated with 353 J/m2 UVB at a rate of 15 J/m2/sec three times a week for 25 wk. It should be noted that in a preliminary experiment, we determined that this dose did not cause erythema over the course of daily irradiation of control mice (n = 6) for 2 wk. Most of the mice were singly housed and where multiply housed there was no evidence of dorsal wounds caused by fighting. The mice were observed three times a week. Digital calipers were used to measure exophytic tumors ≥1 mm in diameter at weekly intervals. The average tumor area per mouse represents the diameter of the largest tumor borne by each mouse. The time to first tumor was determined to be the age when the first measurable tumor (≥1 mm) was observed. All mice were killed 25 wk after initiating the UVB treatment and whole dorsal skin including the tumors was harvested and fixed by incubating in 10% neutral buffered formalin for 24 h.
IHC Techniques.
The fixed skin tissue containing tumors was washed and stored in 80% ethanol at room temperature until processed further. The formalin-preserved tissue was sliced with a scalpel blade and arranged in a cassette containing foam biopsy pads and submitted to the University of North Carolina (UNC) Animal Histopathology Core Facility for processing, embedding, sectioning, and H&E staining. For IHC analysis of XPA protein, mouse skin was collected and formalin fixed, and paraffin-embedded sections were subjected to IHC staining using the antirabbit XPA antibody (lot number IHC-00344-1) from Bethyl Laboratories and the procedure recommended by the vendor. IHC work and analysis for XPA was performed by the Anatomical Pathology Translational Core Lab (APTCL) at UNC Chapel Hill.
Tumor Grading.
H&E-stained sections of tumors/lesions were graded in a blinded manner as reported previously (29, 36). A Nikon Eclipse 80i microscope with Surveyor Mosaic Imaging system at the UNC Neuroscience Confocal and Multi Photon Imaging Facility was used to grade the tumors. Keratoacanthomas appeared as bowl-shaped structures and showed no evidence of stromal invasion. SCCs displayed high heterogeneity in cell morphology, severe anaplastic growth, increased number of mitoses, some apoptotic cells, and increased nuclear-to-cytoplasmic ratio. SCCs were subcategorized on the basis of their penetration into the dermis. SCC in situ remained confined to the epidermis. SCCs were classified as membrane invasive if carcinoma cells invaded the basement membrane and muscle invasive if carcinoma cells invaded the panniculous carnosus of the dermis.
Measurement of CPD and (6-4) PP Repair Rate in Vivo by Slot Blot.
In vivo repair of CPD and (6-4) PP was measured as reported previously (20, 22). Mouse skin was collected at various time points after UVB treatment and immediately flash frozen to an index card and skin patches were stored at −80 °C until processing. Epidermis was peeled off from the skin patches using a freeze–thaw method (37); the skin patch was transferred to a 60 °C water bath for 10 s and immediately transferred to ice-cold water for 15 s. Skin was dehydrated by pressing it between paper towels and epidermis was peeled off gently using blunt ended forceps. See SI Materials and Methods for details.
Preparation of Protein Lysates from Mouse Skin.
The epidermal layer from the skin of SKH-1 mice was removed as described in the previous section. Recovered epidermis was then placed in ice-cold RIPA buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Nonidet P-40, and 1% sodium deoxycholate) with fresh protease and phosphatase inhibitor mixture (Roche). Tissue was disrupted using a glass tissue homogenizer with 10–15 strokes on ice and lysates were sonicated for 10 s on ice and centrifuged at 14,000 rpm in a microfuge for 10 min at 4 °C, and the clear supernatant was stored at −80 °C. For the whole skin protein extract of C57BL/6 mice, tissue was homogenized in liquid nitrogen using a mortar and pestle and then processed the same as the epidermal extract.
Immunoblot Analysis.
See SI Materials and Methods for details.
BrdU Incorporation Assay for Cell Proliferation in Vivo by Slot Blot.
See SI Materials and Methods for details.
Statistical Analysis.
See SI Materials and Methods for details.
Supplementary Material
Acknowledgments
We thank Drs. Songyun Zhu, Johnathan Hall, and Jeanne Burr at North Carolina State University for their help in skin carcinogenesis methodology, and Drs. Nancy Thomas, Joyce Reardon, Laura Lindsey-Boltz, Michael Kemp, Christopher Capp, and Jin Lee at University of North Carolina for their help in project design and comments on the manuscript. We are grateful to Dr. Nana Nikolaishvili-Feinberg and Bentley Midkiff from APTCL core facility of University of North Carolina for IHC staining of mouse skin XPA. We thank Dr. Joseph Ibrahim and Shangbang Rao from the Biostatistics Department at University of North Carolina and Rhishikesh Mandke at North Dakota State University for their help on statistical analysis. This work was supported by National Institutes of Health Grants GM31082, GM32833, and ES014635 (to A.S.) and in part by National Institute of Environmental Health Sciences Grant P30 ES010126 (to S.G.).
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1115249108/-/DCSupplemental.
References
- 1.American Cancer Society Cancer Facts and Figures. 2008. Available at http://www.cancer.org/Research/CancerFactsFigures/cancer-facts-figures2008. Accessed May 5, 2011.
- 2.Fisher DE, James WD. Indoor tanning—science, behavior, and policy. N Engl J Med. 2010;363:901–903. doi: 10.1056/NEJMp1005999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bowden GT. Prevention of non-melanoma skin cancer by targeting ultraviolet-B-light signalling. Nat Rev Cancer. 2004;4:23–35. doi: 10.1038/nrc1253. [DOI] [PubMed] [Google Scholar]
- 4.Narayanan DL, Saladi RN, Fox JL. Ultraviolet radiation and skin cancer. Int J Dermatol. 2010;49:978–986. doi: 10.1111/j.1365-4632.2010.04474.x. [DOI] [PubMed] [Google Scholar]
- 5.Jans J, et al. Powerful skin cancer protection by a CPD-photolyase transgene. Curr Biol. 2005;15:105–115. doi: 10.1016/j.cub.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 6.Brash DE, et al. A role for sunlight in skin cancer: UV-induced p53 mutations in squamous cell carcinoma. Proc Natl Acad Sci USA. 1991;88:10124–10128. doi: 10.1073/pnas.88.22.10124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cleaver JE. Defective repair replication of DNA in xeroderma pigmentosum. Nature. 1968;218:652–656. doi: 10.1038/218652a0. [DOI] [PubMed] [Google Scholar]
- 8.Kraemer KH, Lee MM, Scotto J. Xeroderma pigmentosum. Cutaneous, ocular, and neurologic abnormalities in 830 published cases. Arch Dermatol. 1987;123:241–250. doi: 10.1001/archderm.123.2.241. [DOI] [PubMed] [Google Scholar]
- 9.Reardon JT, Sancar A. Nucleotide excision repair. Prog Nucleic Acid Res Mol Biol. 2005;79:183–235. doi: 10.1016/S0079-6603(04)79004-2. [DOI] [PubMed] [Google Scholar]
- 10.Mu D, et al. Reconstitution of human DNA repair excision nuclease in a highly defined system. J Biol Chem. 1995;270:2415–2418. doi: 10.1074/jbc.270.6.2415. [DOI] [PubMed] [Google Scholar]
- 11.Kang TH, Reardon JT, Kemp M, Sancar A. Circadian oscillation of nucleotide excision repair in mammalian brain. Proc Natl Acad Sci USA. 2009;106:2864–2867. doi: 10.1073/pnas.0812638106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kang TH, Lindsey-Boltz LA, Reardon JT, Sancar A. Circadian control of XPA and excision repair of cisplatin-DNA damage by cryptochrome and HERC2 ubiquitin ligase. Proc Natl Acad Sci USA. 2010;107:4890–4895. doi: 10.1073/pnas.0915085107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reppert SM, Weaver DR. Coordination of circadian timing in mammals. Nature. 2002;418:935–941. doi: 10.1038/nature00965. [DOI] [PubMed] [Google Scholar]
- 14.Lowrey PL, Takahashi JS. Mammalian circadian biology: Elucidating genome-wide levels of temporal organization. Annu Rev Genomics Hum Genet. 2004;5:407–441. doi: 10.1146/annurev.genom.5.061903.175925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sahar S, Sassone-Corsi P. Metabolism and cancer: The circadian clock connection. Nat Rev Cancer. 2009;9:886–896. doi: 10.1038/nrc2747. [DOI] [PubMed] [Google Scholar]
- 16.Antoch MP, Kondratov RV. Circadian proteins and genotoxic stress response. Circ Res. 2010;106:68–78. doi: 10.1161/CIRCRESAHA.109.207076. [DOI] [PubMed] [Google Scholar]
- 17.Benavides F, Oberyszyn TM, VanBuskirk AM, Reeve VE, Kusewitt DF. The hairless mouse in skin research. J Dermatol Sci. 2009;53:10–18. doi: 10.1016/j.jdermsci.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Gruijl FR, Forbes PD. UV-induced skin cancer in a hairless mouse model. Bioessays. 1995;17:651–660. doi: 10.1002/bies.950170711. [DOI] [PubMed] [Google Scholar]
- 19.Kvam E, Tyrrell RM. Induction of oxidative DNA base damage in human skin cells by UV and near visible radiation. Carcinogenesis. 1997;18:2379–2384. doi: 10.1093/carcin/18.12.2379. [DOI] [PubMed] [Google Scholar]
- 20.Wani AA, D'Ambrosio SM, Alvi NK. Quantitation of pyrimidine dimers by immunoslot blot following sublethal UV-irradiation of human cells. Photochem Photobiol. 1987;46:477–482. doi: 10.1111/j.1751-1097.1987.tb04798.x. [DOI] [PubMed] [Google Scholar]
- 21.Mitchell DL. The relative cytotoxicity of (6-4) photoproducts and cyclobutane dimers in mammalian cells. Photochem Photobiol. 1988;48:51–57. doi: 10.1111/j.1751-1097.1988.tb02785.x. [DOI] [PubMed] [Google Scholar]
- 22.Gaddameedhi S, et al. Similar nucleotide excision repair capacity in melanocytes and melanoma cells. Cancer Res. 2010;70:4922–4930. doi: 10.1158/0008-5472.CAN-10-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gao S, Drouin R, Holmquist GP. DNA repair rates mapped along the human PGK1 gene at nucleotide resolution. Science. 1994;263:1438–1440. doi: 10.1126/science.8128226. [DOI] [PubMed] [Google Scholar]
- 24.Vitaterna MH, et al. Differential regulation of mammalian period genes and circadian rhythmicity by cryptochromes 1 and 2. Proc Natl Acad Sci USA. 1999;96:12114–12119. doi: 10.1073/pnas.96.21.12114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van der Horst GT, et al. Mammalian Cry1 and Cry2 are essential for maintenance of circadian rhythms. Nature. 1999;398:627–630. doi: 10.1038/19323. [DOI] [PubMed] [Google Scholar]
- 26.Tanioka M, et al. Molecular clocks in mouse skin. J Invest Dermatol. 2009;129:1225–1231. doi: 10.1038/jid.2008.345. [DOI] [PubMed] [Google Scholar]
- 27.Brown WR. A review and mathematical analysis of circadian rhythms in cell proliferation in mouse, rat, and human epidermis. J Invest Dermatol. 1991;97:273–280. doi: 10.1111/1523-1747.ep12480379. [DOI] [PubMed] [Google Scholar]
- 28.Bjarnason GA, et al. Circadian expression of clock genes in human oral mucosa and skin: Association with specific cell-cycle phases. Am J Pathol. 2001;158:1793–1801. doi: 10.1016/S0002-9440(10)64135-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thompson EA, et al. C/EBPα expression is downregulated in human nonmelanoma skin cancers and inactivation of C/EBPα confers susceptibility to UVB-induced skin squamous cell carcinomas. J Invest Dermatol. 2011;131:1339–1346. doi: 10.1038/jid.2011.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ueda J, Saito H, Watanabe H, Evers BM. Novel and quantitative DNA dot-blotting method for assessment of in vivo proliferation. Am J Physiol Gastrointest Liver Physiol. 2005;288:G842–G847. doi: 10.1152/ajpgi.00463.2004. [DOI] [PubMed] [Google Scholar]
- 31.Hastings MH, Reddy AB, Maywood ES. A clockwork web: Circadian timing in brain and periphery, in health and disease. Nat Rev Neurosci. 2003;4:649–661. doi: 10.1038/nrn1177. [DOI] [PubMed] [Google Scholar]
- 32.Levi F, Schibler U. Circadian rhythms: Mechanisms and therapeutic implications. Annu Rev Pharmacol Toxicol. 2007;47:593–628. doi: 10.1146/annurev.pharmtox.47.120505.105208. [DOI] [PubMed] [Google Scholar]
- 33.de Vries A, et al. XPA-deficiency in hairless mice causes a shift in skin tumor types and mutational target genes after exposure to low doses of U.V.B. Oncogene. 1998;16:2205–2212. doi: 10.1038/sj.onc.1201744. [DOI] [PubMed] [Google Scholar]
- 34.Zanello SB, Jackson DM, Holick MF. Expression of the circadian clock genes clock and period1 in human skin. J Invest Dermatol. 2000;115:757–760. doi: 10.1046/j.1523-1747.2000.00121.x. [DOI] [PubMed] [Google Scholar]
- 35.Akashi M, et al. Noninvasive method for assessing the human circadian clock using hair follicle cells. Proc Natl Acad Sci USA. 2010;107:15643–15648. doi: 10.1073/pnas.1003878107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thomas-Ahner JM, et al. Gender differences in UVB-induced skin carcinogenesis, inflammation, and DNA damage. Cancer Res. 2007;67:3468–3474. doi: 10.1158/0008-5472.CAN-06-3798. [DOI] [PubMed] [Google Scholar]
- 37.House JS, Zhu S, Ranjan R, Linder K, Smart RC. C/EBPalpha and C/EBPbeta are required for Sebocyte differentiation and stratified squamous differentiation in adult mouse skin. PLoS ONE. 2010;5:e9837. doi: 10.1371/journal.pone.0009837. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.