

Abstract
The majority of patients with lung cancer present with metastatic disease. Chronic inflammation and subsequent activation of NF-κB have been associated the development of cancers. The RelA/p65 subunit of NF-κB is typically associated with transcriptional activation. In this report we show that RelA/p65 can function as an active transcriptional repressor through enhanced methylation of the BRMS1 metastasis suppressor gene promoter via direct recruitment of DNMT-1 to chromatin in response to TNF. TNF-mediated phosphorylation of S276 on RelA/p65 is required for RelA/p65-DNMT-1 interactions, chromatin loading of DNMT-1, and subsequent BRMS1 promoter methylation and transcriptional repression. The ability of RelA/65 to function as an active transcriptional repressor is promoter specific as the NF-κB-regulated gene cIAP2 is transcriptionally activated while BRMS1 is repressed under identical conditions. Small molecule inhibition of either of the minimal interacting domains between RelA/p65-DNMT-1 and RelA/p65-BRMS1 promoter abrogates BRMS1 methylation and its transcriptional repression. The ability of RelA/p65 to directly recruit DNMT-1 to chromatin resulting in promoter-specific methylation and transcriptional repression of tumor metastasis suppressor gene BRMS1 highlights a new mechanism through which NF-κB can regulate metastatic disease, and offers a potential target for newer generation epigenetic oncopharmaceuticals.
Keywords: DNMT-1, Phosphorylation, RelA-p65, TNF, Transcription
Introduction
Nuclear factor-κB (NF-κB) is aberrantly activated in malignancies and its activation is essential for tumor proliferation and metastatic process (Baldwin, 1996; Bobrovnikova-Marjon et al., 2004; Bours et al., 1994; Denlinger et al., 2004; Helbig et al., 2003; Huber et al., 2004; Karin and Greten, 2005; Takada et al., 2005). Typically NF-κB is a transcriptional activator after stimulation with pro-inflammatory cytokines, such as tumor necrosis factor (TNF) and interleukin-1 (IL-1) (Chen et al., 1998; Mayo and Baldwin, 2000). In unstimulated cell nuclei RelA/p65 and/or p50 are transcriptionally repressed through interactions on chromatin with histone deacetylases (HDAC) -1 and -3 co-repressor complexes. Following stimulation, derepression of RelA/p65 occurs through phosphorylation on serines 276 (Dong et al., 2008; Zhong et al., 2002) or 536 (Hoberg et al., 2006) which enhances its association with histone acetyltransferases (HATs) allowing RelA/p65 to activate transcription (Chen et al., 1998; Chen et al., 2005).
In 2004, Campbell first described the ability of RelA/65 to function as an “active repressor” under specific cytotoxic stimuli (Campbell et al., 2004). Interestingly, TNF and IL-6, both classical stimuli for NF-κB activation, also have been correlated with enhanced methylation of promoter-CGI resulting in transcriptional repression of tumor suppressor genes (Hodge et al., 2005; Ushijima and Okochi-Takada, 2005; Wehbe et al., 2006). These observations suggest that under specific conditions NF-κB activation may be related to methylation of specific genes, although the mechanisms through which this occurs are unknown.
BRMS1 was originally identified as a tumor metastasis suppressor and its expression levels have been shown to be markedly reduced in various cancers (Liu et al., 2006b; Meehan and Welch, 2003); with the loss of the BRMS1 allele coinciding with increased numbers of metastases and decreased survival in breast cancer and lung cancer patients (Hicks et al., 2006; Smith et al., 2009; Zhang et al., 2006). We have shown that methylation of the specific CGI (−531 to +608) contributes to BRMS1 transcriptional repression in NSCLC cells and human tumors (Nagji et al., 2010).
In this report we provide evidence that RelA/p65 functions as an active transcriptional repressor through enhanced methylation of the BRMS1 promoter via direct recruitment of DNMT-1 to chromatin in response to TNF. Our study offers new insight into an alternative mechanism through which NF-κB can function as an active transcriptional repressor to promote tumor metastasis.
Results
TNF represses BRMS1 transcription via BRMS1 promoter methylation
Chronic inflammation and the pro-inflammatory cytokines are increasingly appreciated as contributing significantly to cancer progression (Coussens and Werb, 2002; Greten and Karin, 2004; Karin and Greten, 2005; Pikarsky et al., 2004). In our model system the pro-inflammatory cytokine TNF significantly increased the invasive potential of NSCLC H157 and H1299 cells (Fig. 1A). Similarly, siRNA knockdown of endogenous BRMS1 resulted in a dramatic increase in the invasive potential of NSCLC cells (Fig. S1).
Fig. 1. TNF reduces BRMS1 expression.
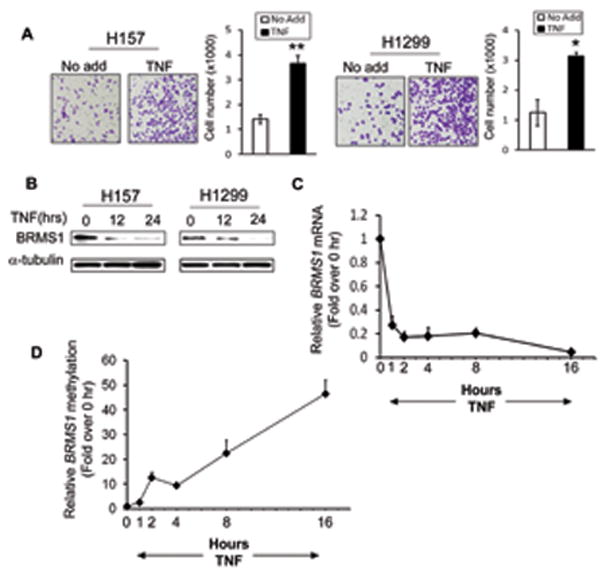
(A) TNF promotes lung cancer cells invasion. NSCLC cells were treated with TNF (40ng/ml) for 48 hrs and the invasion capability was quantified using Boyden chamber assays. Bar graphs show average cell counts per field. Data are presented as mean ± S.D.. (*) p<0.01 and (**) p<0.001 compared to No add. (B) TNF reduces BRMS1 protein level. NSCLC cells were treated with TNF (40ng/ml) at the indicated time points. The protein levels of BRMS1 were evaluated by Western blots. (C) TNF inhibits BRMS1 transcription. NHBE cells were treated with TNF (40ng/ml) and mRNA of endogenous BRMS1 was detected by quantitative RT-PCR at the indicated time points (0, 1, 2, 4, 8 and 16 hrs). (D) TNF induces methylation of the BRMS1 promoter. NHBE cells were treated with TNF as described in (C) and the relative methylation status of BRMS1 was evaluated by quantitative Methylation Specific PCR (MSP).
Since we have shown that BRMS1 is downregulated in NSCLC (Liu et al., 2006b) we next sought to determine whether TNF promotes lung cancer cell invasion through regulation of BRMS1 expression. We observed that TNF reduced both protein (Fig. 1B) and transcription (Fig. 1C) levels of BRMS1 in a time-dependent manner in NSCLC H157 and H1299 cells, and normal bronchial epithelial (NHBE) cells, respectively. These data suggest that repression of BRMS1 transcription is a plausible mechanism through which inflammatory cytokines promotes the metastatic potential of NSCLC.
Given that methylation of the CGI results in BRMS1 transcriptional repression (Nagji et al., 2010), we hypothesized that TNF may be decreasing BRMS1 transcription through enhanced BRMS1 promoter methylation. As we expected, TNF increased BRMS1 methylation in a time -and dose - dependent manner in NHBE cells (Fig. 1D and Fig. S2A). These results establish that TNF enhances BRMS1 transcriptional repression through DNA methylation.
RelA/p65 mediates TNF- induced methylation and transcriptional repression of BRMS1
The biological functions of TNF are realized primarily by activating NF-κB, particularly its active subunit RelA/p65 (Greten et al., 2004). To examine the requirement of endogenous RelA/p65 for enhancing BRMS1 promoter methylation following TNF treatment, p65WT and p65−/− MEF cells were exposed to the demethylating reagent 5-Aza for global hypomethylation of the entire genome. Following removal of 5-Aza, the BRMS1 promoter was re-methylated in the TNF treated p65WT cells. In contrast, in the p65−/− cells the BRMS1 promoter remained hypomethylated despite treatment with TNF (Fig. 2A). Subsequent quantitative RT-PCR studies demonstrated that methylation events observed Fig. 2A correlated with decreased BRMS1 transcription levels in the TNF treated p65 WT MEF cells (Fig. 2B). TNF-mediated repression of BRMS1 transcription results in an approximately 6-fold increase in BRMS1 promoter methylation over basal levels. Importantly, we found that under normal condition the basal expression of RelA/p65 has little impact on the methylation status or transcription of BRMS1 (Figs. 2A and B). This strongly suggests that this process is an inducible event and RelA/p65 is actively involved in TNF-mediated BRMS1 transcriptional repression.
Fig. 2. RelA/p65 mediates TNF-induced BRMS1 transcriptional repression via methylation.
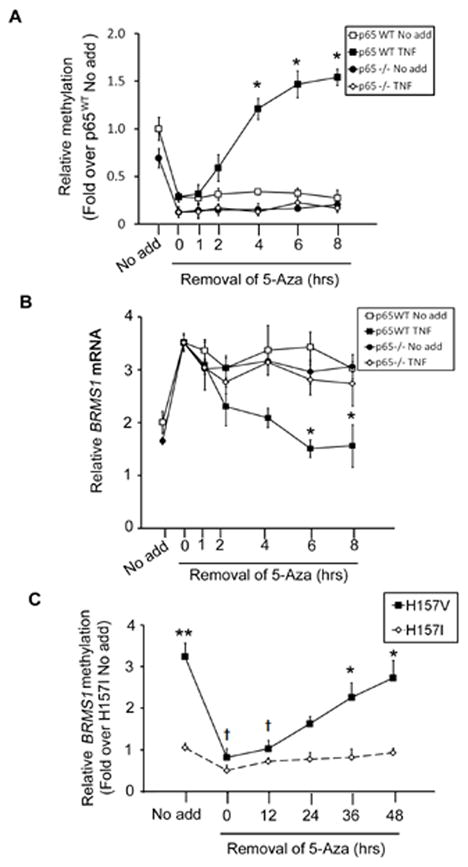
(A) RelA/p65 mediates TNF induced methylation. p65WT and p65−/− MEFs were treated with 5-Aza (5 μM) for 5 days followed by stimulation with TNF (20ng/ml). The relative methylation of BRMS1 was detected by quantitative MSP. The data are plotted as the fold over no add, where results from the group of no add were normalized to 1 for p65WT MEFs. (*) p< 0.05 compared to the same time point in p65WT Control group. (B) RelA/p65 is required for TNF-induced BRMS1 transcriptional repression. p65WT and p65−/− MEFs were treated with 5-Aza and TNF in the same pattern as described in (A). The mRNA of BRMS1 was analyzed by quantitative RT-PCR. (*) p< 0.05 compared to the same time point in p65WT Control group. (C) RelA/p65 mediates methylation of the BRMS1 promoter in NSCLC. H157 V/I cells were treated with 5-Aza (5μM) as described previously. Relative methylation of BRMS1 promoter was determined by quantitative MSP at the indicated time point. (*) p< 0.05 and (**) p<0.01 compared to the same time point in H157I; (†) p<0.05 compared to no add of H157V.
To investigate the effects of RelA/p65 on BRMS1 promoter methylation in NSCLC, we exploited previously generated H157 NSCLC cells which stably express SR-IκB, a dominant-negative inhibitor of NF-κB, (H157I) or a vector control (H157V) (Jones et al., 2000). As shown in Fig. 2C, cells lacking the ability to activate NF-κB (H157I) have a 4-fold decrease in basal BRMS1 promoter methylation levels compared to controls (H157V).
In addition, even with removal of 5-Aza the BRMS1 promoter in the H157I cells cannot be methylated, while H157V cells return to near basal levels at 48 hours. In addition, we also assessed methylation levels of endogenous BRMS1 promoter in H157V and I cells using bisulfite sequencing PCR (BSP). As shown in Fig. S2B, BSP measurements of basal BRMS1 promoter methylation is roughly 3-4-fold in the H157V cells compared to H157I cells which correlates exactly with our MSP results.
Collectively these studies confirm that RelA/p65 is required for TNF-mediated methylation and transcriptional repression of BRMS1 and provide the first evidence of the potential role of RelA/p65 as an epigenetic regulator of DNA methylation.
RelA/p65 directly binds DNA in the CpG island region of the BRMS1 promoter
NF-κB regulates promoter-specific transcription through binding to active -κB binding site(s). (Chen et al., 1998). Analysis of the BRMS1 promoter reveals three putative NF-κB binding sites (arbitrarily named I, II, and III) which are located in the CpG island (Fig. 3A). To determine whether NF-κB can bind these putative -κB binding sites, we performed electrophorectic mobility shift assays (EMSA) using wild-type and mutant -κB binding site as probes. All three putative wild-type -κB binding sites bound NF-κB using the nuclear extracts of NSCLC H157 cells (Fig. 3B) and NHBE cells (Fig. S3A). Subsequent treatment using an anti-RelA/p65 antibody which recognizes the N-terminal region of RelA/p65 confirmed RelA/p65 specificity. Mutation of the last two conserved cytosine (C) to adenine (A) residues in each -κB binding site completely inhibited NF-κB binding to DNA (Fig. 3B and S3A). Moreover, EMSAs indicated that TNF increases nuclear RelA/p65 and enhances NF-κB binding to the -κB binding sites I and II in the BRMS1 promoter, but not III (Fig. 3C and S3B). In summary, like classical TNF-induced NF-κB activated genes, TNF mediates BRMS1 transcriptional repression through enhanced nuclear translocation and DNA binding of RelA/p65.
Fig. 3. A specific -κB binding site in theBRMS1 promoter is required for RelA/p65-mediated transcriptional repression.
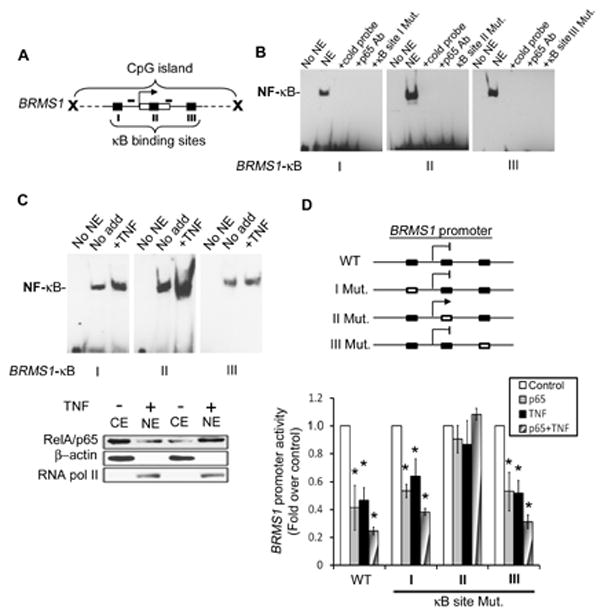
(A) Schematic illustration of the CpG island in BRMS1 promoter. Arrow: the transcription start site; blank box: exon 1; and black squares: the putative NF-κB binding sites and small bars: the primers for MSP. (B) NF-κB binds to the -κB binding sites on the BRMS1 promoter. EMSAs performed using P33-labeled wild-type or mutant -κB binding sites as probe incubated with NSCLC H157 nuclear extracts. (C) TNF enhances NF-κB binding to the NF-κB sites I and II. NSCLC H157 cells were treated with TNF (40ng/ml) for 15 min. EMSAs performed as described in (B). The relative expression of RelA/p65 in nuclear (NE) and cytoplasmic (CE) extracts was evaluated by Western blot. β-actin and RNA pol II were probed as controls for cytoplasmic and nuclear protein, respectively. (D) (Upper) Diagram of wild-type and mutated BRMS1 promoter. Black boxes: wild type -κB binding site and unfilled boxes: the mutant -κB binding site. (Lower) NSCLC H157 cells were co-transfected with indicated BRMS1 promoter reporter genes and RelA/p65 or empty vector. Posttransfection (24 hrs) the cells were treated with or without TNF (40ng/ml) for an additional 18 hrs. Luciferase activity was analyzed. The data are plotted as the fold over control, where results from control in each group were normalized to 1. (*) p< 0.05 compared to control for each group.
To establish which -κB binding site(s) were functional relative to RelA/p65-induced BRMS1 transcriptional repression, we created either individual or combined mutations in the -κB binding sites in the BRMS1 reporter gene. We used site-directed mutagenesis to maintain the conformation of the BRMS1 promoter. These assays demonstrate that RelA/p65 failed to repress the transcriptional activity of the BRMS1 promoter that has the -κB binding site II mutation, while mutation of the other two -κB binding sites individually or in combination (data not shown), had no effect on RelA/p65, TNF, or combined TNF/RelA/p65-induced repression (Fig. 3D). Collectively, these results demonstrate that while all three -κB binding sites in the BRMS1 promoter can bind NF-κB, site II appears to have more functional significance with respect to TNF and/or RelA/p65-mediated repression of BRMS1 transcription.
RelA/p65 is required for assembly of selective DNMTs on the BRMS1 promoter
Given the requirement of DNMT-1 in the methylation process (Bestor, 2000; Okano et al., 1999), we next examined whether RelA/p65 endogenously forms complexes with DNMT-1. As shown in Fig. 4A, RelA/p65 has minimal interaction with DNMT-1 in the absence of TNF. However, there is a robust increase in the endogenous interaction of RelA/p65 and DNMT-1 following TNF treatment. Furthermore, we found that the combination of DNMT-1 and -3b resulted in the most robust repression of BRMS1 transcriptional activity following ectopic expression of RelA/p65 (Fig. S4A).
Fig. 4. RelA/p65 is responsible for DNMT-1 assembling on BRMS1 promoter.
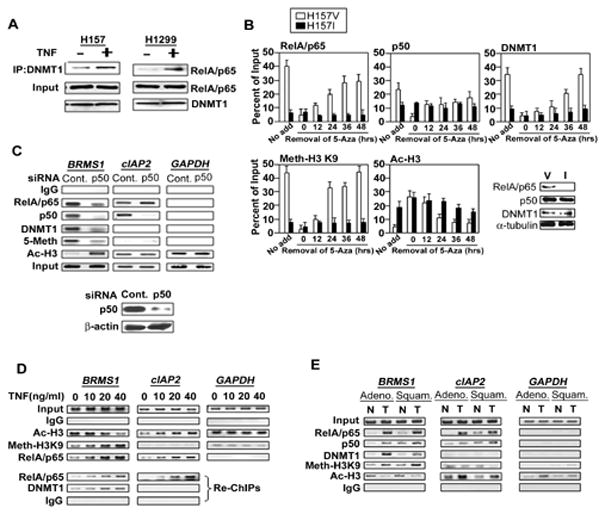
(A) TNF increased the interaction of RelA/p65 and DNMT-1. NSCLC cells were treated with TNF (40ng/ml) or not for 6 hrs. Co-IPs were performed using anti-DNMT-1 antibody, followed by immunoblotting for the presence of RelA/p65. (B) H157V/I cells were treated with 5-Aza as described above. ChIP analysis was performed at the indicated time points after removal of 5-Aza in H157 V/I cells by quantitative PCR specific to the BRMS1 promoter. Western blots show the expression of indicated proteins (nuclear RelA/p65, p50, DNMT1 and α-tubulin) in H157 V/I cells. (C) H157 cells were transiently transfected with siRNA control or p50. Post-transfection 48 hrs, ChIP analysis was performed across the BRMS1 promoter, and the promoters of cIAP2 and GAPDH. The siRNA transfection efficiency was proved by Western blot. (D) ChIP and re-ChIP analyses were performed in HEK 293T cells treated with TNF at the indicated doses for 16 hrs across the BRMS1 promoter, and the promoters of cIAP2 and GAPDH. Re-ChIP analysis used anti-RelA/p65 as the first immunoprecipitating antibody. (E) ChIP analyses were performed using patients’ samples (Adeno: adenocarcinoma, Squam: squamous cell cancer, T: lung tumor, N: matched adjacent non-cancerous lung tissue,) cross the BRMS1 promoter, as well as the promoters of cIAP2 and GAPDH.
To examine the requirement for RelA/p65 in loading of key methylation components on chromatin, chromatin immunoprecipitation (ChIP) assays indicated that removal of 5-Aza resulted in recruitment of DNMT-1 (Fig. 4B) and -3b (Fig. S4B) to the BRMS1 promoter in H157V, but not in the H157I cells in a time-dependent manner. The presence or absence of RelA/p65 had no effect on loading of DNMT-3a (Fig. S4B). Review of the data in Figs. 4B and S4C suggests that the re-recruitment of DNMT-3b on chromatin occurs earlier (~24 hrs), compared to DNMT-1 (~36–48 hrs). Not surprisingly, methylated histone H3-K9 chromatin occupancy was increased on the BRMS1 promoter in the presence of RelA/p65 only. Subsequent Western blot analyses confirmed that the observed decreases of chromatin-associated DNMT-1 and -3b in the H157I cells were not secondary to regulation by RelA/p65 (Figs. 4B and S4B).
Given that heterodimer RelA/p65/p50 is the most common form of NF-κB, siRNA knock down of p50 resulted in significant loss of chromatin-associated RelA/p65, DNMT-1 and methylation on the BRMS1 promoter (Fig. 4C). Interestingly, reducing p50 expression did not affect RelA/p65 loading to the promoter of a known NF-κB-regulated gene, cIAP2 (Liu et al., 2006a). This data suggested that RelA/p65 mediates methylation and transcriptional repression of BRMS1 through heterodimerization with p50.
To determine if TNF-induced chromatin loading of DNMTs is promoter specific we examined the promoter regions of BRMS1, cIAP2, as well as GAPDH. While chromatin occupancy of RelA/p65 increased with TNF concentration (Fig. 4D) and time (Fig. S4C) across both the BRMS1 and cIAP2 promoters, no DNMTs were recruited to the cIAP2 promoter, presumably secondary to an absence of a CpG island in the cIAP2 promoter (Fig. 4D and S4C).
Realizing that DNMTs may be recruited to DNA by factors other than RelA/p65, we performed a re-ChIP assay using RelA/p65 as the first IP antibody. There is a TNF concentration-dependent increase in DNMT-1 recruitment to the BRMS1 promoter, but not on the cIAP2 promoter (Fig. 4D). This analysis strongly suggests that DNMT-1 is part of the methylation complex recruited by RelA/p65 to chromatin. Importantly, this process appears to be gene specific and is likely dependent on the chromatin landscape of each promoter.
To ensure that our cell culture observations were relevant to human lung cancer we performed ChIP analyses in patient specimens using the most common lung cancer histologies -adenocarcinoma (Adeno) and squamous cell (Squam). Identical DNMT-1 and RelA/p65 assembly patterns were uniquely observed across the BRMS1 promoter, but not the cIAP2 or GAPDH promoters (Fig. 4E). Important differences in BRMS1 promoter occupancy are also demonstrated between adjacent non-cancerous tissue (N) and the tumor (T). Collectively, we have demonstrated that RelA/p65 is required for DNMT-1 and -3b binding to BRMS1 chromatin, which is responsible for the subsequent methylation and transcriptional repression of the BRMS1 promoter.
Inhibition of RelA/p65 DNA binding reduces chromatin-associated DNMT-1 and re-activates BRMS1 expression
Crystallography has suggested that the region responsible for RelA/p65 binding to 3’ subsite of -κB binding sites occurs through Arg 33, Arg35, Tyr 36, Glu 39 and Arg 187 (Chen et al., 1998). To better characterize the minimal interacting domain of RelA/p65 that binds to the BRMS1 promoter, we exploited the use of protein transduction domain (PTD) peptide which have been shown to be an effective way of delivering proteins in vitro and in vivo (Schwarze and Dowdy, 2000). We synthesized and expressed three short RelA/p65 peptides including amino acids 31–40, 38–47, and 45–54, linked to an antennapedia- PTD derived from the Drosophila homeotic transcription factor ANTP (Fig.5A). As shown in Fig. 5B, the PTD-RelA/p65 (38–47) peptide blocked RelA/p65 DNA binding in a dose-dependent manner.
Fig. 5. The PTD-p65 (38–47) peptide blocks the recruitment of RelA/p65 and DNMTs to the BRMS1 promoter.
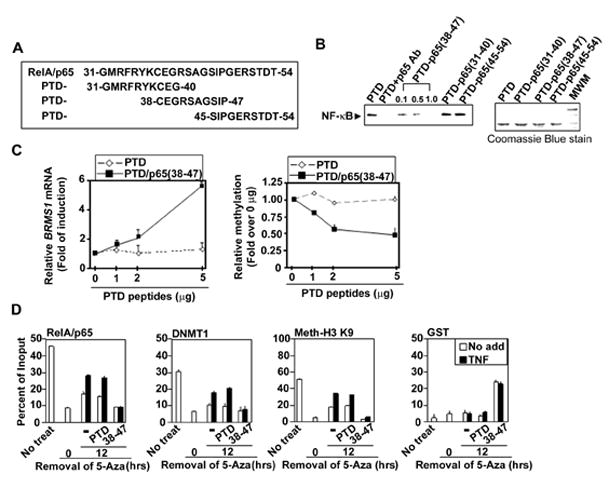
(A) Diagram of protein transduction domain (PTD) peptides conjugated with the putative RelA/p65 DNA binding regions. (B) The PTD-p65 (38–47) peptide inhibits NF-κB binding to the BRMS1 promoter. EMSAs were performed using 293T cell nuclear extracts incubated with indicated peptides (1μg) and 33P-labeled -κB binding site II of BRMS1 promoter as the probe. (C) PTD-p65 (38–47) peptide increases BRMS1 transcripts in NSCLC cells. (Left) BRMS1 mRNA was analyzed by quantitative RT-PCR and (Right) the relative methylation of BRMS1 promoter was evaluated by quantitative MSP in H157 cells treated with PTD peptides at indicated doses for 24 hrs. (D) Inhibition of RelA/p65 DNA binding reduces chromatin-associated DNMT-1 on the BRMS1 promoter. H157 cells were treated with 5-Aza as described previously. After removal of 5-Aza, cells were treated with indicated peptides (5μg/ml) and TNF (20ng/ml) for additional 24 hrs. ChIP analysis was performed.
In order to detect if these short PTD-RelA/p65 peptides could directly bind DNA, in vitro EMSAs were performed. As shown in Fig. S5A, only PTD-RelA/p65 (38–47) directly binds to -κB binding site II. To confirm the specificity of our PTD-RelA/p65 (38–47) peptide we mutated Glu 39 to Ala (E39A); the result is a complete abolishment of DNA binding of the PTD-RelA/p65 (38–47) peptide (Fig. S5A). Data presented in Figs. 5C and S5B highlight the dose-dependent effects of the PTD-RelA/p65 (38–47) on endogenous BRMS1 mRNA, methylation, and protein levels compared to PTD controls. In stark contrast, examination of the known NF-κB-regulated genes, cIAP2 , Bfl/A1 (Liu et al., 2006a) and IκBα demonstrate a robust decrease in these transcripts following PTD/p65(38–47) exposure (Fig. S5C). In aggregate, these data indicate that the PTD-RelA/p65 (38–47) peptide directly binds DNA and functions as a blocking peptide to compete with RelA/p65 binding. Moreover, as shown by others (Chen et al., 1998), E39 is a critical amino acid for RelA/p65 DNA binding. Importantly, the above experiments provide solid evidence that RelA/p65 binds to chromatin to transcriptionally activate some genes (cIAP2, Bfl/1A, IκBα), while in others (BRMS1) RelA/p65 clearly functions as an active transcriptional repressor.
To confirm that RelA/p65 (38–47) was the interacting domain required for RelA/p65 loading to chromatin, ChIP assays were performed. Following removal of 5-Aza, the pan-hypomethylated H157 lung cancer cells were pretreated with PTD-RelA/p65 (38–47) or PTD alone followed by stimulation with TNF (Figs. 5D and S5D). GST-PTD-RelA/p65 (38–47), but not GST-PTD, was found on the BRMS1 promoter suggesting that it endogenously binds to the BRMS1 promoter and functions as a competitive inhibitor for -κB binding sites on chromatin. In the presence of PTD-RelA/p65 (38–47), the recruitment of RelA/p65, methyl-histone H3-K9, and DNMTs were all dramatically decreased on the BRMS1 promoter.
These studies confirm that amino acids 38–47 are necessary for RelA/p65-mediated methylation and active repression of BRMS1. In addition, of the 5 important amino acids, only Glu 39 is located in this region with mutation of Glu 39 completely abolishing the PTD-RelA/p65 (38–47) peptide DNA binding (Fig. 5 and S5).
The RelA/p65 109–120 region is required for DNMT-1 binding
Next, we were interested in whether RelA/p65 binding to DNMT-1 was required for regulating promoter methylation, transcription of BRMS1. To experimentally address this, GST pull-down experiments indicated that the N-terminus region (1–245 aa) of RelA/p65 is responsible for the interaction with DNMT-1 (Fig. 6A). Using a series of N-terminal deletion constructs we found that the first 108 residues in RelA/p65 do not affect RelA/p65-DNMT-1 interactions. However, expanding the deletion inclusive of 119 residues, RelA/p65 failed to bind DNMT-1 (Fig. 6B). We thus hypothesized that the RelA/p65 109–120 aa region was a strong candidate for the minimal interacting domain for RelA/p65 and DNMT-1.
Fig. 6. The region of RelA/p65 interacting with DNMT-1 is located in 109–119 amino acids.
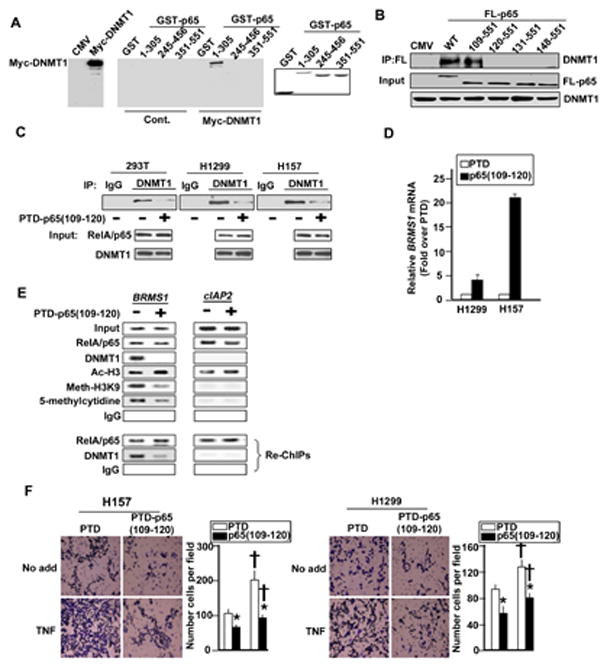
(A) The N-terminal of RelA/p65 is responsible for DNMT-1 binding. HEK 293T cells were transfected with expression vector encoding myc-DNMT1. GST-pull downs were performed using GST-RelA/p65 fusion proteins and the presence of myc-DNMT1 was analyzed by Western blots. Coomassie blue indicates the relative amounts of GST fusion protein. (B) RelA/p65 (109–119) is critical for DNMT-1 binding. HEK 293T cells were transfected with indicated Flag-tagged RelA/p65. IPs were performed using αFlag antibody, followed by immunoblotting for the presence of endogenous DNMT1. (C) The PTD-p65 (109–120) peptide blocks RelA/p65 interacting with DNMT-1. HEK 293T and NSCLC cells treated with the PTD peptides (5μg/ml) for 24 hrs. IPs were performed using antibodies against DNMT-1 or IgG followed by immunoblotting for the presence of RelA/p65. (D) The PTD-p65 (109–120) peptide increases BMRS1 transcription. NSCLC cells were treated with the PTD peptides as described in (C). The mRNA of BRMS1 was analyzed by quantitative RT-PCR. (E) ChIP and Re-ChIP analyses were performed in NSCLC H157 cells treated with the PTD peptide as described in (C) across the BRMS1 promoter and cIAP2 promoter. Re-ChIP analysis used anti-RelA/p65 as the first immunoprecipitating antibody. (F) The PTD-p65 (109–120) peptide inhibits invasion of lung cancer cells. NSCLC cells were treated with the PTD peptides (5μg/ml) and TNF (20ng/ml) for 48 hrs. The invasion capability was quantified as described above. (*) p<0.01 compared to the treatment of PTD group and (†) p<0.01 compared to the No add group.
To ascertain whether inhibition of the endogenous interaction between RelA/p65-DNMT-1 had functional relevance, we synthesized and expressed a short PTD-linked RelA/p65 (109–120 aa) peptide. Following PTD-RelA/p65 (109–120) peptide treatment, the endogenous interaction of RelA/p65-DNMT-1 was significantly decreased in HEK 293T, NSCLC cells (Fig. 6C). Interestingly, both BRMS1 mRNA and protein levels were increased after treated with the PTD-RelA/p65 (109–120) peptide (Fig. 6D and data not shown). To prove the specificity of RelA/p65 (109–120) peptide, a mutant of PTD-RelA/p65 (109–120) peptide failed to affect the interaction of RelA/p65-DNMT-1 and BRMS1 mRNA (Fig. S6A and B). This strongly suggests that DNMT-1 is involved in BRMS1 transcriptional regulation through interactions with RelA/p65.
To investigate if the PTD-RelA/p65 (109–120) peptide blocks the interaction of RelA/p65-DNMT-1 on chromatin resulting in decreased BRMS1 methylation, ChIP and re-ChIP assays were performed cross the BRMS1 and cIAP2 promoters. Inhibition of RelA/p65 and DNMT-1 interaction dramatically reduces the recruitment of DNMT-1 to the BRMS1 promoter, without any effect on RelA/p65 promoter loading. In addition, using 5-methyl-cytidine as well established mark of promoter methylation (Suzuki and Bird, 2008) there is significant inhibition of basal BRMS1 promoter methylation. These results prove that RelA/p65 tethers DNMT-1 on chromatin to promote methylation and transcriptional repression of BRMS1 and that inhibition of RelA/p65-DNMT-1 binding results in an increase in BRMS1 mRNA in NSCLC cells.
In addition, blocking RelA/p65-DNMT-1 binding using the PTD-RelA/p65 (109–120) peptide dramatically inhibited basal and TNF stimulated invasion potential of lung cancer cells compared to the treatment with PTD only (Fig. 6F). This data confirms the biological function of RelA/p65-DNMT-1 binding in NSCLC cells.
Phosphorylation of serine 276 is required for RelA/p65-DNMT-1 interactions on the BMRS1 promoter
Recently, the Ghosh laboratory has demonstrated the importance of phosphorylation of serine 276 as a primary event that governs whether RelA/p65 functions as a transcriptional activator or as a basal transcriptional repressor (Dong et al., 2008; Zhong et al., 2002). In our model system, we observed that S276A mutation in RelA/p65 resulted in a dramatic rescue of basal- and TNF-induced BRMS1 transcriptional repression. Neither K310R nor S536A mutants affected RelA/p65-mediated BRMS1 transcriptional repression (Fig. 7A). These findings suggest that TNF-induced phosphorylation of S276 is important to RelA/p65-mediated BRMS1 transcriptional repression. Importantly the S276A mutant of RelA/p65 fails to bind DNMT-1 (Fig. 7B).
Fig. 7. RelA/p65 serine residue 276 is critical for RelA/p65-mediated BRMS1 transcriptional repression.
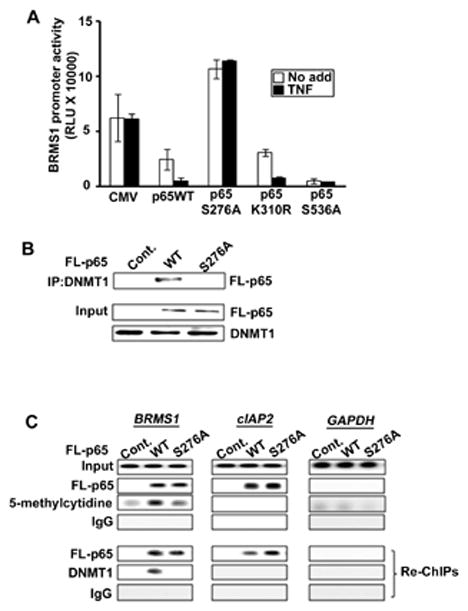
(A) Mutation of RelA/p65 S276 failed to inhibit BRMS1 promoter activity. p65−/−MEFs were transiently co-transfected with BRMS1-luc. reporter and expression vectors encoding indicated Flag-tagged RelA/p65. The luciferase activity was determined as described above. (B) RelA/p65 S276A cannot bind DNMT-1. p65−/− MEFs were transfected with expression vector encoding indicated Flag-tagged RelA/p65. Co-IPs were performed as previously described. (C) Mutation of S276 significantly reduced RelA/p65-mediated DNMT-1 loading to the BRMS1 promoter. HEK 293T cells were transfected with expression vector encoding indicated Flag-tagged RelA/p65. ChIP and re-ChIP analyses were performed across the BRMS1 promoter, and the promoters cIAP2 and GAPDH. Re-ChIP analysis used anti-Flag epitope as the first immunoprecipitating antibody.
Finally, to investigate if phosphorylation of S276 is required for RelA/p65 and subsequent DNMT-1 loading to chromatin ChIP and re-ChIP assays were performed as described above (Fig. 7C). The RelA/p65 S276A mutant had no effect on the recruitment of RelA/p65 to BRMS1 and cIAP2 promoters compared to wild-type RelA/p65. Interestingly, the RelA/p65 S276A mutant dramatically inhibited DNA methylation (5-methyl-cytidine) on the BRMS1 promoter, compared with wild-type RelA/p65. Furthermore, re-ChIP assays indicated that the RelA/p65 S276A mutant cannot assemble DNMT-1 on the BRMS1 promoter. This strongly suggests that phosphorylation of serine 276 is required for DNMT-1 binding to RelA/p65 with subsequent loading of DNMT-1 to chromatin.
Discussion
In this report we show that TNF-induction of the RelA/p65 subunit of NF-κB is required for promoter methylation of BRMS1, a gene we have previously shown to be important in the metastatic movement of lung cancer cells (Smith et al., 2009). RelA/p65 recruits DNMT1 to chromatin resulting in BRMS1 methylation. Moreover, we show that a specific post-translational event, S276 phosphorylation, is required for RelA/p65 binding to DNMT1, the resulting recruitment of DNMT-1 to chromatin, specific CGI methylation, and transcriptional repression (Fig.8). These observations are supported by other studies demonstrating that transcription factors can induce DNA methylation through recruitment of DNMTs to the promoter, such as Stat-3 (Zhang et al., 2005), PML-RAR (Di Croce et al., 2002) and RelB (Puto and Reed, 2008).
Fig. 8. RelA/p65 activates transcription through the dual mechanisms.
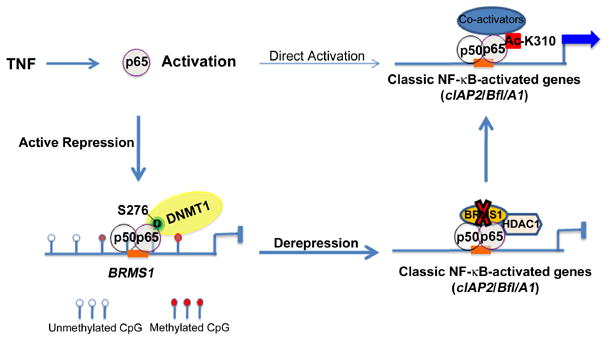
In response to TNF stimulation, RelA/p65 directly activates transcription of classic NF-κB-regulated genes, such as cIAP2 and Bfl/A1. RelA/p65 can also function as an “active repressor” to induce methylation and transcriptional repression of BRMS1, a known co-repressor of RelA/p65, by assembly of DNMT-1 to chromatin. In this process, phosphorylation of serine 276 is required for the interaction of RelA/p65 with DNMT-1. Whether RelA/p65 functions as an inducible transcriptional activator or repressor depends on the chromatin landscape of the target genes. Moreover, the repression of BRMS1 transcription results in derepression of RelA/p65 transactivation through enhanced acetylation of K310, which further enhances RelA/p65-mediated transcriptional activation.
RelA/p65 activation is typically associated with promoting transcription (Baldwin, 1996; Wang et al., 1996). However, depending on the stimuli, the specific gene, and the unique chromatin landscape of that gene, transcription factors can have dual functions in transcriptional regulation. Examples of such dual-functioning transcription factors include p53, Sp1, ERα, NF-Y, and Stat3 (Ceribelli et al., 2008; Hoffman et al., 2002; Mottet et al., 2009; Romano et al.; Sankpal et al., 2009; Zhang et al., 2005).
NF-κB was first identified as an active transcriptional repressor by the Perkin’s laboratory under cytotoxic stimuli such as UV-C and doxorubicin (Campbell et al., 2004). Following TNF treatment, RelA/p65 also has been reported to actively represses transcription of α 2(I) Collagen (Kouba et al., 1999), hypoxia-inducible death factor BNIP3 (Shaw et al., 2006), and tumor suppressor PTEN (Kim et al., 2004; Xia et al., 2007) via directly binding the putative-κB binding sites on chromatin.
Although reported mechanisms through which NF-κB transcriptional inactivation have largely been relegated to the ability of RelA/p65 to associate with HDACs in unstimulated cells (Dong et al., 2008; Zhong et al., 2002), it remains unclear how RelA/p65 functions as a transcriptional repressor in the presence of stimulation, such as TNF. Our studies identify a novel mechanism through which TNF induces RelA/p65 to function as an active repressor. This mechanism involves an inducible RelA/p65/p50-mediated recruitment of DNMTs to chromatin which results in promoter methylation and transcriptional repression. Importantly, in our model system the ability of RelA/p65 to bind DNMT-1 is independent of HDACs activities (Fig. S7).
TNF activates RelA/p65 through inducing phosphorylation of S276 by protein kinase A (PKA) (Zhong et al., 1997), or mitogen and stress-activated protein kinase-1 (MSK1), (Vermeulen et al., 2003). While previous reports suggest that RelA/p65 S276 phosphorylation results in preferential interaction with co-activator complexes for selected genes (Dong et al., 2008; Zhong et al., 2002), we found that the S276 phosphorylation is required for RelA/p65 interacting with DNMT-1, DNMT-1 recruitment to the specific-promoter, and transcriptional repression. We postulate that phosphorylation of S276 may result in a conformational changes of RelA/p65 that unmasks CBP/p300 and DNMT-1 interacting domains. Whether CBP/p300 or DNMT-1 binds RelA/p65 is dependent on the gene-specific chromatin environment. In support of this theory we observed that TNF temporally promotes RelA/p65 binding to the promoters of both the metastasis suppressor gene BRMS1 and the classic NF-κB-regulated gene cIAP2. However, in contrast to BRMS1, there is no CGI in the cIAP2 promoter and thus, there is no chromatin loading of DNMTs. The result is a completely opposite effect on RelA/p65-mediated gene transcription: BRMS1 is transcriptionally repressed and cIAP2 is transcriptionally activated. It is probably because S276 phosphorylation results in differential RelA/p65 affinities to interact with co-activators or co-repressors depending on the underlying chromatin landscape (Chen et al., 2000b). Evidence of differential transcriptional activities following RelA/p65 phosphorylation has been noted by several groups. For instance, MSK1-mediated RelA/p65 S276 phosphorylation transcriptionally activates IL-6, but not another NF-κB-regulated gene NFκB2 (Vermeulen et al., 2003). RelA/p65 S276 phosphorylation by PKA increases the transcription of NF-κB-regulated gene IL-4, but suppresses IL-2 (Neumann et al., 1995).
An additional consideration as to whether RelA/p65 functions as a transcriptional activator or repressor is the location of the -κB-binding site relative to the CGI. As an example, there is differential regulation of GATA4 and SHP1, two Stat3 regulated genes both with a CGI in their proximal promoters, but with different locations of Stat3 binding regions (Snyder et al., 2010; Zhang et al., 2005). We have previously demonstrated the functional significance of specific -κB binding sites located within the promoter-CGI region of BRMS1 (Nagji et al., 2010) confirming this additional layer of chromatin control over this process.
It is well established that RelA/p65 directly activates transcription of classic NF-κB regulated genes (Chen et al., 2000a; Wang et al., 1998). Our previous study indicated that BRMS1 functions as a corepressor to inhibit RelA/p65 transactivation potential by deacetylation of K310. In addition, siRNA knockdown of BRMS1 significantly promotes both basal and TNF-induced RelA/p65 mediated transcription (Liu et al., 2006b). In this study we demonstrate a previously undescribed mechanism through which RelA/p65 and BRMS1 mutually negatively co-regulate one another’s function in lung cancer cells. Following TNF exposure, RelA/p65 actively represses BRMS1 transcription through DNMT-1 mediated promoter methylation. Subsequently this loss of BRMS1 results in derepression of RelA/p65 (via loss of the co-repressor function of BRMS1) (Liu et al., 2006b), thus promoting the transactivation of RelA/p65 (Fig. 8).
In summary, we show that the TNF-induced phosphorylation of S276 of RelA/p65 results in recruitment of DNMT-1 to the BRMS1 promoter. This results in DNA methylation and active transcriptional repression of the metastasis suppressor BRMS1. It is likely that the ability of RelA/p65 to function as a transcriptional repressor is not unique to the BRMS1 promoter, but promoters with similar chromatin landscapes may also be regulated in a similar manner, making our observations broadly applicable. Our studies suggest that in addition to the known post-translational modifications of RelA/p65 that govern its function, promoter specifics such as CGI presence or absence, CGI location, and the location of RelA/p65 binding regions on chromatin are also important determinants of whether RelA/p65 functions as an inducible transcriptional activator or repressor. Our data do suggest a novel mechanism through which NF-κB promotes cancer progression and may allow for the design and development of specific oncopharmacologic compounds that can selectively de-repress important anti-metastatic genes negatively regulated by RelA/p65 through targeting its DNA binding and/or DNMT-1 binding.
Materials and methods
Cells culture and surgical specimens
Human NSCLC cell lines, NHBE, HEK 293T and H157 I/V cells were maintained as previously described (Jones et al., 2000; Liu et al., 2006b). Mouse embryo fibroblast (MEF) cells were grown in DMEM with 10% FBS (Invitrogen, Carlsbad, CA). 5′-aza-2′-deoxycytidine (5-Aza) purchased from Sigma Aldrich (St. Louis, MO). Human specimens were preserved as described previously (Liu et al., 2006b).
Plasmid construction and antibodies
Human BRMS1-Luc promoter and the plasmid encoding Flag-tagged RelA/p65 were generated as described (Jones et al., 2000; Liu et al., 2006b). The oligonucleotides encoding antennapedia PTD (DRQIKIWFQNRRMKWKK) or PTD-RelA/p65 peptides were inserted into pGEX-4T-1 (GE, Uppsala, Sweden) at BamHI/SalI sites. The BRMS1-lucκB-binding site mutants and mutant PTD-RelA/p65 peptides (E39A; 109–120 mutant: C109R/S112R/Q114H/N115K/L116P) were generated using the quick-change mutagenesis kit (Stratagene, La Jolla, CA). The antibodies were used: RelA/p65, p50, DNMT1, DNMT3a, DNMT3b, GST, IκB and RNA Pol II (Santa Cruz Biotech, Santa Cruz, CA); Myc-tag, Acetyl-H3 and pan-methy-H3K9 (Cell signaling, Beverly, MA); Flag-tag and α-tubulin (Sigma Aldrich, St. Louis, MO); 5-methyl-cytidine (Eurogentec, San Diego, CA). siRNA was purchased from Thermo Fisher Scientific (Lafayette, CO).
Total RNA isolation and quantitative reverse transcription-PCR (QRT-PCR)
Total RNA was isolated and QRT-PCR was performed as described previously (Liu et al., 2006b).
Transfection and luciferase reporter assays
Plasmids and reporter genes were transiently transfected and luciferase reporter activity assays were performed as described (Liu et al., 2006b).
Methylation specific PCR (MSP) and bisulfite sequencing PCR (BSP)
The genomic DNA was isolated using DNeasy blood and tissue kit (Qiagen, Valencia CA) and quantitative MSPs and Bisulfite sequencing PCR were performed as desrcibed (Nagji et al., 2010).
Electrophoretic mobility shift assay (EMSA)
The cells nuclear extracts (10μg) were isolated and incubated with the P33-labeled BRMS1-κB binding sites as probes. In vitro EMSAs were performed using indicated GST-PTD peptides (2μg) incubated with the P33-labeled BRMS1-κB binding sites as probes. The specificity of binding was examined by competition with the unlabeled oligonucleotides and antibodies against RelA/p65, Flag-epitope tag or GST.
Expression and purification of peptides
GST-PTD-p65 peptides were expressed and purified as described (Liu et al., 2006b). The GST fragments were cleaved from the peptides by incubating with thrombin (10U/mg, Sigma, MO).
ChIP and re-ChIP assays
ChIP and Re-ChIP assays were performed as previously described (Hoberg et al., 2006; Liu et al., 2006b).
Immunoprecipitation (IP) and Western blot
Immunoprecipitation and western blot were performed as previously described (Liu et al., 2006b).
Transwell migration and invasion assays
Migration and invasion of NSCLC cells was assessed as described (Smith et al., 2009). Parallel experiments were performed in triplicate to assess migration in control chambers and invasion.
Statistical analysis
The results of all experiments represent the means ± SD of three separate experiments performed in duplicate or triplicate. Statistical differences were determined by a two-tailed, unpaired Student t-test when appropriate. P values of < 0.05 were considered significant.
Supplementary Material
Acknowledgments
This work was supported by grants: R01 CA136705 (to DRJ), R01 CA104397 (to MWM), and R01 CA132580 (to MWM). In addition, this project was supported in part by a gift provided to the University of Virginia by Philip Morris USA. The review and approval process was overseen by an independent National External Advisory Board without any affiliation with the University, PM USA, or any other tobacco company.
Footnotes
Conflicts of interest
The authors have no conflicts of interest to declare.
Supplementary information is available at Oncogene’s website
References
- Baldwin AS., Jr The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–683. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- Bestor TH. The DNA methyltransferases of mammals. Hum Mol Genet. 2000;9:2395–2402. doi: 10.1093/hmg/9.16.2395. [DOI] [PubMed] [Google Scholar]
- Bobrovnikova-Marjon EV, Marjon PL, Barbash O, Vander Jagt DL, Abcouwer SF. Expression of angiogenic factors vascular endothelial growth factor and interleukin-8/CXCL8 is highly responsive to ambient glutamine availability: role of nuclear factor-kappaB and activating protein-1. Cancer Res. 2004;64:4858–4869. doi: 10.1158/0008-5472.CAN-04-0682. [DOI] [PubMed] [Google Scholar]
- Bours V, Dejardin E, Goujon-Letawe F, Merville MP, Castronovo V. The NF-kappa B transcription factor and cancer: high expression of NF-kappa B- and I kappa B-related proteins in tumor cell lines. Biochem Pharmacol. 1994;47:145–149. doi: 10.1016/0006-2952(94)90448-0. [DOI] [PubMed] [Google Scholar]
- Campbell KJ, Rocha S, Perkins ND. Active repression of antiapoptotic gene expression by RelA(p65) NF-kappa B. Mol Cell. 2004;13:853–865. doi: 10.1016/s1097-2765(04)00131-5. [DOI] [PubMed] [Google Scholar]
- Ceribelli M, Dolfini D, Merico D, Gatta R, Vigano AM, Pavesi G, et al. The histone-like NF-Y is a bifunctional transcription factor. Mol Cell Biol. 2008;28:2047–2058. doi: 10.1128/MCB.01861-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Edelstein LC, Gelinas C. The Rel/NF-kappaB family directly activates expression of the apoptosis inhibitor Bcl-x(L) Mol Cell Biol. 2000a;20:2687–2695. doi: 10.1128/mcb.20.8.2687-2695.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen FE, Huang DB, Chen YQ, Ghosh G. Crystal structure of p50/p65 heterodimer of transcription factor NF-kappaB bound to DNA. Nature. 1998;391:410–413. doi: 10.1038/34956. [DOI] [PubMed] [Google Scholar]
- Chen LF, Williams SA, Mu Y, Nakano H, Duerr JM, Buckbinder L, et al. NF-kappaB RelA phosphorylation regulates RelA acetylation. Mol Cell Biol. 2005;25:7966–7975. doi: 10.1128/MCB.25.18.7966-7975.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YQ, Sengchanthalangsy LL, Hackett A, Ghosh G. NF-kappaB p65 (RelA) homodimer uses distinct mechanisms to recognize DNA targets. Structure. 2000b;8:419–428. doi: 10.1016/s0969-2126(00)00123-4. [DOI] [PubMed] [Google Scholar]
- Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denlinger CE, Rundall BK, Jones DR. Modulation of antiapoptotic cell signaling pathways in non-small cell lung cancer: the role of NF-kappaB. Semin Thorac Cardiovasc Surg. 2004;16:28–39. doi: 10.1053/j.semtcvs.2003.12.004. [DOI] [PubMed] [Google Scholar]
- Di Croce L, Raker VA, Corsaro M, Fazi F, Fanelli M, Faretta M, et al. Methyltransferase recruitment and DNA hypermethylation of target promoters by an oncogenic transcription factor. Science. 2002;295:1079–1082. doi: 10.1126/science.1065173. [DOI] [PubMed] [Google Scholar]
- Dong J, Jimi E, Zhong H, Hayden MS, Ghosh S. Repression of gene expression by unphosphorylated NF-kappaB p65 through epigenetic mechanisms. Genes Dev. 2008;22:1159–1173. doi: 10.1101/gad.1657408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ, et al. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118:285–296. doi: 10.1016/j.cell.2004.07.013. [DOI] [PubMed] [Google Scholar]
- Greten FR, Karin M. The IKK/NF-kappaB activation pathway-a target for prevention and treatment of cancer. Cancer Lett. 2004;206:193–199. doi: 10.1016/j.canlet.2003.08.029. [DOI] [PubMed] [Google Scholar]
- Helbig G, Christopherson KW, 2nd, Bhat-Nakshatri P, Kumar S, Kishimoto H, Miller KD, et al. NF-kappaB promotes breast cancer cell migration and metastasis by inducing the expression of the chemokine receptor CXCR4. J Biol Chem. 2003;278:21631–21638. doi: 10.1074/jbc.M300609200. [DOI] [PubMed] [Google Scholar]
- Hicks DG, Yoder BJ, Short S, Tarr S, Prescott N, Crowe JP, et al. Loss of breast cancer metastasis suppressor 1 protein expression predicts reduced disease-free survival in subsets of breast cancer patients. Clin Cancer Res. 2006;12:6702–6708. doi: 10.1158/1078-0432.CCR-06-0635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoberg JE, Popko AE, Ramsey CS, Mayo MW. IkappaB kinase alpha-mediated derepression of SMRT potentiates acetylation of RelA/p65 by p300. Mol Cell Biol. 2006;26:457–471. doi: 10.1128/MCB.26.2.457-471.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge DR, Xiao W, Peng B, Cherry JC, Munroe DJ, Farrar WL. Enforced expression of superoxide dismutase 2/manganese superoxide dismutase disrupts autocrine interleukin-6 stimulation in human multiple myeloma cells and enhances dexamethasone-induced apoptosis. Cancer Res. 2005;65:6255–6263. doi: 10.1158/0008-5472.CAN-04-4482. [DOI] [PubMed] [Google Scholar]
- Hoffman WH, Biade S, Zilfou JT, Chen J, Murphy M. Transcriptional repression of the anti-apoptotic survivin gene by wild type p53. J Biol Chem. 2002;277:3247–3257. doi: 10.1074/jbc.M106643200. [DOI] [PubMed] [Google Scholar]
- Huber MA, Azoitei N, Baumann B, Grunert S, Sommer A, Pehamberger H, et al. NF-kappaB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J Clin Invest. 2004;114:569–581. doi: 10.1172/JCI21358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DR, Broad RM, Madrid LV, Baldwin AS, Jr, Mayo MW. Inhibition of NF-kappaB sensitizes non-small cell lung cancer cells to chemotherapy-induced apoptosis. Ann Thorac Surg. 2000;70:930–936. doi: 10.1016/s0003-4975(00)01635-0. discussion 936–937. [DOI] [PubMed] [Google Scholar]
- Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5:749–759. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- Kim S, Domon-Dell C, Kang J, Chung DH, Freund JN, Evers BM. Down-regulation of the tumor suppressor PTEN by the tumor necrosis factor-alpha/nuclear factor-kappaB (NF-kappaB)-inducing kinase/NF-kappaB pathway is linked to a default IkappaB-alpha autoregulatory loop. J Biol Chem. 2004;279:4285–4291. doi: 10.1074/jbc.M308383200. [DOI] [PubMed] [Google Scholar]
- Kouba DJ, Chung KY, Nishiyama T, Vindevoghel L, Kon A, Klement JF, et al. Nuclear factor-kappa B mediates TNF-alpha inhibitory effect on alpha 2(I) collagen (COL1A2) gene transcription in human dermal fibroblasts. J Immunol. 1999;162:4226–4234. [PubMed] [Google Scholar]
- Liu Y, Denlinger CE, Rundall BK, Smith PW, Jones DR. Suberoylanilide hydroxamic acid induces Akt-mediated phosphorylation of p300, which promotes acetylation and transcriptional activation of RelA/p65. J Biol Chem. 2006a;281:31359–31368. doi: 10.1074/jbc.M604478200. [DOI] [PubMed] [Google Scholar]
- Liu Y, Smith PW, Jones DR. Breast cancer metastasis suppressor 1 functions as a corepressor by enhancing histone deacetylase 1-mediated deacetylation of RelA/p65 and promoting apoptosis. Mol Cell Biol. 2006b;26:8683–8696. doi: 10.1128/MCB.00940-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayo MW, Baldwin AS. The transcription factor NF-kappaB: control of oncogenesis and cancer therapy resistance. Biochim Biophys Acta. 2000;1470:M55–62. doi: 10.1016/s0304-419x(00)00002-0. [DOI] [PubMed] [Google Scholar]
- Meehan WJ, Welch DR. Breast cancer metastasis suppressor 1: update. Clin Exp Metastasis. 2003;20:45–50. doi: 10.1023/a:1022542519586. [DOI] [PubMed] [Google Scholar]
- Mottet D, Pirotte S, Lamour V, Hagedorn M, Javerzat S, Bikfalvi A, et al. HDAC4 represses p21(WAF1/Cip1) expression in human cancer cells through a Sp1-dependent, p53-independent mechanism. Oncogene. 2009;28:243–256. doi: 10.1038/onc.2008.371. [DOI] [PubMed] [Google Scholar]
- Nagji AS, Liu Y, Stelow EB, Stukenborg GJ, Jones DR. BRMS1 transcriptional repression correlates with CpG island methylation and advanced pathological stage in non-small cell lung cancer. J Pathol. 2010;221:229–237. doi: 10.1002/path.2707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Grieshammer T, Chuvpilo S, Kneitz B, Lohoff M, Schimpl A, et al. RelA/p65 is a molecular target for the immunosuppressive action of protein kinase A. Embo J. 1995;14:1991–2004. doi: 10.1002/j.1460-2075.1995.tb07191.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, et al. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431:461–466. doi: 10.1038/nature02924. [DOI] [PubMed] [Google Scholar]
- Puto LA, Reed JC. Daxx represses RelB target promoters via DNA methyltransferase recruitment and DNA hypermethylation. Genes Dev. 2008;22:998–1010. doi: 10.1101/gad.1632208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano A, Adriaens M, Kuenen S, Delvoux B, Dunselman G, Evelo C, et al. Identification of novel ER-alpha target genes in breast cancer cells: gene- and cell-selective co-regulator recruitment at target promoters determines the response to 17beta-estradiol and tamoxifen. Mol Cell Endocrinol. 314:90–100. doi: 10.1016/j.mce.2009.08.008. [DOI] [PubMed] [Google Scholar]
- Sankpal NV, Willman MW, Fleming TP, Mayfield JD, Gillanders WE. Transcriptional repression of epithelial cell adhesion molecule contributes to p53 control of breast cancer invasion. Cancer Res. 2009;69:753–757. doi: 10.1158/0008-5472.CAN-08-2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarze SR, Dowdy SF. In vivo protein transduction: intracellular delivery of biologically active proteins, compounds and DNA. Trends Pharmacol Sci. 2000;21:45–48. doi: 10.1016/s0165-6147(99)01429-7. [DOI] [PubMed] [Google Scholar]
- Shaw J, Zhang T, Rzeszutek M, Yurkova N, Baetz D, Davie JR, et al. Transcriptional silencing of the death gene BNIP3 by cooperative action of NF-kappaB and histone deacetylase 1 in ventricular myocytes. Circ Res. 2006;99:1347–1354. doi: 10.1161/01.RES.0000251744.06138.50. [DOI] [PubMed] [Google Scholar]
- Smith PW, Liu Y, Siefert SA, Moskaluk CA, Petroni GR, Jones DR. Breast cancer metastasis suppressor 1 (BRMS1) suppresses metastasis and correlates with improved patient survival in non-small cell lung cancer. Cancer Lett. 2009;276:196–203. doi: 10.1016/j.canlet.2008.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder M, Huang XY, Zhang JJ. Stat3 directly controls the expression of Tbx5, Nkx2.5, and GATA4 and is essential for cardiomyocyte differentiation of P19CL6 cells. J Biol Chem. 2010;285:23639–23646. doi: 10.1074/jbc.M110.101063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki MM, Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet. 2008;9:465–476. doi: 10.1038/nrg2341. [DOI] [PubMed] [Google Scholar]
- Takada Y, Murakami A, Aggarwal BB. Zerumbone abolishes NF-kappaB and IkappaBalpha kinase activation leading to suppression of antiapoptotic and metastatic gene expression, upregulation of apoptosis, and downregulation of invasion. Oncogene. 2005;24:6957–6969. doi: 10.1038/sj.onc.1208845. [DOI] [PubMed] [Google Scholar]
- Ushijima T, Okochi-Takada E. Aberrant methylations in cancer cells: where do they come from? Cancer Sci. 2005;96:206–211. doi: 10.1111/j.1349-7006.2005.00035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeulen L, De Wilde G, Van Damme P, Vanden Berghe W, Haegeman G. Transcriptional activation of the NF-kappaB p65 subunit by mitogen- and stress-activated protein kinase-1 (MSK1) Embo J. 2003;22:1313–1324. doi: 10.1093/emboj/cdg139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CY, Mayo MW, Baldwin AS., Jr TNF- and cancer therapy-induced apoptosis: potentiation by inhibition of NF-kappaB. Science. 1996;274:784–787. doi: 10.1126/science.274.5288.784. [DOI] [PubMed] [Google Scholar]
- Wang CY, Mayo MW, Korneluk RG, Goeddel DV, Baldwin AS., Jr NF-kappaB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science. 1998;281:1680–1683. doi: 10.1126/science.281.5383.1680. [DOI] [PubMed] [Google Scholar]
- Wehbe H, Henson R, Meng F, Mize-Berge J, Patel T. Interleukin-6 contributes to growth in cholangiocarcinoma cells by aberrant promoter methylation and gene expression. Cancer Res. 2006;66:10517–10524. doi: 10.1158/0008-5472.CAN-06-2130. [DOI] [PubMed] [Google Scholar]
- Xia D, Srinivas H, Ahn YH, Sethi G, Sheng X, Yung WK, et al. Mitogen-activated protein kinase kinase-4 promotes cell survival by decreasing PTEN expression through an NF kappa B-dependent pathway. J Biol Chem. 2007;282:3507–3519. doi: 10.1074/jbc.M610141200. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Wang HY, Marzec M, Raghunath PN, Nagasawa T, Wasik MA. STAT3- and DNA methyltransferase 1-mediated epigenetic silencing of SHP-1 tyrosine phosphatase tumor suppressor gene in malignant T lymphocytes. Proc Natl Acad Sci U S A. 2005;102:6948–6953. doi: 10.1073/pnas.0501959102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Yamashita H, Toyama T, Yamamoto Y, Kawasoe T, Iwase H. Reduced expression of the breast cancer metastasis suppressor 1 mRNA is correlated with poor progress in breast cancer. Clin Cancer Res. 2006;12:6410–6414. doi: 10.1158/1078-0432.CCR-06-1347. [DOI] [PubMed] [Google Scholar]
- Zhong H, SuYang H, Erdjument-Bromage H, Tempst P, Ghosh S. The transcriptional activity of NF-kappaB is regulated by the IkappaB-associated PKAc subunit through a cyclic AMP-independent mechanism. Cell. 1997;89:413–424. doi: 10.1016/s0092-8674(00)80222-6. [DOI] [PubMed] [Google Scholar]
- Zhong H, May MJ, Jimi E, Ghosh S. The phosphorylation status of nuclear NF-kappa B determines its association with CBP/p300 or HDAC-1. Mol Cell. 2002;9:625–636. doi: 10.1016/s1097-2765(02)00477-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.