

Introduction
During evolution, multicellular organisms have developed an impressive arsenal of defense and repair mechanisms to counteract threats such as infection and trauma. Such an inflammatory response begins with the detection of the potential life-threatening event by recognizing so-called danger signals. These signal molecules have been classically divided into: i) Exogenous, pathogen-associated molecular patterns (PAMPs) [1], which are conserved motifs on pathogens that are not found in higher eukaryocytes; and ii) endogenous innate danger molecules, also named damage-associated molecular patterns (DAMPs) or alarmins, which are structurally diverse proteins rapidly released by the host itself during infection or (sterile) tissue damage [2].
Known PAMPs include lipopolysaccharide (LPS) from the outer membrane of Gram-negative bacteria, peptidoglycan (present in most bacteria), lipoteichoic acid (in many Gram-positive bacteria), bacterial DNA, viral DNA/RNA and mannans in the yeast cell wall. PAMPs are recognized by pattern recognition receptors (PRRs), in particular Toll-like receptors (TLRs) and Nod-like receptors (NLRs), leading to an inflammatory response via several signaling pathways, including nuclear factor-kappa B (NF-κB) activation and subsequent tumor necrosis factor (TNF)-α production.
Examples of putative DAMPs, the endogenous equivalents of PAMPs, are high-mobility group box 1 (HMGB1), some S100 proteins (S100A8/A9, S100A12), interleukins such as IL-1α, heat-shock proteins (HSPs), and nucleosomes [3]. DAMPs can be secreted either actively or passively following necrosis but are not released by apoptotic cells [4] and have activating effects on receptor-expressing cells engaged in host defense. DAMPs can also be detected by TLRs and NLRs and their engagement induces NF-κB activation as well, suggesting that DAMPs and PAMPs use, at least partially, the same receptors and signaling pathways. Liu et al. [5] however, propose that the immune system treats DAMPs and PAMPs differently; they suggest that DAMPs - but not PAMPs - bring CD24-Siglec G/10 into the proximity of TLRs/NLRs, resulting in repressed DAMP-induced TLR/NLR signaling.
When invaded by pathogens, host defense systems encounter PAMPs from microorganisms and DAMPs that are released from tissues, which are recognized by TLRs and NLRs to warn the host of imminent danger. In addition, the multiligand receptor for advanced glycation endproducts (RAGE) is regarded as a prototypic DAMP receptor that can bind several DAMPs, including HMGB1 and S100A12 [6]. Other known RAGE ligands include amyloid, β-sheet fibrils, S100B and S100P [7]; furthermore, β2 integrins can interact with RAGE [8]. RAGE is expressed at high levels in the lungs and at low levels in normal adult tissues, including on cells involved in the innate immune system, e.g., neutrophils, T and B lymphocytes, monocytes, macrophages, dendritic cells, and endothelial cells [7]. Engagement of RAGE by its ligands leads to receptor-dependent signaling and activation of NF-κB and mitogen-activated protein kinase (MAPK) pathways [7]. Activation of RAGE plays a role in diverse experimentally-induced sterile inflammatory and infectious diseases, including cecal ligation and puncture (CLP)-induced abdominal sepsis [9], diabetic nephropathy, delayed type hypersensitivity, type II collagen induced arthritis, hepatic injury, and diabetic atherosclerosis [7,10-12]. This review focuses on new insights into the pathogenesis of infectious diseases, including sepsis, peritonitis and pneumonia, offered by studies conducted in the RAGE research field.
RAGE: a multiligand receptor
RAGE consists of three immunoglobulin-like regions, a transmembrane domain, and a highly charged short cytosolic tail that is essential for intracellular signaling [13]. The V domain in the extracellular part of RAGE is essential for binding of its ligands. Because of its ability to recognize three-dimensional structures rather than specific amino acid sequences, RAGE can interact with a wide range of ligands. RAGE was first identified as a receptor for advanced glycation endproducts (AGEs), explaining its name. AGEs are products of the non-enzymatic glycation and oxidation of lipids, proteins and other macromolecules that appear, in particular, under conditions of increased availability of reducing sugars and/or enhanced oxidative stress, especially when molecules turn over slowly and aldose levels are elevated. Further investigations showed that RAGE can recognize a diverse array of endogenous molecules that warn the immune system and induce a defensive immune response; the alarmins or DAMPs.
Putative RAGE ligands in infectious diseases
HMGB1
HMGB1 is a non-histone DNA-binding protein that serves as a structural component to facilitate the assembly of nucleoprotein complexes in the nucleus [14]. Extra-cellularly, HMGB1 functions as a cytokine. In response to inflammatory stimuli, including PAMPs, HMGB1 can be actively released into the extracellular environment from a variety of cells including monocytes, macrophages, endothelial cells, enterocytes, pituicytes, dendritic cells, and natural killer cells [14]. HMGB1 can also be passively secreted into the extracellular milieu when cells die in a non-programmed way (necrosis), whereas apoptotic cells modify their chromatin so that HMGB1 binds irreversibly and consequently is not released [4]. During infectious diseases, increased HMGB1 concentrations may be due to active as well as passive release. Detection methods of HMGB1 that are currently used (and published) do not distinguish between these (and possible other) different forms of HMGB1. More studies are necessary to: 1) Report the biological activity of (different forms of) HMGB1; and 2) develop HMGB1 ELISA assays that can distinguish between these (possibly also functionally) different forms of HMGB1. Most investigations on HMGB1 and infection involve sepsis, the second leading cause of death in non-coronary intensive care units (ICUs) and the 10th leading cause of death overall. Patients with severe sepsis display elevated circulating HMGB1 levels [15-17] and HMGB1 is predominantly released at the site of infection; patients with pneumonia and those with peritonitis showed increased concentrations in fluid obtained from the bronchoalveolar space and abdomen, respectively [17]. In an animal model of CLP-induced sepsis, the kinetics of HMGB1 secretion in vivo was delayed and more sustained when compared with the release of pro-inflammatory cytokines, like TNF-α, IL-1β and IL-6 [18,19]. Similarly, various interventions that inhibit HMGB1 activity or production, such as anti-HMGB1 antibodies, the A-box segment of HMGB1, ethyl pyruvate, and nicotine, reduced CLP-induced sepsis and/or LPS lethality even if treatment was delayed for many hours, up to one day after the challenge [20,21]. An implicated crucial event in sepsis patho-physiology is apoptosis of immune cells, playing a major role in immunosuppression and lethality [22]. HMGB1 seems to be a downstream factor of apoptosis in the final common pathway to organ damage in severe sepsis as indicated by observations that prevention of lymphocyte apoptosis improved survival after CLP [23], whereas anti-HMGB1 treatment reduced lethality in the same model without influencing apoptosis [19]. This indicates that HMGB1 secretion is a relatively late event in sepsis that contributes significantly to a worsened outcome. In addition, it has been reported that very pure HMGB1 does not have cytokine-inducing capacity itself, but activates cells indirectly by first acquiring immune stimulating CpG DNA [24], which is released in the blood-stream during bacterial sepsis. However, a recent study reported that HMGB1-mediated induction of macro-phage cytokine production requires binding to TLR4, and that binding and signaling are dependent on a molecular mechanism that requires cysteine in position 106 within the B box [25]. Together these data indicate that HMGB1 may exert pro-inflammatory effects in a direct TLR4-dependent way and an indirect way via binding of DAMPs and other agonistic molecules.
S100A12
S100A12 is a calcium binding protein expressed in the cytoplasm of neutrophils, where it comprises 5% of the total protein content. Furthermore, S100A12 - also known as EN-RAGE (extracellular newly identified ligand of RAGE) or myeloid-related protein (MRP)-6 - is found in monocytes and lymphocytes and provokes pro-inflammatory responses in endothelial cells [26]. Although many RAGE ligands are promiscuous with regard to receptor use, S100A12 has only been shown to bind to RAGE. S100A12 expression is high in inflammatory diseases such as atherosclerosis, rheumatoid arthritis, Crohn's disease, Kawaski disease, and cystic fibrosis. Within the lungs, S100A12 and RAGE are increased during acute lung injury (ALI) [26]. S100A12 expression may reflect activation of neutrophils during pulmonary inflammation and may contribute to endothelial activation via binding to RAGE [26].
β2 integrins
Recruitment of leukocytes to the site of infection is an essential step in host defense during infectious diseases against invading pathogens. RAGE plays a role in the regulation of cell migration in several ways. First of all, RAGE is a counter-receptor for integrins on leukocytes; in particular, RAGE has been identified as a binding partner for the β2 integrins, Mac-1 and p150, 95 [8]. Second, by the interaction of RAGE with β2 integrin-mediated leukocyte recruitment in vivo: RAGE-/- mice displayed a diminished number of adherent inflammatory cells on the peritoneum after CLP [9] and a reduction in neutrophil influx in the peritoneal cavity after thioglycollate peritonitis [8]. Interestingly, HMGB1 can activate lateral (in cis) RAGE-Mac-1 interactions on the leukocyte cell surface, enhancing Mac-1-intercellular adhesion molecule (ICAM)-1-dependent adhesion and migration [27] (Figure 1, indicated by the blue line and blue "+"). Furthermore, a recent report shows that endothelial expressed RAGE acts in concert with ICAM-1 in mediating β2 integrin-dependent leukocyte adhesion during acute trauma-induced inflammation [28] (Figure 1, indicated by the green line and green "+").
Figure 1.
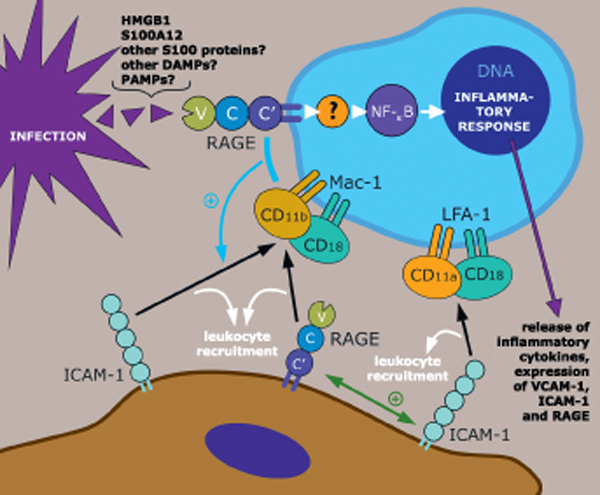
Putative involvement of the receptor for advanced glycation endproducts (RAGE) during infection. The damage-associated molecular patterns (DAMPs), high-mobility group box 1 (HMGB1) and S100A12, are released during infection ([15,17], and unpublished data) and bind to and activate RAGE. It has to be determined whether other S100 proteins and other DAMPs are RAGE ligands (indicated as purple shapes) released during infection. It would be interesting to investigate whether RAGE can directly bind to, become activated and mount a first immune reaction after ligation with specific PAMPs as well. Engagement of RAGE by its ligands results in receptor-dependent signaling and activation of NF-κB leading to a pro-inflammatory response; the signaling pathway is largely unknown. In addition, RAGE interacts as an endothelial (and epithelial) adhesion receptor with the leukocyte integrin, CD11b/CD18 (Mac-1) (lower section) [8]. Furthermore, lateral (in cis) RAGE-Mac-1 interaction on the leukocyte surface is mediated by HMGB1 and activates Mac-1-intercellular adhesion molecule (ICAM)-1 dependent adhesion and migration and augments leukocyte recruitment [27] (indicated by the blue line and blue "+"). Moreover, a recent report shows that endothelially expressed RAGE acts in concert with ICAM-1 in mediating β2 integrin-dependent leukocyte adhesion during acute trauma-induced inflammation [28] (indicated by the green line and green "+").
RAGE: a signal transducing receptor
The signaling cascade(s) of RAGE - induced by engagement of its various ligands - that ultimately activates NF-κB is largely unknown. The predicted cytosolic portion of RAGE, consisting of 43 amino acids, is short compared to other PRRs, the TLRs and IL-1 receptors, and does not include a known signaling domain or motif. A RAGE mutant lacking this intracellular tail does not activate NF-κB and behaves like a dominant negative, preventing pro-inflammatory cytokine release from macrophages. These data indicate a critical role of this cytosolic portion in transducing the signal from the cell surface to the nucleus. One possibility is that RAGE uses as yet unknown adaptors framing a whole 'new' signaling cascade to NF-κB. Another possibility is that the RAGE tail interacts with a Toll/IL-1 receptor (TIR)-containing protein which then recruits the downstream TIR-containing proteins in a way analogous to TLR-mediated signaling pathways. Finally, RAGE could transduce signals from the cell surface to the nucleus by bypassing the TIR-containing adaptor, directly interacting with member(s) of the signaling cascade.
In addition to triggering NF-κB activation, RAGE engagement by its myriad ligands is linked to an array of signaling pathways, including MAPK family members, such as Jun-N-terminal kinase (JNK), p38 and extracellular signal-regulated kinase (ERK), PI3K/Akt, Rho GTPases, Jak/STAT and Src family kinases [7].
This rather extraordinary variety of observed signals may be due to the broad expression of RAGE, its diversity of ligands, and possible contaminating elements in the preparations used in experiments.
Soluble RAGE (sRAGE)
The truncated form of full-length RAGE, soluble RAGE (sRAGE), consists of only the extracellular ligand-binding domain (V-C-C') lacking the cytosolic and transmembrane domains (i.e., the parts that transfer a signal into the cell) and circulates in the bloodstream. sRAGE has been indicated to be involved in inflammatory processes in several ways. First, sRAGE blood concentrations are associated with various inflammatory diseases in patients and in rats with experimentally induced ALI [29]. Furthermore, it is suggested that sRAGE can compete with full length cell-surface RAGE for ligand engagement, preventing these ligands from binding to RAGE or other receptors and/or exerting effects otherwise. Exogenous sRAGE treatment indeed attenuated inflammatory responses in several animal models, including models of type II collagen-induced arthritis, hepatic injury, diabetic atherosclerosis, delayed type hypersensitivity, and experimental auto-immune encephalomyelitis [7]. The involvement of sRAGE during infection is not known. Based on experimental studies in rats and in patients with ALI, sRAGE has been described as a marker of lung injury [29].
RAGE during infection
An increasing amount of research suggests a role for RAGE in the pathogenesis of pneumonia, peritonitis and sepsis, indicating that RAGE (ligand)-directed therapies might offer new treatment opportunities for human disease in the future.
RAGE during pneumonia
Localization and role of RAGE in the lungs
Recent studies point to an important role of RAGE in the pulmonary compartment in physiological as well as in pathological circumstances. Physiologically, RAGE is expressed at high basal levels in the lungs relative to other tissues [26,29-34], suggesting that RAGE may have lung-specific functions distinct from the role of RAGE in other adult tissues. In particular, although the kidney is dramatically affected by microangiopathy and fibrosis - which is substantially attributed to RAGE - in patients with diabetes, the lungs, with a significantly higher baseline RAGE expression than the kidney, remain unaffected. In addition, RAGE has been found to be specifically localized near the basal cell membrane within alveolar pneumocytes [32,35,36]. These two observations raise the question as to whether RAGE has a function in normal healthy lungs. Indeed, Englert et al. documented that aged RAGE-/- mice develop pulmonary fibrosis-like alterations spontaneously; lungs from 19 to 24 month-old RAGE-/- mice showed increased collagen staining and displayed increased levels of hydroxyproline relative to wild type mice [31]. RAGE knockdown in pulmonary fibroblasts increased their proliferation and migration in vitro, suggesting an important protective function of RAGE in the lungs and that loss of RAGE may be related to functional changes of pulmonary cell types resulting in fibrotic disease [34]. Another study demonstrated that RAGE on epithelial cells promoted their adherence to human collagen (a major component of the alveolar basal lamina) and a spreading morphology, which may facilitate gas exchange and alveolar stability in vivo [34,35]. Together, these data suggest that RAGE plays a role in maintaining lung homeostasis in normal, healthy lungs. Further studies are needed to unravel the function(s) of pulmonary RAGE in physiology in more detail.
This putative functional role of RAGE in healthy lungs may be the explanation for the finding that the inhibition of RAGE signaling attenuates pathological sterile inflammatory responses in diverse non-pulmonary experimental studies [9-12], whereas in pulmonary non-infectious pathological inflammatory conditions, somewhat conflicting results emerge. Lung injury induced by either bleomycin or hyperoxia is diminished in RAGE-/- mice [37,38], suggesting a deteriorating attribution of RAGE. In contrast, Englert et al. showed that RAGE-/- mice developed more severe lung fibrosis after asbestos administration as measured by histological scoring and total lung hydroxyproline quantification [31]. Of note, in all these studies, the mice were much younger at the time of sacrifice than the aged (19-24 month-old) RAGE-/- mice that developed pulmonary fibrosis spontaneously in the experiment by Englert et al. [31]. Interestingly, lung homogenates and bronchoalveolar lavage (BAL) fluid from patients suffering from idiopathic pulmonary fibrosis reveal reduced membrane bound (and soluble) RAGE protein levels compared to healthy donor samples [31,34].
RAGE expression during pneumonia
Community-acquired pneumonia (CAP) is distinguished from hospital-acquired pneumonia (HAP) according to the time of acquisition of pneumonia and the pathogens involved. The Gram-positive bacterium, Streptococcus pneumoniae, is the single most frequent pathogen causing CAP, responsible for up to 60% of cases; Klebsiella pneumoniae, Haemophilus influenzae, Staphylococcus aureus, and viruses are isolated in about 10% of cases each and Mycobacterium tuberculosis is more prevalent in developing countries. Knowledge of the expression and role of RAGE in host defense during pneumonia is limited. Morbini et al. observed increased RAGE expression in patients with interstitial and post-obstructive pneumonia [32]; this report left unanswered whether patients with bacterial pneumonia were included in the analysis. Notably, two other studies showed that constitutively present RAGE was not upregulated during pulmonary inflammation associated with ALI or acute respiratory distress syndrome (ARDS): First, rats with ALI induced by intratracheally administered LPS displayed no change in the distribution of RAGE-expressing cells [29]; and second, patients with ARDS did not have increased pulmonary expression of RAGE [26]. sRAGE has been suggested to be a lung injury marker based on studies in patients with ALI and on experimental studies in rats [29]. sRAGE was increased in pulmonary edema fluid and serum from patients with either ALI or ARDS and with hydrostatic pulmonary edema, and in BAL fluid from rats with either LPS- or hydrochloric acid-induced ALI [29].
Considering the ubiquitous expression of RAGE in the lungs, its putative involvement in the regulation of lung inflammation and the somewhat inconsistent findings which currently exist in the literature, we investigated its role during pneumonia, a major cause of morbidity and mortality world-wide. Recently, we reported that murine pneumonia induced by S. pneumoniae and by influenza A virus was associated with an upregulation of intra-alveolar (membrane bound) RAGE expression [39,40]. Furthermore, lung tissue of mice intranasally infected with the K. pneumoniae or with M. tuberculosis also showed increased RAGE expression (unpublished data). These clinically very different types of pulmonary infection and the involvement of RAGE therein will be discussed below.
Role of RAGE in pneumonia caused by different pathogens
Levels of the high-affinity RAGE ligand, HMGB1, were higher in BAL fluid from patients with pneumonia compared to BAL fluid from healthy controls [17]. In experimentally induced pneumococcal pneumonia, the presence of RAGE was detrimental: Mice lacking RAGE had a better survival rate together with a lower pulmonary bacterial load and decreased dissemination of S. pneumoniae to blood and spleen compared to wildtype mice [39]. The difference was possibly partially due to an increased killing capacity of RAGE-/- alveolar macrophages. Additionally, lung injury and neutrophil recruitment were reduced in the RAGE-/- mice, which parallels findings on RAGE as an endothelial counter receptor for the β2 integrin, Mac-1, [8] and the interplay between RAGE and Mac-1 on leukocytes, required for HMGB1-mediated inflammatory cell recruitment [27]. In addition, blockade of the RAGE-HMGB1 interaction and prevention of the subsequent pro-inflammatory stimulus might be an explanation for the less severe pulmonary damage in the RAGE-/- mice during S. pneumoniae pneumonia.
Interestingly, in contrast to Gram-positive pneumonia, preliminary data from our laboratory reveal that RAGE plays a beneficial role in mice during the host response to Gram-negative pneumonia (unpublished data). Indeed, RAGE deficiency was associated with increased mortality and increased bacterial outgrowth and dissemination after K. pneumoniae inoculation (unpublished data). Relative to wild type mice, lung inflammation was similar and cytokine and chemokine levels were slightly - if at all - elevated. Moreover, RAGE-/- mice showed an unaltered response to intranasally instilled Klebsiella LPS with respect to pulmonary cell recruitment and local release of cytokines and chemokines. Together, these findings indicate that RAGE contributes to an effective antibacterial host response during K. pneumoniae pneumonia, whereas RAGE plays an insignificant part in the lung inflammatory response to either intact Klebsiella or Klebsiella LPS.
It is unclear whether RAGE can also interact with ligands from pathogens. If so, this could be part of the explanation for our observation that RAGE involvement during Gram-positive and -negative pneumonia had such opposite effects on mortality. In addition, RAGE-mediated effects on other first-line defense mechanisms, such as chemotaxis, phagocytosis, killing (including respiratory burst), may depend on the pathogen and may contribute to the observed effects in Gram-positive and -negative pneumonia models. However, this remains speculative until investigations have been performed to analyze this interesting issue.
In addition to its potential to cause pandemics, seasonal influenza A virus infection causes over 200,000 hospitalizations and approximately 41,000 deaths in the United States annually, being the 7th leading cause of mortality. We demonstrated that RAGE deficiency resulted in a better outcome from pulmonary influenza A virus infection as indicated by a relative protection from influenza A virus-induced lethality in mice [40]. This was accompanied by improved viral clearance and enhanced cellular T cell response and activation of neutrophils, suggesting that endogenous RAGE impairs the cellular immunity against respiratory tract infection with influenza A virus. RAGE ligand, HMGB1, as well as sRAGE were upregulated in BAL fluid during influenza A virus pneumonia. Hence, similar to pneumonia induced by the Gram-positive bacterium S. pneumoniae, RAGE is detrimental during pneumonia caused by influenza A virus. This is of particular interest, since it has been suggested that the greatest proportion of the mortality associated with influenza A virus infection is due to secondary bacterial pneumonia, with S. pneumoniae as the most frequent pathogen of the superinfection. Therefore, RAGE is a potential treatment target in postinfluenza pneumococcal pneumonia and further research is warranted to investigate this.
RAGE during abdominal sepsis
The role of RAGE in abdominal sepsis has been investigated in a limited number of studies so far. RAGE-deficient mice showed decreased mortality after induction of polymicrobial sepsis induced by CLP in two reports [9,41]. Moreover, anti-RAGE antibody yielded a better survival even when the anti-RAGE therapy was delayed up to 24 hours after CLP in mice receiving antibiotics [41]. The protective effect provided by the absence of RAGE was related to a firm inhibition of NF-κB activation, suggesting that the lack of excessive NF-κB activation in RAGE-/- mice might have contributed to their reduced mortality [9]. In addition, RAGE deficiency resulted in fewer inflammatory cells in the peritoneum [9], which parallels the results of an earlier investigation by the same group of authors identifying RAGE as a counter-receptor for the β2 integrin, Mac-1 (CD11b/CD18), and thereby as a mediator of leukocyte recruitment and adhesion [8]. Furthermore, the protective effect of RAGE inhibition in this CLP model could at least in part be the consequence of the inhibition of one of its ligands, HMGB1. Indeed, HMGB1 is secreted into the circulation after CLP and anti-HMGB1 antibody led to increased survival after CLP-induced peritonitis [18].
In the same surgically (CLP)-induced model of sepsis, RAGE deficiency and anti-RAGE therapy were reported not to affect bacterial outgrowth in the peritoneum, liver, or spleen [41]. Notwithstanding, a possible role of RAGE in antibacterial defense cannot be easily evaluated from this study because host defense against CLP depends, at least in part, on the extent of intestinal necrosis and the formation of a local abscess. Also, all mice in this experiment received broad spectrum antibiotics and bacterial outgrowth was only determined in mice that survived (i.e., not at predefined time points after CLP). For this reason, we used our model of abdominal sepsis induced by injection of the Gram-negative bacterium Escherichia coli into the peritoneum [42,43] to study whether RAGE affects antibacterial defense. This model is a relevant tool to investigate the role of receptors/mediators in limiting the growth and dissemination of bacteria after a primary intra-abdominal infection and to assess the contribution of these proteins to specific immune responses. RAGE expression was upregulated during E. coli induced sepsis [42]. RAGE deficiency (either pharmacologically using anti-RAGE IgG anti-bodies or genetically using RAGE knock out mice) was related to a higher bacterial load and dissemination [42]. These data indicate that RAGE signaling contributes to an effective antibacterial response during abdominal sepsis. RAGE exerted this effect probably indirectly and not via direct interaction with E. coli, considering the observation that leukocytes from RAGE-/- mice had an unaltered capacity to phagocytose and kill E. coli in vitro. Furthermore, the finding that deficiency of RAGE in general was associated with an exaggerated host response during E. coli sepsis [42] on the one hand, and with an attenuated inflammatory response and better survival in (other) sterile models of intraperitoneal injection of LPS derived from E. coli [42,44] on the other hand, suggests that although RAGE is involved in the immune reaction to E. coli, this function can be compensated for by other receptors in the presence of a growing bacterial load. The high-affinity RAGE ligand, HMGB1, is secreted into the circulation systemically during clinical sepsis [16-18] as well as in our experimental sepsis model of E. coli [43]. Importantly, HMGB1 has been shown to transduce cellular signals in vitro and in vivo by interacting with at least three other receptors, i.e., TLR2, TLR4 and TLR9 when HMGB1 is complexed with CPG DNA [24,44,45]. One possible explanation for the increased response in the RAGE lacking mice during E. coli sepsis is, therefore, that the absence of RAGE could facilitate the interaction between HMGB1 and TLR2, TLR4 and/or TLR9.
Evidence of involvement of ligands of RAGE and HMGB1 in host defense in E. coli abdominal sepsis was recently published by our laboratory [43]. Inhibition of multiple RAGE ligands (by the administration of sRAGE) and inhibition of HMGB1 (by the administration of anti-HMGB1 antibodies) led to an enhanced bacterial dissemination of E. coli, denoting an advantageous role of RAGE ligands, including HMGB1, in the antibacterial response during Gram-negative sepsis.
Interestingly, we recently found that S100A12, another high-affinity ligand of RAGE, is released systemically in patients during (abdominal) sepsis and also locally during peritonitis (unpublished data). Additionally, intravenous injection of LPS in healthy humans raised circulating S100A12 levels, implying that LPS might partially contribute to this upregulation during Gram-negative infection. Payen et al. reported that, in patients with septic shock, mRNA S100A12 expression by circulating leukocytes was decreased during the recovery phase [46]. One possible function of S100A12 in host defense during infection and sepsis is its role as a DAMP. NF-κB mediated expression of pro-inflammatory cytokines and upregulation of ICAM-1 and vascular cell adhesion molecule (VCAM)-1 on endothelium has been documented in vitro after S100A12 stimulation [47]. Further-more, S100A12 could be of benefit for the host during infection and sepsis due to its (more direct) antibacterial activity. Cole et al. determined that S100A12 has activity primarily against Gram-negative bacteria, including E. coli [48]. Because of the absence of S100A12 in rodents, a potential functional role of S100A12 during sepsis cannot be easily investigated by inhibiting/deleting S100A12 in animals. Altogether, the role of S100A12 during sepsis has yet to be evaluated using non-rodent models.
Bopp et al. documented that septic patients have elevated circulating sRAGE levels and that non-survivors show higher plasma sRAGE concentrations than survivors, suggesting that sRAGE is related to severity and clinical outcome in sepsis [49]. Knowledge on the role of endogenous sRAGE in sepsis is scarce. Hudson et al. demonstrated that sRAGE levels might represent an early marker of microvascular dysfunction, a phenomenon also present in sepsis [50]. Furthermore, increased sRAGE concentrations in sepsis might represent the acute inflammatory status as splice variants of RAGE or as split off variants of the cell surface RAGE, the latter analogous to ICAM-1, another member of the immunoglobulin superfamily, which is a marker of cellular damage during sepsis. Another possibility is that systemic sRAGE levels might be elevated in parallel with HMGB1/S100A12 levels as a counter-system against HMGB1/S100A12 elicited tissue effects. More research is needed to clarify the functional role of sRAGE in sepsis and its putative role as a new sepsis marker.
Conclusion
The innate immune response is the first line of defense against pathogens. The experimental studies described herein provide further insight into the role of RAGE and its ligands in host defense during clinically important infections, which eventually may contribute to better therapies against specific pathogens. While interpreting the results from preclinical investigations, one has to keep in mind that a careful balance between the inflammatory and anti-inflammatory response is vital in order to survive or recover from a severe infection.
The observation that lack of RAGE is of benefit in one pneumonia model and detrimental in another, clearly adds to the notion that the way in which RAGE mediates host defense against different pathogens relies on distinct mechanisms. It would be highly interesting to evaluate whether RAGE can directly bind to, become activated, and mount a first immune reaction after ligation with specific PAMPs. Furthermore, RAGE-mediated effects on other first-line defense mechanisms, such as chemo-taxis, killing, phagocytosis and respiratory burst could depend on the pathogen. As such, targeting RAGE may be ineffective or even harmful in some infectious conditions. Therefore, more studies are necessary to justify clinical trials targeting RAGE in patients with severe infections. In this respect one could think of research on RAGE inhibition in pneumococcal and influenza A viral pneumonia. Additionally, experiments in which RAGE targeting is delayed until after bacterial/viral infection and combined with antibiotic/antiviral therapy should be considered. Moreover, more studies need to be conducted on the role of RAGE in critical organ derangements involved in the pathogenesis of severe infection, including activation of the coagulation system and the complement system. RAGE remains a potential yet promising therapeutic target that awaits further research.
Competing interests
The authors declare that they have no competing interests.
List of abbreviations used
AGE: advanced glycation endproducts; ALI: acute lung injury; BAL: bronchoalveolar lavage; CAP: community-acquired pneumonia; CLP: cecal ligation and puncture; DAMPS: damage-associated molecular patterns; ERK: extracellular signal-related kinase; HAP: hospital-acquired pneumonia; HMGB1: high-mobility group box 1; HSP: heat shock protein; JNK: Jun-N-terminal kinase; MAPK: mitogen-activated protein kinase; MRP: myeloid-related protein; NF-κB: nuclear factor-kappa B; NLRs: Nod-like receptors; PAMP: pathogen-associated molecular patterns; PRRs: pattern recognition receptors; RAGE: receptor for advanced glycation endproducts; TIR: Toll/IL-1 receptor; TLRs: Toll-like receptors; TNF: tumor necrosis factor; sRAGE: soluble RAGE; VCAM: vascular cell adhesion molecule.
Acknowledgements
This article is one of eleven reviews selected from the Annual Update in Intensive Care and Emergency Medicine 2011 (Springer Verlag) and co-published as a series in Critical Care. Other articles in the series can be found online at http://ccforum.com/series/annual. Further information about the Annual Update in Intensive Care and Emergency Medicine is available from http://www.springer.com/series/8901
References
- Janeway CA Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- Bianchi ME. DAMPs, PAMPs and alarmins: All we need to know about danger. J Leukoc Biol. 2007;81:1–5. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- Harris HE, Raucci A. Alarmin(g) News about danger: Workshop on innate danger signals and HMGB1. EMBO Rep. 2006;7:774–778. doi: 10.1038/sj.embor.7400759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–195. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- Liu Y, Chen GY, Zheng P. CD24-Siglec G/10 discriminates danger-from pathogen-associated molecular patterns. Trends Immunol. 2009;30:557–561. doi: 10.1016/j.it.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, Mori S, Wake H. et al. Establishment of in vitro binding assay of high mobility group box-1 and S100A12 to receptor for advanced glycation endproducts: heparin's effect on binding. Acta Med Okayama. 2009;63:203–211. doi: 10.18926/AMO/31812. [DOI] [PubMed] [Google Scholar]
- Bierhaus A, Stern DM, Nawroth PP. RAGE in inflammation: a new therapeutic Target? Curr Opin Investig Drugs. 2006;7:985–991. [PubMed] [Google Scholar]
- Chavakis T, Bierhaus A, Al Fakhri N. et al. The pattern recognition receptor (RAGE) is a counterreceptor for leukocyte integrins: a novel pathway for inflammatory cell recruitment. J Exp Med. 2003;198:1507–1515. doi: 10.1084/jem.20030800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liliensiek B, Weigand MA, Bierhaus A. et al. Receptor for advanced glycation end products (RAGE) regulates sepsis but not the adaptive immune response. J Clin Invest. 2004;113:1641–1650. doi: 10.1172/JCI18704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goova MT, Li J, Kislinger T. et al. Blockade of receptor for advanced glycation end-products restores effective wound healing in diabetic mice. Am J Pathol. 2001;159:513–525. doi: 10.1016/S0002-9440(10)61723-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldegirmen G, Zeng S, Feirt N. et al. RAGE limits regeneration after massive liver injury by coordinated suppression of TNF-alpha and NF-kappaB. J Exp Med. 2005;201:473–484. doi: 10.1084/jem.20040934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park L, Raman KG, Lee KJ. et al. Suppression of accelerated diabetic atherosclerosis by the soluble receptor for advanced glycation endproducts. Nat Med. 1998;4:1025–1031. doi: 10.1038/2012. [DOI] [PubMed] [Google Scholar]
- Schmidt AM, Yan SD, Yan SF, Stern DM. The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J Clin Invest. 2001;108:949–955. doi: 10.1172/JCI14002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotze MT, Tracey KJ. High-mobility group box 1 protein (HMGB1): Nuclear weapon in the immune arsenal. Nat Rev Immunol. 2005;5:331–342. doi: 10.1038/nri1594. [DOI] [PubMed] [Google Scholar]
- Ueno H, Matsuda T, Hashimoto S. et al. Contributions of high mobility group box protein in experimental and clinical acute lung injury. Am J Respir Crit Care Med. 2004;170:1310–1316. doi: 10.1164/rccm.200402-188OC. [DOI] [PubMed] [Google Scholar]
- Wang H, Bloom O, Zhang M. et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285:248–251. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- van Zoelen MA, Laterre PF, van Veen SQ. et al. Systemic and local high mobility group box 1 concentrations during severe infection. Crit Care Med. 2007;35:2799–2804. doi: 10.1097/01.CCM.0000287588.69000.97. [DOI] [PubMed] [Google Scholar]
- Yang H, Ochani M, Li J. et al. Reversing established sepsis with antagonists of endogenous high-mobility group box 1. Proc Natl Acad Sci USA. 2004;101:296–301. doi: 10.1073/pnas.2434651100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin S, Wang H, Yuan R. et al. Role of HMGB1 in apoptosis-mediated sepsis lethality. J Exp Med. 2006;203:1637–1642. doi: 10.1084/jem.20052203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suda K, Kitagawa Y, Ozawa S. et al. Anti-high-mobility group box chromosomal protein 1 antibodies improve survival of rats with sepsis. World J Surg. 2006;30:1755–1762. doi: 10.1007/s00268-005-0369-2. [DOI] [PubMed] [Google Scholar]
- Wang H, Liao H, Ochani M. et al. Cholinergic agonists inhibit HMGB1 release and improve survival in experimental sepsis. Nat Med. 2004;10:1216–1221. doi: 10.1038/nm1124. [DOI] [PubMed] [Google Scholar]
- Hotchkiss RS, Nicholson DW. Apoptosis and caspases regulate death and inflammation in sepsis. Nat Rev Immunol. 2006;6:813–822. doi: 10.1038/nri1943. [DOI] [PubMed] [Google Scholar]
- Hotchkiss RS, Chang KC, Swanson PE. et al. Caspase inhibitors improve survival in sepsis: a critical role of the lymphocyte. Nat Immunol. 2000;1:496–501. doi: 10.1038/82741. [DOI] [PubMed] [Google Scholar]
- Tian J, Avalos AM, Mao SY. et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol. 2007;8:487–496. doi: 10.1038/ni1457. [DOI] [PubMed] [Google Scholar]
- Yang H, Hreggvidsdottir HS, Palmblad K. et al. A critical cysteine is required for HMGB1 binding to toll-like receptor 4 and activation of macrophage cytokine release. Proc Natl Acad Sci USA. 2010;107:11942–11947. doi: 10.1073/pnas.1003893107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittkowski H, Sturrock A, van Zoelen MA. et al. Neutrophil-derived S100A12 in acute lung injury and respiratory distress syndrome. Crit Care Med. 2007;35:1369–1375. doi: 10.1097/01.CCM.0000262386.32287.29. [DOI] [PubMed] [Google Scholar]
- Orlova VV, Choi EY, Xie C. et al. A novel pathway of HMGB1-mediated inflammatory cell recruitment that requires Mac-1-integrin. EMBO J. 2007;26:1129–1139. doi: 10.1038/sj.emboj.7601552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frommhold D, Kamphues A, Hepper I. et al. RAGE and ICAM-1 Cooperate in mediating leukocyte recruitment during acute inflammation in vivo. Blood. 2010;116:841–849. doi: 10.1182/blood-2009-09-244293. [DOI] [PubMed] [Google Scholar]
- Uchida T, Shirasawa M, Ware LB. et al. Receptor for advanced glycation end-products is a marker of type i cell injury in acute lung injury. Am J Respir Crit Care Med. 2006;173:1008–1015. doi: 10.1164/rccm.200509-1477OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng C, Tsuneyama K, Kominami R. et al. Expression profiling of endogenous secretory receptor for advanced glycation end products in human organs. Mod Pathol. 2005;18:1385–1396. doi: 10.1038/modpathol.3800450. [DOI] [PubMed] [Google Scholar]
- Englert JM, Hanford LE, Kaminski N. et al. A role for the receptor for advanced glycation end products in idiopathic pulmonary fibrosis. Am J Pathol. 2008;172:583–591. doi: 10.2353/ajpath.2008.070569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morbini P, Villa C, Campo I, Zorzetto M, Inghilleri S, Luisetti M. The receptor for advanced glycation end products and its ligands: a new inflammatory pathway in lung disease? Mod Pathol. 2006;19:1437–1445. doi: 10.1038/modpathol.3800661. [DOI] [PubMed] [Google Scholar]
- Buckley ST, Ehrhardt C. The receptor for advanced glycation end products (RAGE) and the lung. J Biomed Biotechnol. 2010. p. 917108. [DOI] [PMC free article] [PubMed]
- Queisser MA, Kouri FM, Konigshoff M. et al. Loss of RAGE in pulmonary fibrosis: molecular relations to functional changes in pulmonary cell types. Am J Respir Cell Mol Biol. 2008;39:337–345. doi: 10.1165/rcmb.2007-0244OC. [DOI] [PubMed] [Google Scholar]
- Demling N, Ehrhardt C, Kasper M, Laue M, Knels L, Rieber EP. Promotion of cell adherence and spreading: a novel function of RAGE, the highly selective differentiation marker of human alveolar epithelial type I cells. Cell Tissue Res. 2006;323:475–488. doi: 10.1007/s00441-005-0069-0. [DOI] [PubMed] [Google Scholar]
- Fehrenbach H, Kasper M, Tschernig T, Shearman MS, Schuh D, Muller M. Receptor for advanced glycation endproducts (RAGE) exhibits highly differential cellular and subcellular localisation in rat and human lung. Cell Mol Biol. 1998;44:1147–1157. [PubMed] [Google Scholar]
- He M, Kubo H, Ishizawa K. et al. The role of the receptor for advanced glycation end-products in lung fibrosis. Am J Physiol Lung Cell Mol Physiol. 2007;293:L1427–L1436. doi: 10.1152/ajplung.00075.2007. [DOI] [PubMed] [Google Scholar]
- Reynolds PR, Schmitt RE, Kasteler SD. et al. Receptors for advanced glycation end-products targeting protect against hyperoxia-induced lung injury in mice. Am J Respir Cell Mol Biol. 2010;42:545–551. doi: 10.1165/rcmb.2008-0265OC. [DOI] [PubMed] [Google Scholar]
- van Zoelen MA, Schouten M, de Vos AF. et al. The receptor for advanced glycation end products impairs host defense in pneumococcal pneumonia. J Immunol. 2009;182:4349–4356. doi: 10.4049/jimmunol.0801199. [DOI] [PubMed] [Google Scholar]
- van Zoelen MA, van der Sluijs KF, Achouiti A. et al. Receptor for advanced glycation end products is detrimental during influenza A virus pneumonia. Virology. 2009;391:265–273. doi: 10.1016/j.virol.2009.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutterloh EC, Opal SM, Pittman DD. et al. Inhibition of the RAGE products increases survival in experimental models of severe sepsis and systemic infection. Crit Care. 2007;11:R122. doi: 10.1186/cc6184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Zoelen MA, Schmidt AM, Florquin S. et al. Receptor for advanced glycation end products facilitates host defense during Escherichia coli-induced abdominal sepsis in mice. J Infect Dis. 2009;200:765–773. doi: 10.1086/604730. [DOI] [PubMed] [Google Scholar]
- van Zoelen MA, Achouiti A, Schmidt AM. et al. Ligands of the receptor for advanced glycation end products, including high-mobility group box 1, limit bacterial dissemination during escherichia coli peritonitis. Crit Care Med. 2010;38:1414–1422. doi: 10.1097/CCM.0b013e3181de18bc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Zoelen MA, Yang H, Florquin S. et al. Role of Toll-Like receptors 2 and 4, and the receptor for advanced glycation end products in high-mobility group box 1-induced inflammation in vivo. Shock. 2009;31:280–284. doi: 10.1097/SHK.0b013e318186262d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JS, Svetkauskaite D, He Q. et al. Involvement of Toll-Like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem. 2004;279:7370–7377. doi: 10.1074/jbc.M306793200. [DOI] [PubMed] [Google Scholar]
- Payen D, Lukaszewicz AC, Belikova I. et al. Gene profiling in human blood leucocytes during recovery from septic shock. Intensive Care Med. 2008;34:1371–1376. doi: 10.1007/s00134-008-1048-1. [DOI] [PubMed] [Google Scholar]
- Foell D, Wittkowski H, Vogl T, Roth J. S100 Proteins expressed in phagocytes: a novel group of damage-associated molecular pattern molecules. J Leukoc Biol. 2007;81:28–37. doi: 10.1189/jlb.0306170. [DOI] [PubMed] [Google Scholar]
- Cole AM, Kim YH, Tahk S. et al. Calcitermin, a novel antimicrobial peptide isolated from human airway secretions. FEBS Lett. 2001;504:5–10. doi: 10.1016/S0014-5793(01)02731-4. [DOI] [PubMed] [Google Scholar]
- Bopp C, Hofer S, Weitz J. et al. SRAGE is elevated in septic patients and associated with patients outcome. J Surg Res. 2008;147:79–83. doi: 10.1016/j.jss.2007.07.014. [DOI] [PubMed] [Google Scholar]
- Hudson BI, Harja E, Moser B, Schmidt AM. Soluble levels of receptor for advanced glycation endproducts (SRAGE) and coronary artery disease: the next C-reactive protein? Arterioscler Thromb Vasc Biol. 2005;25:879–882. doi: 10.1161/01.ATV.0000164804.05324.8b. [DOI] [PubMed] [Google Scholar]