

Abstract
Increase of portal venous vascular resistance is counteracted by decrease of hepatic arterial vascular resistance (hepatic arterial buffer response). This process is mediated by adenosine in normal livers. In cirrhosis, hepatic arterial vascular resistance is decreased but the involvement of adenosine in this process is unknown. The aim of our study was to identify the signalling pathway responsible for the decreased hepatic arterial resistance in cirrhotic livers.
Methods
Cirrhosis was induced by CCl4. Using a bivascular liver perfusion dose–response curves to adenosine of the HA were performed in the presence and the absence of pan-adenosine blocker (8-SPT), A1 blocker (caffeine) or nitric oxide synthase-blocker (L-NMMA) after preconstriction with an α1-agonist (methoxamine). Western blot of the HA were used to measure the density of the A1 and A2a receptors.
Results
Adenosine caused a dose dependent relaxation of the hepatic artery of both cirrhotic and control animals that were blocked in both groups by 8-SPT (P<0.02). The response to adenosine was greater in cirrhotic rats (P = 0.016). Both L-NMMA (P = 0.003) and caffeine reduced the response to adenosine in cirrhotic but not in control animals. Western blot analysis showed a higher density of A1 and a lower density of A2a receptor in cirrhotic animals (P < 0.05).
Conclusion
The adenosine-induced vasodilatation of the HA is increased in cirrhotic rats suggesting a role for adenosine-NO in the decreased hepatic arterial vascular resistance found in cirrhosis. This significantly greater response in cirrhosis by the A1 receptor follows the same pathway that is seen in hypoxic conditions in extra-hepatic tissues.
Keywords: adenosine, cirrhosis, hepatic artery, liver perfusion
The liver is unique in having an arterial and venous blood supply. Liver perfusion is a function of combination of these, and the two blood supplies are inter-regulated (1, 2). In the healthy liver an experimental reduction in portal blood flow results in a reduction in the arterial vascular resistance with increase in arterial flow (hepatic arterial buffer response) and vice versa. The signalling pathway for this response is local, with the reduction of liver perfusion resulting in an increase in concentration of the vasodilatator adenosine (2–4). In general, vasodilatatory effects of adenosine are mediated by four different adenosine receptors but mainly by A1 and A2 receptors (5). The responsible receptor for adenosine-mediated vasodilatation in the hepatic artery of normal livers is the adenosine A2 receptor (6).
The situation in the cirrhotic liver is partially analogous to the experimental reduction of portal blood flow in that there is a reduction in portal flow, and a corresponding decrease in hepatic vascular resistance and increase in arterial blood flow (7, 8). The increased hepatic arterial blood flow in cirrhosis can be inhibited by a pan-adenosine receptor antagonist suggesting that the mechanism present in the healthy liver is operative in the cirrhotic liver (9, 10). This is further suggested by adenosine-mediated vasodilatation of the hepatic artery in cirrhotic patients (11).
An understanding of the pathways regulating vascular resistance and flow in the cirrhotic liver are of great interest. The assumption that the adenosine A2 receptor mechanism identified as responsible for the hepatic arterial buffer response in the healthy liver is also responsible for the increased hepatic arterial blood flow in the cirrhotic liver has not been directly tested. Furthermore, in other vascular beds different pathways come into play in disease states (5, 12, 13). The aim of our study was to identify the signalling pathway responsible for the increased hepatic arterial flow in the cirrhotic liver.
Methods
Fifty-nine male Sprague–Dawley rats (Harlan Laboratories, Indianapolis, IN, USA) were included in this study. The American Physiological Society guide principles for the care and use of animals were followed. The appropriate Institutional Animal Care and Use committee previously approved all procedures involving animals.
Induction of CCl4-cirrhosis
Rats underwent inhalation exposure of carbon tetrachloride (CCl4) three times a week. Phenobarbital (0.35 g/L) was added to the drinking water as described previously (8). Treatment was given for at approximately 12 weeks. Perfusions were performed 6–10 days after the last doses of CCl4 and phenobarbital. Age-matched rats were used as control group.
In situ rat liver perfusion
Rats were anaesthetized with ketamine hydrochloride (Ketaset, Fort Dodge Animal Health, Fort Dodge, IA, USA; 100mg/kg body wt) and xylazine (Rompun, Bayer, Shawne Mission, KS, USA; 40mg/animal). A bivascular liver perfusion was performed as described before (7, 14). Briefly, after the abdomen was opened loose ligatures were placed around the aorta cranial to the celiac artery, around the superior mesenteric artery immediately after branching from the aorta, and the aorta caudal to the mesenteric artery. Left gastric and splenic arteries were tied at its origin of the celiac artery and a loose ligature placed around the oesophagus. Left and right renal arteries as well as gastroduodenal artery (branch of the common hepatic artery) were ligated. The bile duct was cannulated with a polyethylene tube (PE 10). The portal vein was cannulated with a 14G teflon catheter and the perfusion with 32 ml/min of oxygenated (carbon gas, 95% O2, 5% CO2) Krebs–Henseleit solution containing dextrose (11mM) in a nonrecirculating mode was started. The inferior vena cava was cut immediately. The aorta was cannulated with an 18G teflon catheter and the ligatures around the superior mesenteric artery and the oesophagus were closed. The perfusion of the hepatic artery with 8 ml/min of oxygenated (carbon gas, 95% O2, 5% CO2) Krebs–Henseleit solution containing dextrose (11mM) in a non-recirculating mode was started. The tip of the catheter was placed close to the branch of the celiac artery and all ligatures around the aorta were closed. A 14G catheter was introduced in the inferior vena cava and the thorax was opened.
In order to measure the sinusoidal pressure, a PE-60 catheter was guided from the right atrium, through the thoracic segment of the inferior vena cava into the left hepatic lobe and wedged in the hepatic vein (7). The ligature around the inferior vena cava was closed to secure the wedged catheter. The preparation was transferred to a temperature-controlled (37 °C) Plexiglas perfusion chamber (Yale University Medical Instrument, New Haven, CT, USA) initiating the stabilization period.
During the stabilization and the experimental period the perfusion pressure of the portal vein and the hepatic artery were measured constantly using two independent strain-gauge transducers (P23XL, Spectramed, Oxnard, CA, USA) respectively. The wedged pressure was measured during the experimental period using a third independent strain-gauge transducers (P23XL, Spectramed). Before each experiment, all pressure measurement systems were calibrated with the zero point at the level of the hepatic hilum. Perfusion and sinusoidal pressure were continuously recorded by CHART 3.6 program using MacLab/4e hardware (AD instruments Inc., Colorado Springs, CO, USA). During the stabilization and experimental period the perfusate was oxygenated using a Silastic tubing lung interposed between the perfusate reservoir and the peristaltic pump (15).
Experimental design
All livers were perfused with constant flows during the stabilization period and the flow through the wedged catheter was maintained. The stabilization period was performed in a recirculating mode in absence or presence of the NO-production inhibitor L-NMMA (4×10−4 M; Sigma Chemicals Co., St Louis, MO, USA) or panadenosine receptor inhibitor 8-sulphophenyltheophylline (8-SPT; 10−5 M; Sigma Chemicals Co.). After the stabilization period the wedged catheter outflow was interrupted, allowing the measurement of the wedged pressure and the perfusion was changed to an open mode in presence and absence of L-NMMA or 8-SPT respectively. In an additional set of rats (n = 8) the same experimental setting was used but instead of L-NMMA or 8-SPT the adenosine A1 receptor blocker caffeine (10−4 M; Sigma Chemicals Co.) was administered during the entire perfusion.
This open mode was kept until the end of the experiment allowing the selective measurement of the drugs effect in the hepatic artery, the portal vein, and the sinusoidal area. The liver perfusion system is known as a vasodilatated system. To investigate the effects of vasodilatators a preconstriction is needed. Therefore, after the stabilization period a preconstriction with the α1-agonist methoxamine (10−4 M; Sigma Chemicals Co.) was performed followed by a dose–response curve using three consecutive doses of adenosine (10−6–10−4 M; Sigma Chemicals Co.) infused in the hepatic artery.
Liver global viability was assessed by gross appearance of the liver, stable perfusion curves and bile production during the stabilization period (> 0.4 μl/min/g liver). After the experiment liver and spleen were removed and weighed. Liver tissue sample were collected and fixed in formalin.
Western blot of the hepatic artery
Additional animals (n = 8) were used for collection of the extrahepatic part of the hepatic artery. Rats were anaesthetized with ketamine hydrochloride (Ketaset, Fort Dodge Animal Health; 100 mg/kg body wt) and xylazine (Rompun, Bayer; 40 mg/animal). The abdomen was opened and the extrahepatic part of the hepatic artery as well as the upper part of celiac artery was carefully released from the surrounding tissue. The vessel was dissected and washed in Krebs solution and immediately frozen using liquid nitric oxygen, and stored at −80 °C. Samples were homogenized in an appropriate lysis buffer containing 50mM Tris–HCl, 1mmol 4-(2-aminoethyl)-benzenesulphonyl fluoride, protease inhibitor cocktail tablet (Roche Diagnostics GmbH, Mannheim, Germany), and 1% (v/v) Nonidet PK40, pH 7.5. Protein content in the supernatants was quantified using the Lowry method with bovine serum albumin as standard. The supernatants were subjected to the SDS-PAGE gel electrophoresis of protein (20 mg), and Western blotting was performed using antibodies that recognized adenosine A1 (Sigma Chemicals Co.), Adenosine A2a (Sigma Chemicals Co.), and eNOS (Transduction Laboratories, Lexington, KY, USA). Enhanced chemiluminescence was used for protein detection. Intensity of the bands corresponding to the protein of interest was measured using densitometry.
Calculations of vascular resistances
Hepatic arterial vascular resistance (HAR) was calculated from the hepatic arterial flow and the hepatic arterial perfusion pressure. Portal venous vascular resistance (PVR) was calculated from the portal venous perfusion pressure and portal venous flow. Sinusoidal vascular resistance (SiVR) was calculated from the wedge pressure and the total flow, i.e. portal venous and hepatic arterial flow.
Statistics
Data are presented as means ± SEM. Mann–Whitney test was used for comparisons of two different groups and one-way analysis of variance for comparison of more than two groups followed by a preplanned contrast test to compare cirrhotic and control groups. Comparison for repeated measurements was assessed using multivariate analysis of repeated measurements followed by Bonferroni’s test to detect differences between groups. P-values ≤ 0.05 were considered significant.
Results
Liver perfusion
Liver weight was equal (11.7 ± 0.6 vs. 11.6 ± 0.2 g) and spleen weight higher (2.1 ± 0.09 vs. 1.2 ± 0.02 g; P<0.001) in cirrhotic compared with control animals. Basal PVR (0.28 ± 0.008 vs. 0.24 ± 0.005 mmHg/ml/min; P<0.001) and SiVR (0.13 ± 0.009 vs. 0.08 ± 0.005 mmHg/ml/min; P<0.001) were higher and, in contrast, basal HAR (6.46 ± 0.25 vs. 8.62 ± 0.37 mmHg/ml/min) lower in cirrhotic compared with control animals. PVR, SiVR and HAR did not change during incubation neither with 8-SPT nor with L-NMMA.
Effect of adenosine on hepatic arterial resistance
Incubation with methoxamine in absence of 8-SPT and L-NMMA lead to a lower increase of HAR in cirrhotic animals compared with control animals (24.52 ± 1.70 vs. 32.87 ± 0.87 mmHg/ml/min; P = 0.004). Because the response to methoxamine was different in both groups the response to adenosine is shown as percentage of the methoxamine-induced increase.
Adenosine caused a dose-dependent decrease in HAR in cirrhotic and control livers and, moreover, this decrease was significantly higher in cirrhosis (P = 0.005; Fig. 1). Presence of 8-SPT inhibited significantly the response to adenosine in both cirrhosis (P<0.001) as well as controls (P = 0.014). Interestingly, presence of L-NMMA inhibited the response to adenosine in cirrhotic (P = 0.003) but not in control livers (Fig. 1). Furthermore, in presence of L-NMMA the response to adenosine was not different between cirrhotic and control livers (Fig. 1).
Fig. 1.

Dose–response curves to adenosine of the hepatic artery in cirrhotic (diamond; n = 13) and normal (circle; n = 14) rats in absence (solid line) and presence (dashed lines) of the nitric oxide production inhibitor L-NMMA. The vasodilatatatory effect of adenosine in cirrhotic livers in the absence of L-NMMA was significantly higher compared with normal livers and to the presence of L-NMMA (P < 0.005). It is concluded that the greater response in cirrhotic livers is because of the nitric oxide dependent adenosine A1 receptor.
The adenosine A1 receptor blocker caffeine decreased the response to adenosine in hepatic arteries of cirrhotic livers but not in hepatic arteries of normal livers. Therefore, the previously detected different response to adenosine in cirrhotic and normal animals was not observed in the presence of caffeine as there were no significant different in the dose–response curves between cirrhotic and normal animals (Fig. 2).
Fig. 2.
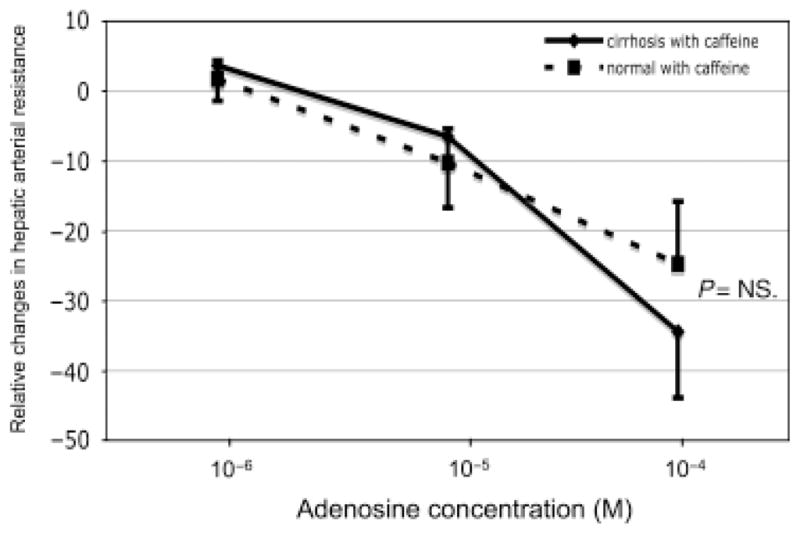
Dose–response curve to adenosine of the hepatic artery in cirrhotic (solid line; n = 4) and normal (dashed; n = 4) line in presence of caffeine (adenosine A1 receptor blocker). The effect of caffeine was only present in cirrhosis and the curves were not significant different.
Effects of adenosine of the portal venous and sinusoidal resistance
The vascular resistance of the portal vein is determined in the sinusoids and the confluence of the hepatic artery is located in zone 1 of the sinusoids (16). Therefore, infusion of vasoactive drugs into the hepatic artery could also change the sinusoidal and portal venous resistance.
Presence of 8-SPT did not influence the methoxamine-induced increase in PVR in cirrhotic nor in control livers compared with the methoxamine-induced increased in absence of 8-SPT. In contrast, presence of L-NMMA caused a greater increase in PVR because of methoxamine in cirrhotic (P = 0.007) as well as control livers (P = 0.003). Interestingly, adenosine infusion via the hepatic artery caused an increase in PVR in control livers but not in cirrhotic livers (P = 0.001; Fig. 3). This effect was blocked with 8-SPT in controls (P = 0.001). However, L-NMMA did not inhibit the response to adenosine (via hepatic artery) in controls and neither did the presence of 8-SPT nor L-NMMA modify the effect to adenosine in cirrhosis (Fig. 3). The presence of caffeine did not change the response to adenosine in control nor in cirrhotic livers.
Fig. 3.

Dose–response curves to adenosine (administered into the hepatic artery) of the portal vein in cirrhotic (diamond) and normal (circle) rats in absence (line) and presence (dashed lines) of the nitric oxide production inhibitor L-NMMA. The effect of adenosine was only present in normal livers.
As seen in PVR, presence of 8-SPT did not change the response to methoxamine in SiVR of cirrhotic nor of control livers compared with absence of 8-SPT. In contrast, presence of L-NMMA increased the response to methoxamine in cirrhotic (P = 0.007) but not in control livers. Adenosine administration via the hepatic artery did not changed SiVR in cirrhosis and controls neither in presence nor in absence of 8-PST, caffeine or L-NMMA.
Western blot analyses of the adenosine receptors
Hepatic arteries from cirrhotic rats showed a significant higher eNOS expression compared with the control group (P<0.05; Fig. 4). In both, cirrhotic and control animals, adenosine A2a receptors and adenosine A1 receptors were present. However, cirrhotic animals showed a significant lower relative density of adenosine A2a receptors (P<0.01) and, in contrast, expressed significantly more adenosine A1 receptors compared with control animals (P<0.05, Fig. 4).
Fig. 4.

Results of the Western blots (n = 8). (A) eNOS expression of the hepatic artery of normal and cirrhotic rats. eNOS expression was significantly higher in hepatic arteries of cirrhotic animals compared with normal animals (P<0.05). (B) Adenosine A1 expression of the hepatic artery of normal and cirrhotic rats. The adenosine A1 expression was significantly higher in cirrhotic animals compared with normal animals (P<0.05). (C) Adenosine A2a expression of the hepatic artery of normal and cirrhotic rats. The adenosine A2a expression was significantly higher in normal animals compared with cirrhotic animals (P<0.01).
Discussion
The hepatic arterial resistance in cirrhosis is decreased because of increased nitric oxide production and vascular remodeling (8). The vasodilatation of the hepatic artery in cirrhosis leads to an absolute and relative increase of the proportion of the hepatic arterial perfusion on the total liver perfusion and knowledge about the mediators of the hepatic artery are necessary for possible future therapies. The results of the present study suggest that adenosine could be an additional mediator of the hepatic arterial vasodilatation in cirrhosis. Previous studies showed that adenosine is a potent vasodilatator of the hepatic artery in normal livers (2, 17). This adenosine-mediated vasodilatation in normal livers is mediated by the adenosine A2 receptor in a nitric oxide independent fashion (6, 18). Our results in normal livers with vasodilatation because of adenosine, inhibition of this effect by the pan-adenosine blocker 8-SPT, and the lack of any effect of both, the adenosine A1 receptor blocker caffeine and the nitric oxide blocker L-NMMA, support these results. Although we also found in normal hepatic arteries expression of the adenosine A1 receptor, a nitric oxide-dependent receptor, the lack of any effect of blocking nitric oxide synthesis and the lack of any effect of the adenosine A1 blocker caffeine suggests that the main effect in normal livers is mediated through the nitric oxide independent adenosine A2 receptor.
In this study adenosine shows a significantly greater vasodilatatory effect in hepatic arteries of cirrhotic compared with control livers. Inhibition of nitric oxide production corrected the response to adenosine in cirrhosis to a level of normal livers. That implicates that the additional effect of adenosine in cirrhosis is mediated by an additional receptor because the adenosine A2 receptor is nitric oxide independent (6, 18). Indeed, our results with lower effect of adenosine during nitric oxide inhibition, the effect of the adenosine A1 receptor blocker that was present only in cirrhotic hepatic arteries and the greater expression of the nitric oxide dependent adenosine A1 receptor in cirrhotic hepatic arteries suggest that this receptor is the responsible receptor for mediating the effect of adenosine in cirrhosis. Furthermore, we have previously shown in two different models of cirrhosis that nitric oxide production is upregulated (8). The adenosine A1 receptor is located on the endothelium and since we also found an upregulated eNOS expression in the hepatic arteries of cirrhotic animals this further suggests a greater role of that receptor in cirrhosis (12, 19). Taken together, our results implicate that in cirrhotic animals the hepatic arterial vasodilatation caused by adenosine is mainly mediated by the adenosine A1 receptor.
Adenosine is an important mediator involved in the regulation of the intrahepatic circulation that is the main mediator of the hepatic arterial buffer response in normal livers (2). The hepatic arterial buffer response is a phenomenon that counteracts changes in portal venous blood flow. Decrease in portal venous blood flow leads to increase in hepatic arterial flow (3). It has been proposed that the decreased portal venous flow caused accumulation of adenosine and therefore hepatic arterial vasodilatation (3, 20). The functional role of the hepatic arterial buffer response seems mainly to maintain part of the oxygen supply to the liver than to stabilize the blood supply (10, 17). Adenosine is produced in tissues under hypoxic conditions and the increase of high-oxygenated arterial blood would counteract therefore hypoxia.
In this regard, vasodilatatory response to adenosine under systemic hypoxia in skeletal muscle is mediated by the adenosine A1 receptor although both, the adenosine A1 as well as the adenosine A2 receptor are present (12, 21). Inhibition of nitric oxide production diminished the response to adenosine under hypoxic conditions demonstrating also an involvement of nitric oxide (12, 21). Therefore, our findings of a greater influence of the adenosine A1 receptor in cirrhosis and lowering of the effect with inhibition of nitric oxide production are comparable with findings under systemic hypoxic conditions in other tissues. Hypoxaemia is a well-known condition in cirrhosis and the presence of the hepatic arterial buffer response in cirrhosis has been shown previously (9, 21). In that context our results clarify the responsible receptor of the adenosine-mediated vasodilatation and implicate a role of adenosine in mediating vasodilatation in cirrhosis. Furthermore, we speculate that hypoxia could be the initial trigger leading to a greater influence of the adenosine A1 receptors in hepatic arteries of cirrhotic livers (22–24). This is further supported by previous results showing that adenosine is an excellent vasodilator of the hepatic artery in cirrhotic patients and that this leads to an improvement of the oxygen-dependent liver function (11, 25).
On the other side we found a vasoconstriction because of adenosine on the portal venous side in normal livers. This vasoconstriction was not blocked by L-NMMA nor by caffeine. However, we infused the adenosine to the hepatic artery and not to the portal vein. To draw conclusions from these findings one should perfuse the adenosine through the portal vein to achieve higher concentration.
In conclusion, we demonstrated a greater vasodilatatory effect of adenosine in hepatic arteries of cirrhotic livers. Both, the over-expression of the adenosine A1 receptor in hepatic arteries of cirrhotic animals as well as decreased response to adenosine because of inhibition of nitric oxide production in cirrhotic animals but not in control animals, lead to the conclusion that the response of adenosine in hepatic arteries of cirrhotic livers is mediated mainly by the adenosine A1 receptor.
Acknowledgments
A. Zipprich was supported by the Wilhelm-Roux- Stipendium of the Martin-Luther-University of Halle-Wittenberg, Germany. Cristina Ripoll was supported by the Fondo de Investigaciones Sanitarias (Instituto de Salud Carlos III) (CM 03/00037). R. Groszmann was supported by the VA Merit Review. W. Mehal was supported by %R01DK076674-4.
References
- 1.Lautt WW, Legare DJ, Ezzat WR. Quantitation of the hepatic arterial buffer response to graded changes in portal blood flow. Gastroenterology. 1990;98:1024–8. doi: 10.1016/0016-5085(90)90029-z. [DOI] [PubMed] [Google Scholar]
- 2.Lautt WW. Mechanism and role of intrinsic regulation of hepatic arterial blood flow: hepatic arterial buffer response. Am J Physiol. 1985;249:G549–56. doi: 10.1152/ajpgi.1985.249.5.G549. [DOI] [PubMed] [Google Scholar]
- 3.Lautt WW, Legare DJ, d’Almeida MS. Adenosine as putative regulator of hepatic arterial flow (the buffer response) Am J Physiol. 1985;248:H331–8. doi: 10.1152/ajpheart.1985.248.3.H331. [DOI] [PubMed] [Google Scholar]
- 4.Mathie RT, Alexander B. The role of adenosine in the hyperaemic response of the hepatic artery to portal vein occlusion (the ‘buffer response’) Br J Pharmacol. 1990;100:626–30. doi: 10.1111/j.1476-5381.1990.tb15857.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tabrizchi R, Bedi S. Pharmacology of adenosine receptors in the vasculature. Pharmacol Ther. 2001;91:133–47. doi: 10.1016/s0163-7258(01)00152-8. [DOI] [PubMed] [Google Scholar]
- 6.Mathie RT, Alexander B, Ralevic V, Burnstock G. Adenosine- induced dilatation of the rabbit hepatic arterial bed is mediated by A2-purinoceptors. Br J Pharmacol. 1991;103:1103–7. doi: 10.1111/j.1476-5381.1991.tb12307.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zipprich A, Loureiro-Silva MR, D’Silva I, Groszmann RJ. The role of hepatic arterial flow on portal venous and hepatic venous wedged pressure in the isolated perfused CCl4-cirrhotic liver. Am J Physiol Gastrointest Liver Physiol. 2008;295:G197–202. doi: 10.1152/ajpgi.00190.2007. [DOI] [PubMed] [Google Scholar]
- 8.Zipprich A, Loureiro-Silva MR, Jain D, D’Silva I, Groszmann RJ. Nitric oxide and vascular remodeling modulate hepatic arterial vascular resistance in the isolated perfused cirrhotic rat liver. J Hepatol. 2008;49:739–45. doi: 10.1016/j.jhep.2008.06.027. [DOI] [PubMed] [Google Scholar]
- 9.Richter S, Mucke I, Menger MD, Vollmar B. Impact of intrinsic blood flow regulation in cirrhosis: maintenance of hepatic arterial buffer response. Am J Physiol Gastrointest Liver Physiol. 2000;279:G454–62. doi: 10.1152/ajpgi.2000.279.2.G454. [DOI] [PubMed] [Google Scholar]
- 10.Mucke I, Richter S, Menger MD, Vollmar B. Significance of hepatic arterial responsiveness for adequate tissue oxygenation upon portal vein occlusion in cirrhotic livers. Int J Colorectal Dis. 2000;15:335–41. doi: 10.1007/s003840000247. [DOI] [PubMed] [Google Scholar]
- 11.Kleber G, Steudel N, Behrmann C, et al. Hepatic arterial flow volume and reserve in patients with cirrhosis: use of intra-arterial Doppler and adenosine infusion. Gastroenterology. 1999;116:906–14. doi: 10.1016/s0016-5085(99)70074-0. [DOI] [PubMed] [Google Scholar]
- 12.Bryan PT, Marshall JM. Cellular mechanisms by which adenosine induces vasodilatation in rat skeletal muscle: significance for systemic hypoxia. J Physiol. 1999;514:163–75. doi: 10.1111/j.1469-7793.1999.163af.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frobert O, Haink G, Simonsen U, et al. Adenosine concentration in the porcine coronary artery wall and A2A receptor involvement in hypoxia-induced vasodilatation. J Physiol. 2006;570:375–84. doi: 10.1113/jphysiol.2005.100115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gardemann A, Strulik H, Jungermann K. A portal-arterial glucose concentration gradient as a signal for an insulin-dependent net glucose uptake in perfused rat liver. FEBS Lett. 1986;202:255–9. doi: 10.1016/0014-5793(86)80697-4. [DOI] [PubMed] [Google Scholar]
- 15.Hamilton RL, Berry MN, Williams MC, Severinghaus EM. A simple and inexpensive membrane “lung” for small organ perfusion. J Lipid Res. 1974;15:182–6. [PubMed] [Google Scholar]
- 16.Takasaki S, Hano H. Three-dimensional observations of the human hepatic artery (arterial system in the liver) J Hepatol. 2001;34:455–66. doi: 10.1016/s0168-8278(00)00058-1. [DOI] [PubMed] [Google Scholar]
- 17.Richter S, Vollmar B, Mucke I, Post S, Menger MD. Hepatic arteriolo-portal venular shunting guarantees maintenance of nutritional microvascular supply in hepatic arterial buffer response of rat livers. J Physiol. 2001;531:193–201. doi: 10.1111/j.1469-7793.2001.0193j.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Macedo MP, Lautt WW. Potentiation to vasodilators by nitric oxide synthase blockade in superior mesenteric but not hepatic artery. Am J Physiol. 1997;272:G507–14. doi: 10.1152/ajpgi.1997.272.3.G507. [DOI] [PubMed] [Google Scholar]
- 19.Fahim M, Hussain T, Mustafa SJ. Role of endothelium in adenosine receptor-mediated vasorelaxation in hypertensive rats. Fundam Clin Pharmacol. 2001;15:325–34. doi: 10.1046/j.1472-8206.2001.00042.x. [DOI] [PubMed] [Google Scholar]
- 20.Morimoto Y, Wettstein M, Haussinger D. Hepatocyte heterogeneity in response to extracellular adenosine. Biochem J. 1993;293:573–81. doi: 10.1042/bj2930573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bryan PT, Marshall JM. Adenosine receptor subtypes and vasodilatation in rat skeletal muscle during systemic hypoxia: a role for A1 receptors. J Physiol. 1999;514:151–62. doi: 10.1111/j.1469-7793.1999.151af.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moreau R, Lee SS, Soupison T, Roche-Sicot J, Sicot C. Abnormal tissue oxygenation in patients with cirrhosis and liver failure. J Hepatol. 1988;7:98–105. doi: 10.1016/s0168-8278(88)80512-9. [DOI] [PubMed] [Google Scholar]
- 23.Colle I, Langlet P, Barriere E, et al. Evolution of hypoxemia in patients with severe cirrhosis. J Gastroenterol Hepatol. 2002;17:1106–9. doi: 10.1046/j.1440-1746.2002.02849.x. [DOI] [PubMed] [Google Scholar]
- 24.Moller S, Hillingso J, Christensen E, Henriksen JH. Arterial hypoxaemia in cirrhosis: fact or fiction? Gut. 1998;42:868–74. doi: 10.1136/gut.42.6.868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zipprich A, Steudel N, Behrmann C, et al. Functional significance of hepatic arterial flow reserve in patients with cirrhosis. Hepatology. 2003;37:385–92. doi: 10.1053/jhep.2003.50065. [DOI] [PubMed] [Google Scholar]