

Abstract
Transcriptional activity of the androgen receptor (AR) is crucial for growth and survival of prostate cancer even upon development of resistance to androgen ablation and antiandrogen therapies. Therefore, novel therapies that can suppress AR transcriptional activity when conventional hormone therapies fail are needed. Here, we show that histone deacetylase (HDAC) inhibitors, including SAHA (vorinostat) and LBH589, which are currently being tested in clinic, could be such a therapy. HDAC inhibitors block the AR-mediated transcriptional activation of many genes, including the TMPRSS2 gene involved in fusion with ETS family members in a majority of prostate cancers. Genetic knockdown of either HDAC1 or HDAC3 can also suppress expression of AR-regulated genes, recapitulating the effect of HDAC inhibitor treatment. Whereas HDAC inhibitor treatment can lower androgen receptor protein levels in prostate cancer cells, we show that independent of AR protein levels, HDAC inhibitors block AR activity through inhibiting the assembly of coactivator/RNA polymerase II complex after AR binds to the enhancers of target genes. Failed complex assembly is associated with a phase shift in the cyclical wave of AR recruitment that typically occurs in response to ligand treatment. HDAC inhibitors retain the ability to block AR activity in castration-resistant prostate cancer models and, therefore, merit clinical investigation in this setting. The HDAC-regulated AR target genes defined here can serve as biomarkers to ensure sufficient levels of HDAC inhibition.
Introduction
Current hormone therapy for prostate cancer consists of two classes of drugs: those that lower serum testosterone and androgen receptor (AR) antagonists that target the ligand-binding domain (LBD) of the receptor. Although initially effective at blocking tumor growth, these therapies eventually fail, leading to a lethal drug-resistant stage called castration-resistant prostate cancer (CRPC). Evidence suggests that CRPC continues to depend on AR function for growth, and the progression from castration sensitive to castration-resistant state involves reactivation of AR in low androgen milieu (1). Our laboratory previously used microarray profiling of seven isogenic xenograft models to show that the transition from castration-sensitive to castration-resistant disease is associated with overexpression of AR protein, which proved necessary and sufficient to confer drug resistance (2). Expression profiling and AR mutagenesis studies suggested a mechanism whereby the modest increase in AR protein level hypersensitizes cells to residual levels of ligand remaining during hormone therapy and restores the transcription of key AR-regulated genes (ARG). This increase in AR protein can also convert the AR antagonist bicalutamide into an agonist. Therefore, novel agents that can disrupt AR function in the setting of overexpression are needed.
Transcription of AR target genes is regulated by the assembly of a multiprotein transcription factor complex. Agonists promote recruitment of AR and coactivators that have histone acetyltransferase activity to promoters of AR target genes, leading to histone acetylation and active transcription (3). In contrast, AR bound to antagonists, such as bicalutamide, recruits corepressors, such as NCoR or SMRT, that complex with histone deacetylases (HDAC) and repress gene expression (4). This and other evidence correlate histone acetylation with active gene transcription. Consistent with this model, HDAC inhibitors can relieve transcriptional repression mediated by nuclear receptors (5-7). By analogy, the HDAC inhibitor trichostatin A (TSA) has been reported to augment AR activity, as measured by androgen-dependent reporters and PSA (3, 8). However, two lines of evidence suggest that HDACs may be required for active transcription of ARGs. First, HDACs are overexpressed in prostate cancer and overexpression is associated with poor outcome (9). Second, HDAC inhibitors have greater antiproliferative activity against steroid receptor–positive prostate and breast cancer models compared with prostate and breast cancer models that are steroid receptor–negative (10-12). If HDACs function solely as repressors of hormone receptor signaling, then HDAC inhibitors should augment steroid receptor signaling and stimulate growth.
We addressed this complexity in prostate cancer by examining the effect of HDAC inhibitors on AR function. Our data show that HDAC inhibitors decrease AR protein levels by inhibiting transcription of AR without significantly affecting AR protein stability, as previously reported (13, 14). In addition, independent of their effect on AR protein levels, HDAC inhibitors directly inhibit transcription of AR target genes. Through expression profiling, we defined a subset of AR target genes (~50%) that are HDAC-dependent. The HDAC-dependent AR target genes include TMPRSS2, the 5′ partner of a series of ETS fusion genes detected in 50% of human prostate cancers (15, 16). Expression profiling of cells with knockdown of HDAC1, HDAC2, HDAC3, and HDAC8 shows that the effects of pharmacologic HDAC inhibition on AR function can be partially recapitulated by knockdown of HDAC1 and HDAC3. Although HDAC inhibitors can lower AR protein levels through inhibition of AR transcription, we show that the predominant mechanism for interference with AR function is by blockade of RNA polymerase II (Pol II) recruitment to the promoters of HDAC-dependent AR target genes.
Materials and Methods
Materials
TSA, sodium butyrate, and cycloheximide were obtained from Sigma. LBH589 was kindly given by Novartis. SAHA was obtained from CTEP. Immunoblot assays were done using the following antibodies: AR N-20 (Santa Cruz); acetyl-α-tubulin (Sigma); α-tubulin (Santa Cruz); acetyl-H3 (Upstate); HDAC1, HDAC2, and HDAC8 (Upstate); HDAC3 (Santa Cruz); β-actin AC15 (Sigma); and PSA C-19 (Santa Cruz). Quantitative reverse transcription–PCR (RT-PCR) was performed on an Eppendorf Realplex machine. Reverse transcription and Sybr Green real-time PCR were performed using reagents from Applied Biosystems.
DNA constructs and PCR primers
AR2Pb-luciferase and ARE-luciferase were kindly provided by Robert Matusik (Vanderbilt) and Michael Rosenfeld (San Diego), respectively. Short hairpin RNA (shRNA) contructs against HDACs in the lentiviral pLKO.1 backbone were purchased from Open-biosystems (shHDAC1, TRCN0000004814; shHDAC2, TRCN0000004819; shHDAC3, TRCN0000004825; shHDAC8, TRCN0000004849). Control shRNA against firefly luciferase in the pLKO.1 backbone was a generous gift from David Sabatini (Whitehead Institute, Massachusetts Institute of Technology). Real-time primers against KLK2, NKX3.1, TMPRSS2, HDAC1, HDAC2, HDAC3, and HDAC8 were purchased from Superarray. Primers for PSA (F, 5′-CATCAGGAACAAAAGCGTGA-3′; R, ATATCGTAGAGCGGGTGTGG) and actin (F, 5′-TGTCACCAACTGGGACGACA; R, GGGGTGTTGAAGGTCTCAAA) were generated using PRIMER3 (17). Primers for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were 5′-GAAGGTGAAGGTCGGAGTC-3′ and 5′-GAAGATGGTGATGGGATTTC-3′. Primers for PSA enhancer were 5′-GCCTGGATCTGAGAGAGATATCATC-3′ and 5′-ACACCTTTTTTTTTCTGGATTGTTG-3 (18).
In vivo tumor growth
Animal studies were carried out under protocols approved by the Institutional Animal Care and Use Committee, and institutional guidelines for the proper human use of animals in research were followed. Nude females were obtained from National Cancer Institute Frederick Cancer Research and Development Center and maintained in ventilated caging. LBH589 was dissolved in 5% dextrose water.
Microarray experiments and data analysis
For HDAC inhibitor studies, LNCaP cells used for the microarray study were charcoal-stripped for 5 d and then stimulated for 18 h in the presence of 10 μg/mL cycloheximide with no HDAC inhibitors and two concentrations of TSA (0.1 and 1.0 μmol/L), SAHA (0.5 and 5.0 μmol/L), and LBH589 (10 and 100 nmol/L), each with or without 1.0 nmol/L R1881. RNA was isolated using RNAeasy kit (Qiagen), labeled according to manufacturer’s instructions (Affymetrix), and hybridized to GeneChip U133A 2.0 Chips. Labeling and hybridization were performed by the Memorial Sloan-Kettering Cancer Center Genomics Core Facility. All experiments were performed in duplicate, and raw CEL data can be downloaded from National Center for Biotechnology Information Gene Expression Omnibus accession GSE9000 and GSE12438. Data analysis was performed using GeneSpring software (Agilent Technologies). Expression changes were considered significant if the absolute expression was >250 in all samples and the fold-change was >1.7. Scatter plots were generated using SigmaPlot (Systat software), and heat map was generated using HeatMap Builder (Stanford University).
For HDAC knockdown studies, LNCaP cells were infected with lentiviral shRNA against individual HDACs or control (luciferase) at multiplicity of infection of ~5, and no selection was performed. Three days after infection, cells were plated in charcoal-stripped serum, and 6 d after infection, cells were treated in duplicate with vehicle or 1 nmol/L R1881 for 18 h in the presence of cyclohexamide, as above.
Chromatin immunoprecipitation
LNCaP cells were grown in 15-cm plates with 5% charcoal-stripped/dextran-treated fetal bovine serum (CS-FBS) for 5 d (90% confluency) and then stimulated as indicated. At the indicated time, cells were cross-linked with 1% formaldehyde and chromatin immunoprecipitation (ChIP) was performed, as previously described, using primers previously published for the PSA enhancer (19). Quantification was performed using real-time PCR of the immunoprecipated template. AR (N-20), SRC1 (M-371), and p300 (N-20) antibodies were from Santa Cruz Biotechnology, and Pol II (8WG16) was from Covance.
Results
HDAC inhibitors suppress AR transcriptional activity in castration-sensitive and castration-refractory prostate cancer
Previous studies using reporter assays rather than endogenous target genes have found that HDAC inhibitors enhance AR transcriptional activity (20, 21). As expected, we also observed that TSA potentiates AR activity when assessed using transfected templates, such as a multimerized ARE reporter or the modified probasin promoter (Supplementary Fig. S1). This potentiation occurred in transient assays, wherein the reporter plasmids are episomal, or in stable transfectants, wherein the reporter is presumably integrated into chromatin. However, TSA blocked androgen-induced PSA production in the LNCaP prostate cancer cell line at the same concentration required to induce acetylation of tubulin and histone H3 (Fig. 1A). Other HDAC inhibitors, including sodium butyrate (Fig. 1B) and clinically relevant compounds SAHA and LBH589 (Fig. 1C), all inhibit R1881-mediated PSA stimulation at concentrations that stimulated histone H3 acetylation. In a separate AR-positive cell line, LAPC4, R1881 stimulated PSA transcription at ~2.6-fold and is not significantly affected by the low concentration of SAHA or LBH589. SAHA (5 μmol/L) and LBH589 (100 nmol/L), which inhibited PSA in LNCaP cells, profoundly inhibited PSA transcription to a level even below the nonstimulated state (Fig. 1D).
Figure 1.
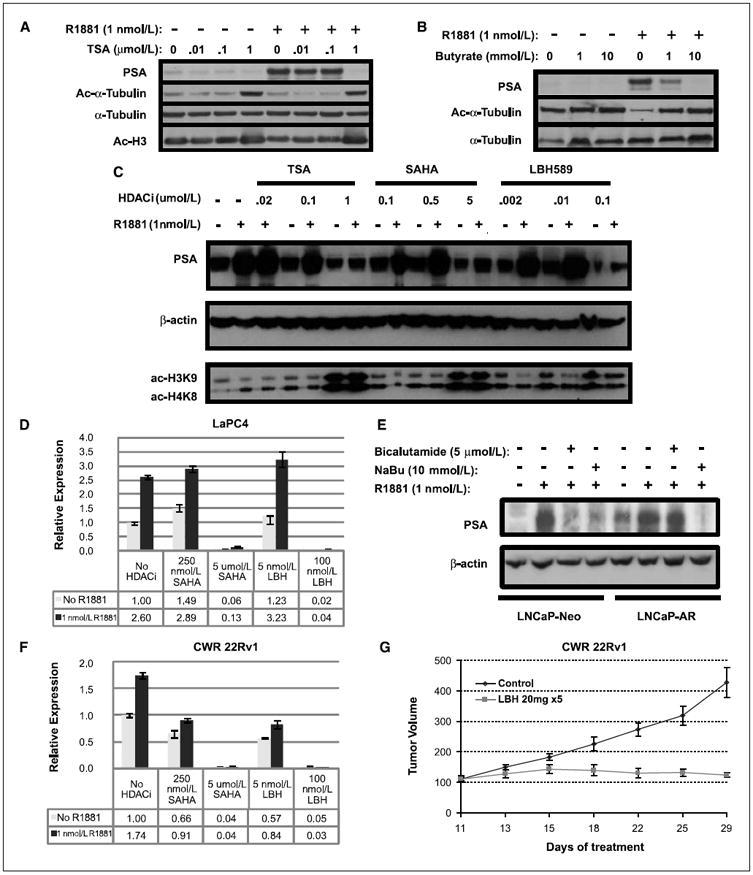
HDAC inhibition decreases AR activity in hormone-sensitive and hormone-refractory prostate cancer. A-C, LNCaP cells were starved of androgen for 5 d in 1% CS-FBS and then stimulated with or without 1.0 nmol/L R1881 in the presence of increasing doses of TSA (A), sodium butyrate (B), or TSA, SAHA, and LBH589 (C). After 24 h, lysates were blotted for PSA as indicator of AR function, acetyl-tubulin, acetyl-H3, or acetyl-H4 as markers of HDAC inhibition, and α-tubulin or actin as loading control. D, LaPC4 cells starved of androgen were treated with or without 1 nmol/L R1881 in the presence of HDAC inhibitors, as indicated for 16 h. Quantitative RT-PCR against PSA was normalized using GAPDH. E, LNCaP-Neo and LNCaP-AR cells were starved and treated with 1 nmol/L R1881 in the presence or absence of 10 mmol/L sodium butyrate or 5 μmol/L bicalutamide for 48 h. They were then lysed and immunoblotted for PSA and β-actin. F, RT-PCR of PSA of CWR22Rv1 cells performed as in D. G, CWR22Rv1 cells were mixed with Matrigel and inoculated s.c. in the right flank of four 6-wk-old mice. When tumors reached a minimum diameter of 5 mm, mice (n = 5–10 per treatment group) were randomly assigned to treatment with LBH589 (20 mg/kg) by i.p. injection. Twice a week, mice were weighed and tumor volumes were measured with vernier calipers.
Because residual AR activity, despite low androgen levels, underlies castration resistance, the ability of HDAC inhibitors to suppress PSA below that in charcoal-stripped serum suggests that they may be particularly useful in CRPC. We, thus, directly asked whether HDAC inhibitors are also able to block AR function in CRPC models. LNCaP-AR cells, generated by over-expression of AR, can grow in castrate mice and are resistant to conventional antiandrogens, such as bicalutamide (2). As expected, compared with LNCaP-Neo cells, LNCaP-AR cells produce more PSA in the uninduced state (Fig. 1E, lane 1 versus lane 5) and are resistant to bicalutamide. In contrast, sodium butyrate completely inhibited PSA production in both LNCaP-Neo and LNCaP-AR cells.
Previous work has established that HDAC inhibitors impair the growth of hormone-dependent prostate cancer xenografts, such as CWR22 (11), but the effect on CRPC is unknown. CWR22Rv1 is a castration-resistant xenograft derived by selection of CWR22 in castrate mice (22). CWR22Rv1 expresses high levels of PSA at baseline, which is only modestly stimulated by R1881, indicating high basal AR activity. Yet, both SAHA and LBH589 can significantly inhibit PSA transcription well below baseline (Fig. 1F). Next, we examined the effect of LBH589 on the growth of CWR22Rv1 propagated in female nude mice. LBH589 (20 mg/d given for 5 days) completely blocked the growth of established tumors, whereas tumors in control animals increased 4-fold in size over 18 days (Fig. 1G), thereby demonstrating that HDAC inhibitors have antitumor activity against CRPC.
HDAC inhibitors lower AR protein levels by blocking AR mRNA production
We next investigated the mechanism by which HDAC inhibitors interfere with AR activity. We explored three possible mechanisms: (a) increased degradation of AR by inhibition of HSP90 (14, 23), (b) inhibition of AR transcription (24), and (c) direct inhibition of transcription of AR target genes. We observed a decline in AR protein level when LNCaP cells were treated with TSA or sodium butyrate (Fig. 2A), consistent with either increased AR degradation or decreased transcription. To distinguish between the possibilities, we first measured AR mRNA and found decreased levels after TSA treatment (Fig. 2B). We then treated LNCaP cells with cycloheximide to block new protein synthesis and followed the turnover of AR over time. The rate of AR decline over 24 hours was not appreciably affected by TSA, suggesting no significant increase in the rate of degradation (Fig. 2C). Next, we used LNCaP-AR cells, in which the long terminal repeat (LTR) promoter, whose transcription is not suppressed by HDAC inhibitors, drives expression of exogenous AR. Whereas endogenous AR levels in LNCaP-Neo cells declined in response to TSA treatment, AR levels in LNCaP-AR cells did not (Fig. 2D). These data indicate that transcriptional suppression of AR, but not enhanced protein degradation, is the dominant mechanism by which AR protein levels are lowered by HDAC inhibitors. Despite the rescue of AR protein levels by exogenous AR expression in LNCaP-AR cells, sodium butyrate and TSA blocked the synthesis of PSA mRNA as efficiently as in parental LNCaP cells (Fig. 2E; see also Fig. 1E), indicating that HDAC inhibitors are also PSA expression independent from the lowering of AR mRNA and protein levels.
Figure 2.
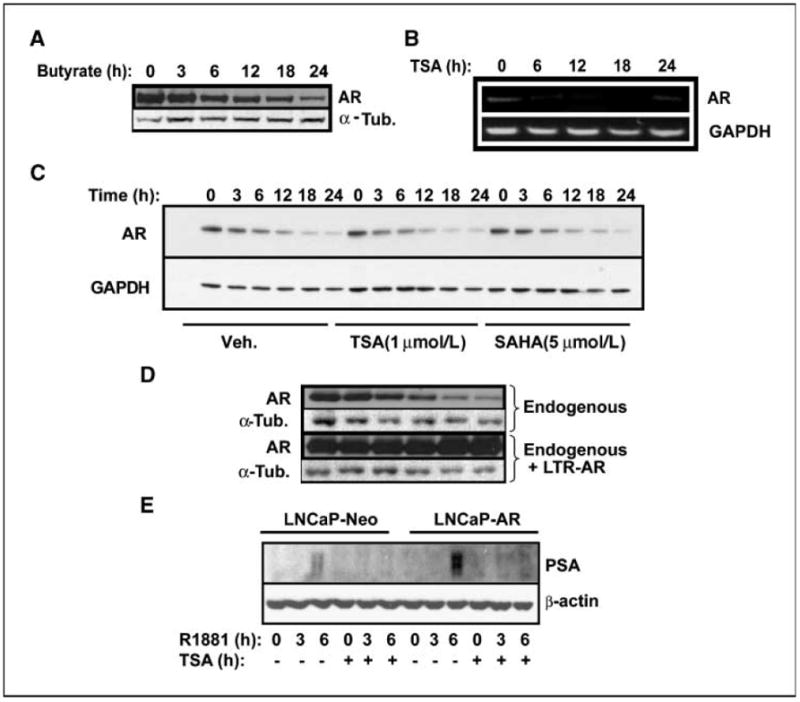
HDAC inhibition decreases endogenous AR levels by effects on transcription and not stability. A, LNCaP cells were treated with 10 mmol/L sodium butyrate up to 24 h. At each of the time points, cells were lysed and immunoblotted for AR and α-tubulin. B, LNCaP cells were treated with 1 μmol/L TSA for up to 24 h. At each time point, total RNA was extracted and AR and GAPDH mRNA were measured by semiquantitative (22 cycles) RT-PCR. C, LNCaP cells were treated with vehicle, 1 μmol/L TSA, or 5 μmol/L SAHA in the presence of 10 μg/mL cycloheximide for up to 24 h. Protein was lysed at each time point and immunoblotted for AR and GAPDH. D, LNCaP cells expressing empty plasmid or lentiviral LTR-driven exogenous AR were treated with TSA, lysed at indicated times, and blotted against AR and α-tubulin. E, LNCaP-Neo and LNCaP-AR cells were starved and treated with 1 nmol/L R1881 in the presence or absence of 1 μmol/L TSA. Cells were lysed and immunoblotted for PSA at each time point.
Defining a set of HDAC-dependent AR target genes
Having established that the activity of HDAC inhibitors on PSA expression is not solely due to lowering AR levels, we sought to characterize the effect of HDAC inhibition on AR target genes more broadly. We conducted microarray-based expression profiling studies of charcoal-stripped LNCaP cells treated with two doses of TSA (0.1 and 1 μmol/L), SAHA (0.5 and 5 μmol/L), or LBH589 (10 and 100 nmol/L) with and without androgen stimulation. The low and high concentrations inhibit PSA transcript by ~30% and 100%, respectively (Supplementary Fig. S2). Cycloheximide was included to block new protein synthesis and, thereby, bias the analysis toward primary AR target genes. In addition, cyclohexamide should negate the suppressive effect of HDAC inhibitors on AR transcription and ensure similar AR protein levels in all conditions.
We identified 159 (1.6%) genes whose expression was induced 1.7-fold by 1 nmol/L R1881, which we refer to as ARGs (Fig. 3A; Supplementary Table S1). This list shows substantial overlap with previously published gene lists in LNCaP cells (25, 26). Next, we examined the effects of HDAC inhibition on gene expression. As expected, treatment with higher dose of HDAC inhibitors induced a substantial change in expression profile, changing the expression of 23% genes over 1.7-fold (up or down in roughly equal distribution) in the case of SAHA and LBH589 and 27% in case of TSA (Fig. 3B for SAHA). Although this number is higher than quoted historically (27, 28), it is consistent with recent work using more sensitive microarray technology (29).
Figure 3.
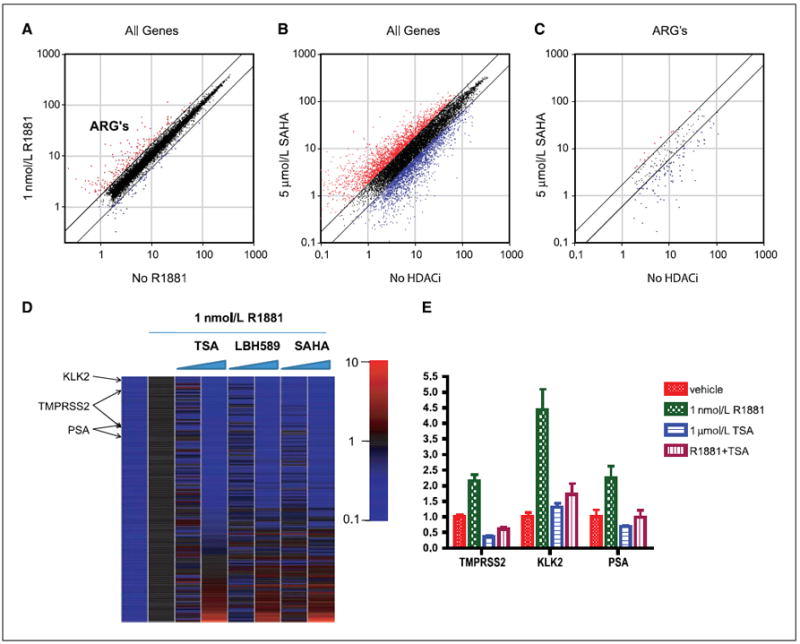
Effect of HDAC inhibitors on transcription of ARGs. LNCaP cells were starved for 3 d in charcoal-stripped serum and subsequently treated with the combination of HDAC inhibitor and R1881 in duplicate, as described in Materials and Methods. RNA was isolated, and expression profiling was performed. A, scatter plot of expression of all expressed genes at baseline (x axis) versus 1 nmol/L R1881 (y axis). Trend lines indicate 1.7-fold change; red dots are AR-induced genes, whereas blue dots are AR-suppressed genes. B, scatter plot of no HDAC inhibitor (HDACi) versus high-dose SAHA (5 μmol/L), which is representative of all three HDAC inhibitors at high dose. C, scatter plot of expression of ARGs (red dots from A) in cells treated with R1881 with or without 5 μmol/L SAHA. D, the expression levels of ARGs were normalized to no HDAC inhibitor and with 1 nmol/L R1881, and expression of genes is sorted from TSA suppression to TSA induction. First column represents uninduced expression and various shades of blue by definition. TMPRSS2, PSA, and KLK2 are indicated as shown. E, quantitative RT-PCR verification for PSA, KLK2, and TMPRSS2.
We then restricted analysis to the 159 ARGs defined above. Whereas high-dose SAHA treatment did not significantly affect the uninduced expression level of these genes, it inhibited R1881-induced expression by >1.7-fold in 72 of 159 (45%) of the ARGs (Fig. 3C). The majority of the remaining ARGs were affected by <1.7-fold, and only nine genes (5.6%) were induced. These percentages are substantially different from the effects seen on global gene expression.
To examine potential dose-dependent and compound-dependent effects of the three HDAC inhibitors, we generated a heat map of up-regulated ARGs ranked in order of effect by 1.0 μmol/L TSA (most inhibited to most induced; Fig. 3D) and scatter plots comparing the different conditions (Supplementary Fig. S3). Several conclusions arise from this analysis. First, the effects on AR target gene expression are consistent across all three compounds, with SAHA and LBH589 showing the greatest similarity. Second, the effects on ARGs are not seen until the higher dose. This finding is relevant because prior work has raised the possibility of a biphasic effect on PSA expression, with stimulation at low doses (3, 21) and suppression at high doses (our work). Third, the ARGs at either end of the heat map show substantial changes in gene expression and are candidates for pharmacodynamic biomarkers of HDAC inhibition in prostate cancer. HDAC-dependent expression of a subset of these genes was confirmed by quantitative RT-PCR in cells treated independently with TSA (Fig. 3E). Notably, this list includes the androgen-regulated gene TMPRSS2, which drives the expression of the oncoproteins ERG or ETV1 in human prostate cancers with TMPRSS2 translocations (16, 30).
HDAC1 and HDAC3 are required for optimal AR transcriptional activity
Having shown that HDAC inhibitors block expression of many ARGs, we asked if shRNA knockdown of a single HDAC can recapitulate the transcriptional effects of pharmacologic HDAC inhibition. TSA, SAHA, and LBH589 inhibit both classes I and II HDACs, whereas sodium butyrate and MS-275 (which similarly blocks PSA expression; data not shown) are class I–specific. Furthermore, HDAC1, HDAC2, and HDAC3 can be recruited to the PSA promoter by androgen stimulation (3). Therefore, we focused our analysis on the four class I HDACs (HDAC1, HDAC2, HDAC3, and HDAC8).
LNCaP cells expressing individual shRNAs against each of these four HDACs were generated and showed selective protein and mRNA knockdown of the appropriate HDAC (Fig. 4A and Supplementary Fig. S4). The microarray-based mRNA expression profiling of HDAC knockdown cells treated with or without R1881 revealed that HDAC1 knockdown, and to a lesser degree HDAC3 knockdown, caused suppression of ARGs, whereas HDAC2 and HDAC8 knockdown caused more modest changes in ARG expression (Fig. 4B). HDAC1 knockdown suppressed 42% of ARGs by >1.7-fold and induced 8%, which is very similar to SAHA. Examination of individual ARGs that was suppressed by SAHA treatment (black columns) revealed that KLK2 and PSA were significantly suppressed by both HDAC1 and HDAC3 knockdown; NKX3.1 was suppressed by HDAC1 knockdown, whereas TMPRSS2 was unaffected (gray columns; Fig. 4C). This result suggests that some ARGs, such as KLK2 and PSA, require activity of both HDAC1 and HDAC3 for optimal expression, whereas others, such as NKX3.1, only require HDAC1. TMPRSS2, although suppressed by HDAC inhibitors, is unaffected by any single HDAC knockdown, raising the possibility of redundancy or transcriptional control by another less abundantly expressed HDAC that we have not directly tested.
Figure 4.
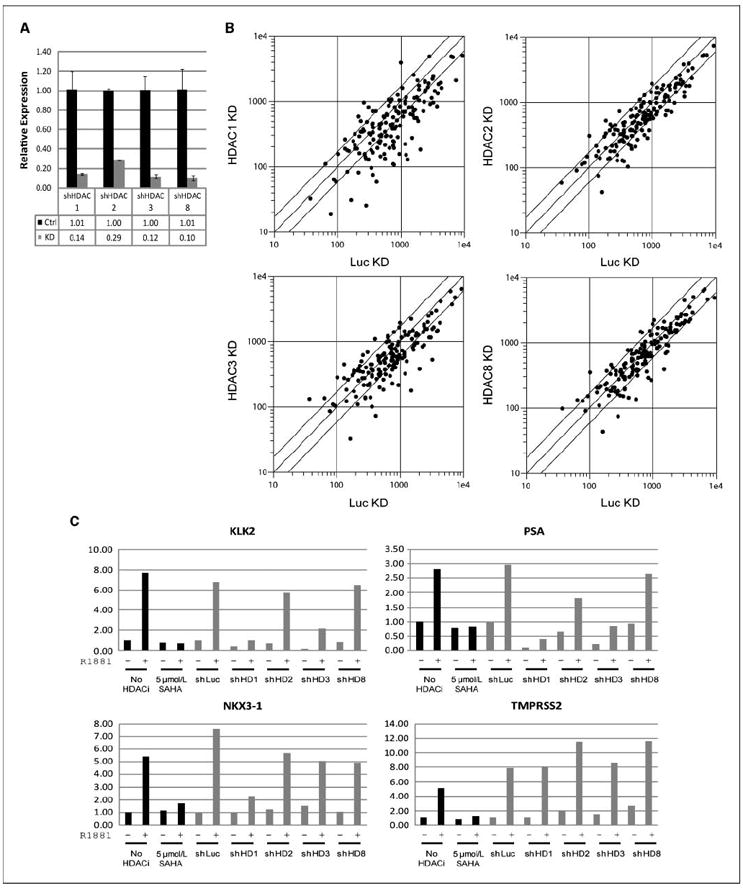
Effect of HDAC knockdown on transcription of ARGs. LNCaP cells were infected with lentiviral shRNA against HDAC1, HDAC2, HDAC3, HDAC8, or control (luciferase). After 3 d, cells were starved. After 6 d, cells were treated with 1 nmol/L R1881 or vehicle in the presence of cyclohexamide for 18 h, and expression profiling was performed. A, quantitative RT-PCR of HDAC1, HDAC2, HDAC3, and HDAC8 in LNCaP cells infected with shRNA against each of the HDACs. B, scatter plot of expression of ARGs (defined in Fig. 2A) in R1881-induced control shRNA infected (x axis) and HDAC shRNA infected (y axis) for HDAC1, HDAC2, HDAC3, and HDAC8. C, microarray expression levels of KLK2, PSA, NKX3.1, and TMPRSS2. The leftmost four columns (black) are from the 5 μmol/L SAHA, and no HDAC inhibitor conditions from the microarray experiment of HDAC inhibitors (see Fig. 3) as reference. The next 10 columns (gray) are from the knockdown microarray experiment. −, treated with vehicle; +, treated with 1 nmol/L R1881.
HDAC inhibitors block the assembly of an AR/RNA Pol II transcription complex
Our data that HDAC inhibitors block PSA production in LNCaP-AR cells despite preserved AR protein level (Fig. 2D) and that ARGs are down-regulated in the presence of cyclohexamide indicate that HDAC inhibitors directly block transcription of ARGs. To investigate the mechanism, we first examined recruitment of AR and RNA Pol II to the AR binding site in the PSA enhancer using ChIP (31). AR was recruited to the PSA enhancer 4 hours after R1881 treatment, and this was not significantly altered by TSA. In contrast, Pol II was recruited to the PSA enhancer by R1881, and this recruitment was inhibited by TSA treatment (Fig. 5A). This further validates that HDAC inhibitors directly block transcription of PSA. In addition, this suggests that HDAC inhibitor–induced alterations of chromatin structure impair the ability of AR to recruit the transcriptional complex. Because assembly of p160 (SRC1, GRIP1, and AIBP) and p300 (p300, CREB) class of coactivators downstream of AR binding help recruit Pol II (3), we asked if these factors are properly recruited in the presence of TSA. At 4 hours after R1881 addition, recruitment of SRC1 and p300 was inhibited by TSA. This indicates that HDAC inhibitors block coactivator recruitment by AR (Fig. 5B).
Figure 5.
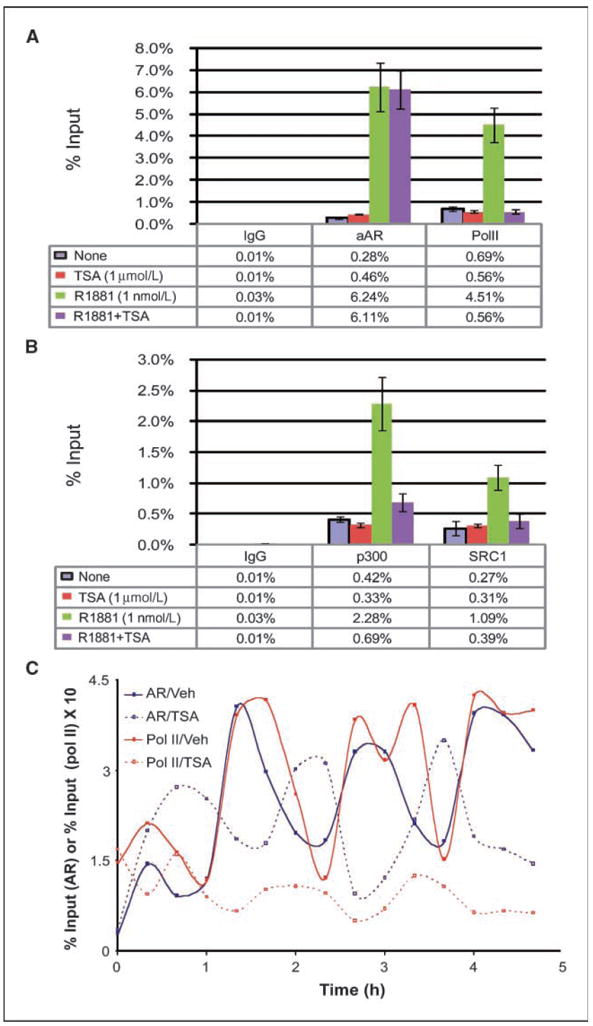
HDAC inhibitors impair recruitment of Pol II by AR and alter the phase of transcriptional cycling. A, LNCaP cells were starved for 72 h and treated with vehicle, 1 nmol/L R1881, 1 μmol/L TSA, or the combination of the two. Cells were harvested after 4 h, and ChIP was performed using antibodies against either AR or Pol II. B, ChIP was performed as above against SRC1 and p300. C, LNCaP cells were treated with R1881, in the presence or absence of TSA, over a 4 h 40 min time course. At 20-min intervals, ChIP was performed.
The assembly of hormone receptor transcription factor complexes on chromatin is dynamic, with evidence that both estrogen receptor and AR cycle on and off their binding sites in a cyclical manner (32, 33). Therefore, we examined the effect of TSA on the time course of AR and Pol II recruitment to the PSA enhancer. AR was recruited to the PSA enhancer in a cyclic fashion (with a periodicity of 80–90 minutes) with the lagging end of each wave recruiting Pol II (Fig. 5C). Consistent with estrogen receptor activity on the pS2 promoter, the first recruitment wave was diminutive compared with subsequent ones (32, 34). In the presence of TSA, AR binding was observed throughout the time course but the phase of recruitment was dramatically offset. The first cycle of AR binding was abruptly terminated after 40 minutes, and a new cycle was observed, which was 180° out-of-phase relative to non–TSA-treated cells. Throughout the time course, this new AR cycle was unable to efficiently recruit Pol II, consistent with the decreased transcription of PSA in the presence of HDAC inhibitors.
Discussion
HDACs are traditionally associated with transcriptional repression, particularly suppressing gene expression by a number of nuclear receptors (35). Hormone receptor antagonists, such as bicalutamide for AR and tamoxifen for the estrogen receptor, promote the assembly of HDAC complexes at hormone receptor binding sites to block ligand-induced gene expression. Here, we show, using genetic and pharmacologic approaches, that HDACs are also paradoxically required for activation of a substantial fraction of AR target genes including the transcriptional driver of ETS fusion mRNAs (TMPRSS2) implicated in ~70% of human prostate cancers. Our data illustrate the important clinical implications for the development of HDAC inhibitors as potential prostate cancer drugs.
Using shRNA, we showed that knockdown of individual HDACs recapitulates what is seen with treatment of HDAC inhibitors on transactivation of nearly all the androgen receptor target genes and AR mRNA levels. This was specifically observed with loss of HDAC1 and, to a lesser extent, loss of HDAC3. Indeed, knockdown of these two HDACs can account for the majority of the antiandrogen effect. However, neither loss of HDAC1 nor loss of HDAC3 phenocopies the effect of HDAC inhibitors on androgen-induced TMPRSS2 expression. This suggests that androgen regulation of TMPRSS2 is not dependent on a single class I HDAC but rather a combination of them. This question can be addressed by performing combinatorial knockdown studies of the individual histone deacetylases and ChIP on individual promoters.
Further investigation is required to discern precisely how HDAC inhibition interferes with AR complex assembly on chromatin. The cyclical nature of AR (and ER) binding to target genes is well established, including the nonproductive first wave of receptor binding that terminates in the presence of HDACs at the promoter/enhancer. Correspondingly, each wave of transcription is associated with a round of acetylation and deacetylation of histone H3 and H4. HDAC inhibitors could interfere with a proposed, albeit controversial, action of HDACs in resetting the promoter for assembly of a competent transcriptional complex (32). Our data, showing that TSA treatment shifts the phase of cyclical AR binding to the PSA enhancer, are consistent with this model. ChIP experiments indicate that HDAC inhibitor treatment rapidly and substantially enhances histone H3 acetylation at the PSA enhancer,5 indicating the constitutive presence of strong HAT activity at the loci. Therefore, one possibility is that hyperacetylated chromatin is incompatible with recruitment of coactivators and RNA Pol II.
Prior work has shown that HDAC inhibitors potentiate AR transcription, consistent with the transcriptional repression model. We reconcile our findings with these earlier reports in two ways. First, hyperactivation of AR function by HDAC inhibitors was observed using AR-dependent reporter constructs, which are unlikely to reflect the chromatinized state of endogenous genes. Second, the stimulatory effect of HDAC inhibitors on endogenous genes, such as PSA, was seen at doses (100 nmol/L TSA) below those required for achieving an increase in global histone acetylation. At this dose, we did not see a significant affect on PSA (Fig. 1A) or ARGs in expression profile (Fig. 3D). Higher doses are required to suppress AR function and may be indicative of a biphasic response to HDAC inhibition in certain contexts that could have clinical relevance if traditional end points, such as serum PSA levels, are used to monitor response to treatment. The HDAC-dependent and HDAC-repressed AR target genes identified in our gene array studies could serve as biomarkers for selecting those that ensure sufficient levels of HDAC inhibition in future clinical trials of these agents.
The most immediate clinical implication of our data is that HDAC inhibitors may have activity in prostate cancer. Conventional antiandrogens, such as bicalutamide, are highly effective as initial therapy but inevitably fail due to the emergence of drug-resistant disease. Here, we show that HDAC inhibitors remain potent inhibitors of AR function even in the setting of bicalutamide resistance and have antitumor activity in hormone refractory xenograft models.
Collectively, these data justify a careful examination of the therapeutic potential of HDAC inhibitors in prostate cancer. However, the magnitude of HDAC inhibition required to inhibit AR activity is critical because the effects we observed are clearly dose-dependent. Pharmacokinetic studies of the HDAC inhibitor SAHA suggest that the levels required for maximal AR inhibition are unlikely to be achieved using the oral regimen currently approved for lymphoma therapy (36). Indeed, phase I studies of SAHA, LBH589, and depsipeptide in CRPC, conducted by us and others, have been disappointing, with zero of eight PSA responders to LBH589 (37) and 2 of 31 PSA responders to depsipeptide (38). However, doses capable of AR inhibition should be possible with i.v. delivery or possibly through high dose of intermittent oral therapy, and the HDAC-regulated AR target genes defined here could be useful biomarkers to guide dose selection. This has led us to initiate a phase I study of intermittent i.v. LBH589.
Supplementary Material
Acknowledgments
Grant support: U.S. National Cancer Institute, University of California at Los Angeles (UCLA) Prostate Specialized Programs of Research Excellence seed grant, UCLA Medical Scientist Training Program (D.S. Welsbie), Ruth L. Kirschstein National Research Service Awards T32CA09056 (J. Xu) and 5T32CA009207 (Y. Chen), AACR–Bristol-Myers Squibb Oncology Fellowship (Y. Chen), and ASCO Young Investigator Award (Y. Chen).
C.L. Sawyers is a Doris Duke distinguished clinical scientist and an investigator of the Howard Hughes Medical Institute.
We thank Sawyers laboratory members Jennifer King and Phillip Watson for helpful discussions.
Footnotes
Y. Chen, unpublished data.
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest H.I. Scher: Commercial research grant and honoraria, Novartis. N. Rosen: Honoraria, Novartis. C.L. Sawyers: Honoraria, Merck and Novartis. The other authors disclosed no potential conflicts of interest.
References
- 1.Chen Y, Sawyers CL, Scher HI. Targeting the androgen receptor pathway in prostate cancer. Curr Opin Pharmacol. 2008;8:440–80. doi: 10.1016/j.coph.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen CD, Welsbie DS, Tran C, et al. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10:33–9. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- 3.Shang Y, Myers M, Brown M. Formation of the androgen receptor transcription complex. Mol Cell. 2002;9:601–10. doi: 10.1016/s1097-2765(02)00471-9. [DOI] [PubMed] [Google Scholar]
- 4.Rosenfeld MG, Lunyak VV, Glass CK. Sensors and signals: a coactivator/corepressor/epigenetic code for integrating signal-dependent programs of transcriptional response. Genes Dev. 2006;20:1405–28. doi: 10.1101/gad.1424806. [DOI] [PubMed] [Google Scholar]
- 5.Heinzel T, Lavinsky RM, Mullen TM, et al. A complex containing N-CoR, mSin3 and histone deacetylase mediates transcriptional repression. Nature. 1997;387:43–8. doi: 10.1038/387043a0. [DOI] [PubMed] [Google Scholar]
- 6.Nagy L, Kao HY, Chakravarti D, et al. Nuclear receptor repression mediated by a complex containing SMRT, mSin3A, and histone deacetylase. Cell. 1997;89:373–80. doi: 10.1016/s0092-8674(00)80218-4. [DOI] [PubMed] [Google Scholar]
- 7.Jepsen K, Hermanson O, Onami TM, et al. Combinatorial roles of the nuclear receptor corepressor in transcription and development. Cell. 2000;102:753–63. doi: 10.1016/s0092-8674(00)00064-7. [DOI] [PubMed] [Google Scholar]
- 8.List HJ, Smith CL, Rodriguez O, Danielsen M, Riegel AT. Inhibition of histone deacetylation augments dihydrotestosterone induction of androgen receptor levels: an explanation for trichostatin A effects on androgen-induced chromatin remodeling and transcription of the mouse mammary tumor virus promoter. Exp Cell Res. 1999;252:471–8. doi: 10.1006/excr.1999.4638. [DOI] [PubMed] [Google Scholar]
- 9.Weichert W, Roske A, Gekeler V, et al. Histone deacetylases 1, 2 and 3 are highly expressed in prostate cancer and HDAC2 expression is associated with shorter PSA relapse time after radical prostatectomy. Br J Cancer. 2008;98:604–10. doi: 10.1038/sj.bjc.6604199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Margueron R, Licznar A, Lazennec G, Vignon F, Cavailles V. Oestrogen receptor α increases p21(WAF1/CIP1) gene expression and the antiproliferative activity of histone deacetylase inhibitors in human breast cancer cells. J Endocrinol. 2003;179:41–53. doi: 10.1677/joe.0.1790041. [DOI] [PubMed] [Google Scholar]
- 11.Butler LM, Agus DB, Scher HI, et al. Suberoylanilide hydroxamic acid, an inhibitor of histone deacetylase, suppresses the growth of prostate cancer cells in vitro and in vivo. Cancer Res. 2000;60:5165–70. [PubMed] [Google Scholar]
- 12.Rokhlin OW, Glover RB, Guseva NV, Taghiyev AF, Kohlgraf KG, Cohen MB. Mechanisms of cell death induced by histone deacetylase inhibitors in androgen receptor-positive prostate cancer cells. Mol Cancer Res. 2006;4:113–23. doi: 10.1158/1541-7786.MCR-05-0085. [DOI] [PubMed] [Google Scholar]
- 13.Bali P, Pranpat M, Bradner J, et al. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: a novel basis for antileukemia activity of histone deacetylase inhibitors. J Biol Chem. 2005;280:26729–34. doi: 10.1074/jbc.C500186200. [DOI] [PubMed] [Google Scholar]
- 14.Chen L, Meng S, Wang H, et al. Chemical ablation of androgen receptor in prostate cancer cells by the histone deacetylase inhibitor LAQ824. Mol Cancer Ther. 2005;4:1311–9. doi: 10.1158/1535-7163.MCT-04-0287. [DOI] [PubMed] [Google Scholar]
- 15.Tomlins SA, Mehra R, Rhodes DR, et al. TMPRSS2:ETV4 gene fusions define a third molecular subtype of prostate cancer. Cancer Res. 2006;66:3396–400. doi: 10.1158/0008-5472.CAN-06-0168. [DOI] [PubMed] [Google Scholar]
- 16.Tomlins SA, Rhodes DR, Perner S, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–8. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 17.Rosen S, Skaletsky HJ. Primer3 on the WWW for general users and for biologist programmers. In: Krawetz S, Misener S, editors. Bioinformatics Methods and Protocols: Methods in Molecular Biology. Totowa (NJ): Humana Press; 2000. pp. 365–86. [DOI] [PubMed] [Google Scholar]
- 18.Jia L, Shen HC, Wantroba M, et al. Locus-wide chromatin remodeling and enhanced androgen receptor-mediated transcription in recurrent prostate tumor cells. Mol Cell Biol. 2006;26:7331–41. doi: 10.1128/MCB.00581-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Q, Carroll JS, Brown M. Spatial and temporal recruitment of androgen receptor and its coactivators involves chromosomal looping and polymerase tracking. Mol Cell. 2005;19:631–42. doi: 10.1016/j.molcel.2005.07.018. [DOI] [PubMed] [Google Scholar]
- 20.Fu M, Rao M, Wang C, et al. Acetylation of androgen receptor enhances coactivator binding and promotes prostate cancer cell growth. Mol Cell Biol. 2003;23:8563–75. doi: 10.1128/MCB.23.23.8563-8575.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Korkmaz CG, Fronsdal K, Zhang Y, Lorenzo PI, Saatcioglu F. Potentiation of androgen receptor transcriptional activity by inhibition of histone deacetylation-rescue of transcriptionally compromised mutants. J Endocrinol. 2004;182:377–89. doi: 10.1677/joe.0.1820377. [DOI] [PubMed] [Google Scholar]
- 22.Sramkoski RM, Pretlow TG, II, Giaconia JM, et al. A new human prostate carcinoma cell line, 22Rv1. In vitro Cell Dev Biol Anim. 1999;35:403–9. doi: 10.1007/s11626-999-0115-4. [DOI] [PubMed] [Google Scholar]
- 23.Solit DB, Zheng FF, Drobnjak M, et al. 17-Allylamino-17-demethoxygeldanamycin induces the degradation of androgen receptor and HER-2/neu and inhibits the growth of prostate cancer xenografts. Clin Cancer Res. 2002;8:986–93. [PubMed] [Google Scholar]
- 24.Marrocco DL, Tilley WD, Bianco-Miotto T, et al. Suberoylanilide hydroxamic acid (vorinostat) represses androgen receptor expression and acts synergistically with an androgen receptor antagonist to inhibit prostate cancer cell proliferation. Mol Cancer Ther. 2007;6:51–60. doi: 10.1158/1535-7163.MCT-06-0144. [DOI] [PubMed] [Google Scholar]
- 25.Febbo PG, Lowenberg M, Thorner AR, Brown M, Loda M, Golub TR. Androgen mediated regulation and functional implications of fkbp51 expression in prostate cancer. J Urol. 2005;173:1772–7. doi: 10.1097/01.ju.0000155845.44729.ba. [DOI] [PubMed] [Google Scholar]
- 26.Hendriksen PJ, Dits NF, Kokame K, et al. Evolution of the androgen receptor pathway during progression of prostate cancer. Cancer Res. 2006;66:5012–20. doi: 10.1158/0008-5472.CAN-05-3082. [DOI] [PubMed] [Google Scholar]
- 27.Glaser KB, Staver MJ, Waring JF, Stender J, Ulrich RG, Davidsen SK. Gene expression profiling of multiple histone deacetylase (HDAC) inhibitors: defining a common gene set produced by HDAC inhibition in T24 and MDA carcinoma cell lines. Mol Cancer Ther. 2003;2:151–63. [PubMed] [Google Scholar]
- 28.Mariadason JM, Rickard KL, Barkla DH, Augenlicht LH, Gibson PR. Divergent phenotypic patterns and commitment to apoptosis of Caco-2 cells during spontaneous and butyrate-induced differentiation. J Cell Physiol. 2000;183:347–54. doi: 10.1002/(SICI)1097-4652(200006)183:3<347::AID-JCP7>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 29.Peart MJ, Smyth GK, van Laar RK, et al. Identification and functional significance of genes regulated by structurally different histone deacetylase inhibitors. Proc Natl Acad Sci U S A. 2005;102:3697–702. doi: 10.1073/pnas.0500369102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tomlins SA, Laxman B, Dhanasekaran SM, et al. Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature. 2007;448:595–9. doi: 10.1038/nature06024. [DOI] [PubMed] [Google Scholar]
- 31.Huang W, Shostak Y, Tarr P, Sawyers C, Carey M. Cooperative assembly of androgen receptor into a nucleoprotein complex that regulates the prostate-specific antigen enhancer. J Biol Chem. 1999;274:25756–68. doi: 10.1074/jbc.274.36.25756. [DOI] [PubMed] [Google Scholar]
- 32.Metivier R, Penot G, Hubner MR, et al. Estrogen receptor-α directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell. 2003;115:751–63. doi: 10.1016/s0092-8674(03)00934-6. [DOI] [PubMed] [Google Scholar]
- 33.Kang Z, Pirskanen A, Janne OA, Palvimo JJ. Involvement of proteasome in the dynamic assembly of the androgen receptor transcription complex. J Biol Chem. 2002;277:48366–71. doi: 10.1074/jbc.M209074200. [DOI] [PubMed] [Google Scholar]
- 34.Shang Y, Hu X, DiRenzo J, Lazar MA, Brown M. Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell. 2000;103:843–52. doi: 10.1016/s0092-8674(00)00188-4. [DOI] [PubMed] [Google Scholar]
- 35.Karagianni P, Wong J. HDAC3: taking the SMRT-N-CoRrect road to repression. Oncogene. 2007;26:5439–49. doi: 10.1038/sj.onc.1210612. [DOI] [PubMed] [Google Scholar]
- 36.Kelly WK, Richon VM, O’Connor O, et al. Phase I clinical trial of histone deacetylase inhibitor: suberoylanilide hydroxamic acid administered intravenously. Clin Cancer Res. 2003;9:3578–88. [PubMed] [Google Scholar]
- 37.Rathkopf DE, Wong BY, Ross RW, et al. A phase I study of oral panobinostat (LBH589) alone and in combination with docetaxel (Doc) and prednisone in castration-resistant prostate cancer (CRPC) J Clin Oncol. 2008;26 doi: 10.1007/s00280-010-1289-x. 2008 ASCO Annual Meeting Proceedings (abstract 5152) [DOI] [PubMed] [Google Scholar]
- 38.Parker C, Molife R, Karavasilis V, et al. Romidepsin (FK228), a histone deacetylase inhibitor: Final results of a phase II study in metastatic hormone refractory prostate cancer (HRPC) J Clin Oncol. 2007;25 2007 ASCO Annual Meeting Proceedings Part I (abstract 15507) [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.