

Abstract
The molecular signaling events leading to protection from oxidative stress-induced apoptosis upon contact inhibition have not been fully investigated. Previous research has indicated a role for mitogen-activated protein kinases (MAPKs) in the regulation of contact inhibition, and these proteins have also been associated with cell cycle regulation and stress-induced apoptosis. The potential role of the MAPK JNK-1 in the stress-response of actively proliferating and contact-inhibited cells was investigated. Actively proliferating normal fibroblasts (BJ) and fibrosarcoma cells (HT-1080) were stressed with H2O2, and levels of activated JNK-1 and cleaved PARP were ascertained. Similarly, these results were compared to levels of activated JNK-1 and cleaved PARP detected in H2O2-stressed confluent fibrosarcoma or contact-inhibited fibroblast cells. Contact-inhibited fibroblasts were protected from apoptosis in comparison to subconfluent fibroblasts, concurrent with decreased JNK-1 activation. Increased culture density of fibrosarcoma cells was not protective against apoptosis, and these cells did not demonstrate density-dependent alterations in the JNK-1 stress response. This decreased activation of JNK-1 in stressed, contact-inhibited cells did not appear to be dependent upon increased expression of MKP-1; however over-expression of MKP-1 was sufficient to result in a slight decrease in H2O2-stimulated PARP cleavage. Increasing the antioxidant capacity of fibroblasts through NAC-treatment lessened H2O2-stimulated JNK-1 activation, but also did not influence the expression of MKP-1. Taken together, these results suggest that regulation of negative regulation of JNK-1 upon contact inhibition is protective against apoptosis, and that this regulation is independent of MKP-1.
Keywords: JNK-1, contact inhibition, apoptosis, oxidative stress, MKP-1
Introduction
Mitogen-activated protein kinases (MAPKs) mediate the response of the cells to many external stimuli, ranging from growth factors to cellular stresses. Several families of MAPKs have been identified, including extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), and p38, whose activity is tightly controlled by the phosphorylation of conserved threonine and tyrosine residues within their kinase domains by specific MAP kinase kinases (MKKs) triggered by external stimuli. Once activated, MAPKs allow the cell to respond to external signals by phosphorylating a variety of substrates, including other downstream protein kinases, cytoskeletal proteins, and transcription factors that regulate gene expression [1, 2]. The activity of MAPKs is kept in check through negative regulation by mitogen-activated protein kinase phosphatases (MKPs). MKPs are dual specificity phosphatases, capable of dephosphorylating MAPKs on both phosphothreonine and phosphotyrosine residues, inactivating them. Over 10 MKPs have been characterized, though their specificities for different MAPKs vary. MKP-1 and -2 selectively bind and inhibit ERK, JNK, and p38 kinases, while MKP-3 is a specific inactivator of ERK MAPKs [3,4].
MAPK pathways have been shown to play critical roles in the regulation of both cellular proliferation and apoptosis [5–7]. Likewise, the importance of protein phosphatases in MAPK regulation has become clear [8]. The mRNAs of many MKPs are induced following stimulation with a variety of mitogens and stresses, which also activate MAP kinases, and are undetectable in quiescent cells, suggesting a role for MKPs in the feedback regulation of MAP kianses after extracellular stimulation [9, 10]. Tight interactions between MAP kinases and their substrates and regulators are critical for the control of signaling pathways. Docking sites have been identified on MAP kinases that facilitate their interactions with MKPs [11]. Additionally, scaffold proteins have also been shown to act as tethers, allowing complexes of signaling molecules to be assembled in response to specific stimuli, permitting MAP kinases, their regulatory proteins, and their substrates, to become localized for rapid and specific pathway regulation [11, 12].
Modulation of the activity of MAPKs has been implicated in contact-inhibited growth control. Many primary cultured cells will demonstrate contact inhibition upon the formation of a confluent monolayer, regardless of the presence of growth factors. This density-dependent negative regulation is believed to be caused by a combination of signaling through soluble polypeptides in the environment of the cell, and by cell-cell contact [13]. Contact inhibition of normal human fibroblasts results in increased expression of MKP-1, -2, and -3, and attenuation of ERK activity [14]. Similarly, p38 activity is attenuated upon contact inhibition in normal fibroblasts, while fibrosarcoma cells, which lack contact-inhibited growth control, do not demonstrate density-dependent alterations in p38 or ERK activity [14, 15]. Upon contact inhibition both in normal fibroblasts and in endothelial cells, there is modulation to a less reducing redox state [16]. Redox state changes have been shown to influence the activity of a variety of proteins, including kinases, phosphatases, and transcription factors [17, 18]. Oxidants stimulate the transcription of a variety of phosphatases, including MKP-1 [19]. In addition, obtaining a confluent state has been demonstrated to be protective against stress-induced apoptosis [20, 21]. It is likely that the natural redox state changes seen in normal fibroblast cells undergoing the transition to a contact-inhibited state may influence the activity of MAP kinases or MKPs, and the resulting modulation of MAPK pathways may play a role in the ability of cells to respond to exogenous oxidative stress. In this study, the activity of MAP kinases and the expression of MKPs following treatment with oxidative stress were measured in actively proliferating cell cultures in comparison to those in a contact inhibited state. Concurrently, the ability of cell cultures at different densities to survive stress was assessed. Contact inhibition of normal human fibroblasts (BJ) was found to be protective against oxidative-stress induced apoptosis, and was correlated with decreased JNK activity. Increased culture density was not protective of fibrosarcoma cells (HT-1080), which lack contact-inhibited growth control.
Materials and Methods
Cell lines and culture conditions
Cells were maintained as anchorage-dependent monolayers in Dulbecco’s Modified Eagle’s Medium (DMEM) (Life Technologies, Gaithersburg, MD, USA) supplemented with 10% fetal bovine serum (Atlanta Biologicals, Lawrenceville, GA, USA) at 37 C in a humidified atmosphere containing 5% CO2. Medium was replaced every three days to prevent serum-starvation. The BJ cell line, normal human foreskin fibroblasts, and HT-1080 fibrosarcoma cells were purchased from ATCC (Manassas, VA, USA). Culture density was determined by direct counting with a hemocytometer, utilizing trypan blue dye exclusion. Confluent BJ cells achieved an average density of 3.4×106 cells/dish (100 mm dish); subconfluent BJ cells averaged 1.7×106 cells/dish. Confluent HT-1080 cells achieved a density of 14.1×106 cells/dish (100 mm dish); subconfluent HT-1080 cells averaged 4.1×106 cells/dish. For stress treatment, BJ or HT-1080 cell cultures (70% confluent) were serum-starved overnight or pre-treated with 5 mM NAC in serum-free DMEM overnight prior to treatment with 300 μM H2O2 for 10 min.
Antioxidant capacity assay
The total antioxidant capacity of normal human fibroblasts was measured using an antioxidant capacity kit (Cayman Chemical) that measures the ability of cellular lysates to inhibit ABTS oxidation. The protocol was followed per the manufacturer’s specifications, and the results were normalized for cell number.
Antibodies and western blot analysis
Polyclonal JNK-1 (C-17), MKP-1 (C-19), and MKP-3 (N-antibodies were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Monoclonal phospho-SAPK/JNK (p46, p54) antibodies were purchased from Cell Signaling (Danvers, MA, USA). Monoclonal FLAG-M2 and polyclonal actin antibodies were purchased from Sigma (St. Louis, MO, USA). Western blot analysis was performed as previously described [14]. Briefly, cell cultures were washed twice with ice-cold phosphate-buffered saline (PBS) and lysed in 800 μl of lysis buffer (20 mM N-2-hydroxyethylpiperazine-N1-2-ethane sulfonic acid (HEPES), pH 7.4, 50 mM β-glycerophosphate, 1% Triton X-100, 10% glycerol, 2 mM ethylene bis (oxyethylenenitrilo) tetraacetic acid (EGTA), 1 mM DTT, 10 mM sodium fluoride, 1 mM sodium orthovanadate, 2 μM leupeptin, μ2 M aprotinin, and 1 mM phenylmethylsulfonyl fluoride (PMSF). The lysates were clarified by centrifugation at 14,000 rpm for 10 min. 15 μg of each protein sample was resolved by electrophoresis through 10% Tris-glycine-SDS gels. Proteins were transferred onto polyvinylidene difluoride (PVDF) membranes (Millipore, Bedford, MA, USA) and enhanced chemiluminescence reagent (ECL Plus, Amersham, Uppsala, Sweden) was used for the detection of the immunoreactive bands. For reprobing, blots were stripped by incubation for 15 min at 50 °C in a buffer containing 50 mM Tris-HCl pH 6.8, 2% sodium dodecyl sulfate (SDS), and 0.1 M β-mercaptoethanol. Densitometry was performed on reactive bands utilizing the public domain NIH Image program (developed at the U.S. National Institutes of Health and available on the Internet at http://rsb.info.nih.gov/nih-image/).
Plasmids and transfection
The construction of the pSR α-FLAG and pSR α-FLAG-MKP-1 vectors have previously been described [22]. For transfection, 1.3 × 105 HT-1080 fibrosarcoma cells were plated into 100 mm dishes. After 24 hrs, 4 μg of pSR α-FLAG or pSR α-FLAG-MKP-1 were transfected using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA).
Results
Subconfluent fibroblasts have an increased antioxidant capacity
Antioxidant capacity has been linked to enhanced survival from oxidative stress stimulus. The antioxidant defense system of cells is composed of enzymes, such as superoxide dismutase, catalase, and glutathione peroxidase, as well as small molecules, such as glutathione, that scavenge free radicals [23]. Subconfluent normal fibroblasts have increased amounts of glutathione [16]. To determine if these cells also have an overall enhanced antioxidant capacity, the antioxidant capacity of both subconfluent and confluent normal fibroblasts (BJ) was compared using an antioxidant capacity detection system that measures the ability of cellular lysates to inhibit ABTS oxidation. Subconfluent normal fibroblasts were found to have a much greater antioxidant capacity (Fig. 1).
Fig. 1.
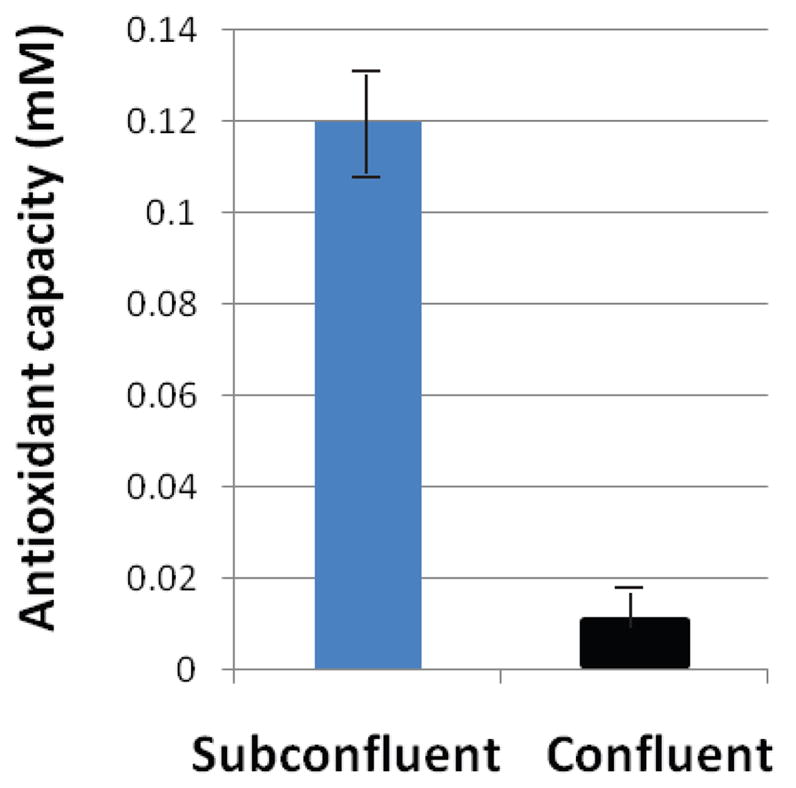
Subconfluent fibroblasts (BJ) have an increased capacity of antioxidants in comparison to confluent, contact-inhibited cultures. Antioxidant capacity was measured by the ability of cellular lysates to inhibit ABTS oxidation (Antioxidant Assay Kit, Cayman). Results shown are an average of 8 experiments, and are normalized for cell number.
H2O2-induced activation of JNK is dampened in contact-inhibited fibroblasts
JNK has been shown to be activated upon treatment with H2O2, and plays a role in oxidative-induced apoptosis [19]. The activation of JNK following H2O2 treatment was assessed in both subconfluent and confluent, contact-inhibited fibroblasts (BJ). Although pJNK levels were almost undetectable in both control subconfluent and contact inhibited fibroblasts, pJNK levels were elevated following oxidative stress treatment with 300 μM H2O2 for 10 minutes. However, pJNK expression was higher in the subconfluent treated cells, despite their increased antioxidant capacity, than in the confluent treated cells (Fig. 2a). Densitometric analysis was used to quantify detected bands, and the differences in pJNK expression were found to be statistically significant through ANOVA (Fig. 2b). The expression of total JNK in treated and untreated cultures did not change with increased culture density (Fig. 2a). Equivalency of loading was shown by the Coomassie blue stained membranes and re-probing with actin (Fig. 2a). In comparison, increased culture density does not influence the H2O2-induced activation of JNK in fibrosarcoma cells (HT-1080). Fibrosarcoma cells do not demonstrate contact-inhibited growth control, and previously, it was found that these cells maintain elevated activation of JNK and ERK despite reaching confluency [14, 15]. When treated fibrosarcoma cells were treated with 300 μM H2O2, pJNK levels were elevated to equal levels in both subconfluent and confluent treated cultures. The overall JNK levels, however, remained constant (Fig. 3a). Equivalency of loading is shown by the Coomassie blue stained membranes and re-probing with actin (Fig. 3b).
Fig. 2.
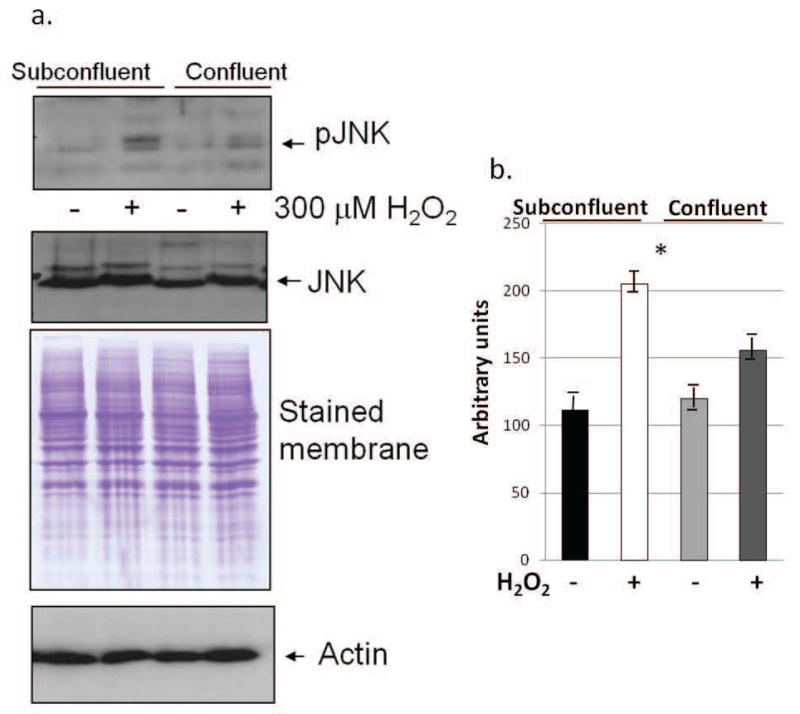
pJNK levels are higher in H2O2-treated subconfluent fibroblasts (BJ) than in treated confluent, contact-inhibited BJ cells, although JNK levels are constant. (a) Western blot analysis in H2O2-treated and control subconfluent and confluent cultures. Equivalency of loading is shown by the Coomassie blue stained membrane and reprobing with actin. Results shown are representative of 10 independent experiments. (b) Average densitometry of pJNK results is shown (from all independent experiments and normalized to actin). S = subconfluent, C = confluent. Results were found to be significant (F<0.001) through ANOVA.
Fig. 3.
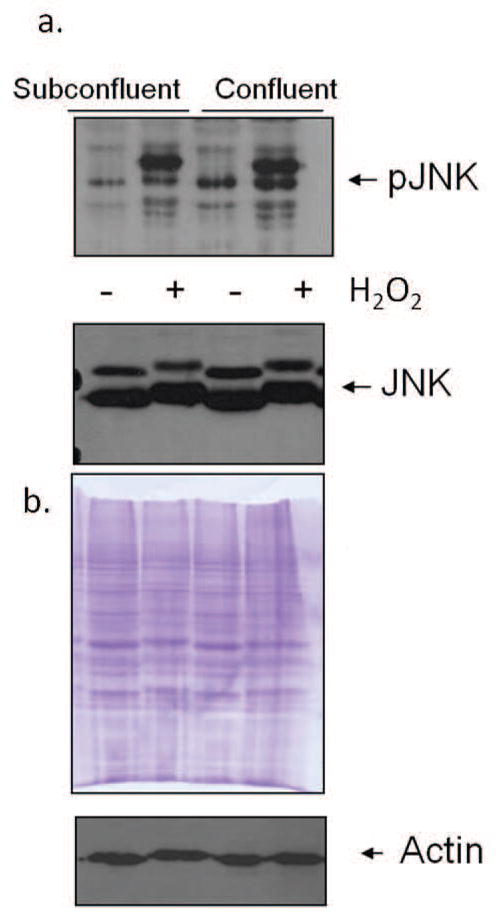
pJNK and total JNK levels are similar in H2O2-treated subconfluent and confluent fibrosarcoma (HT-1080) cells. (a) Western blot analysis in H2O2-treated and control subconfluent and confluent cultures. Results shown are representative of 10 independent experiments. (b) Equivalency of loading is shown by the Coomassie blue stained membrane and reprobing with actin.
Expression of MKP-1 is increased upon H2O2 treatment in both subconfluent and confluent cells
MKP-1 is involved in feedback regulation of MAPK activity. Stressing subconfluent or confluent cells fibroblasts (BJ) with H2O2 resulted in a slight increase in the level of MKP-1 (Fig. 4a). MKP-1 did not significantly increase upon H2O2 treatment in subconfluent or confluent fibrosarcoma cells (HT-1080) (Fig. 4b). Equivalency of loading is shown by detection of actin and the Coomassie blue stained membranes.
Fig. 4.
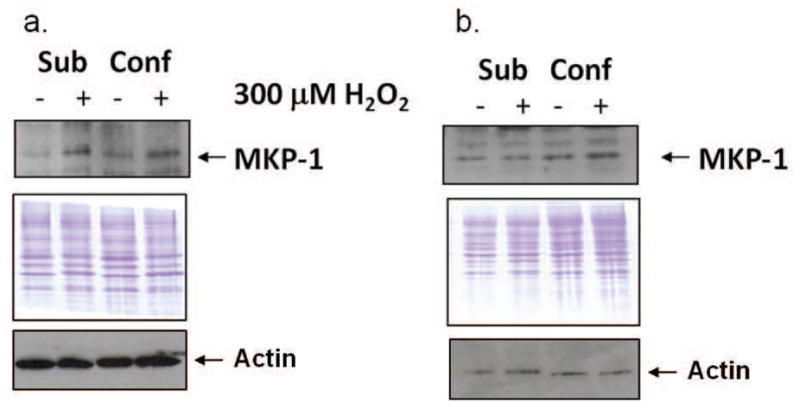
Expression of MKP-1 increases in both subconfluent and confluent, contact-inhibited fibroblasts (BJ), but not fibrosarcoma cells, upon treatment with H2O2. Western blot analysis in H2O2-treated and control subconfluent and confluent cultures of normal fibroblasts (a) and fibrosarcoma cells (b). Results shown are representative of 4 independent experiments. Equivalency of loading is shown by the Coomassie blue stained membranes. Sub = subconfluent; Conf = confluent.
Contact-inhibited fibroblast cells have less H2O2-stimulated apoptosis
Poly(ADP-ribose) polymerase (PARP) is cleaved early during apoptosis by caspase-3 [24]. Cleaved PARP (85 kDa) was detected in untreated and H2O2-treated subconfluent and confluent fibroblasts and fibrosarcoma cells. H2O2-stimulation of subconfluent fibroblasts resulted in a substantial increase in cleaved PARP in comparison to control cultures. Contact-inhibited fibroblast cultures showed only a slight increase in cleaved PARP upon stress treatment, reflecting less apoptosis (Fig. 5a). This was also confirmed through visual inspection of the cultures, with apoptotic cultures exhibiting over 80% of the cells appearing thinner and more loosely attached to the culture dishes. These results were found to be statistically significant through ANOVA (Fig. 5b). In contrast, increased culture density of fibrosarcoma cells (HT-1080) did not appear to be protective against H2O2-stimulated apoptosis. Cleavage of PARP was robustly seen in both confluent and subconfluent cultures following H2O2-treatment (Fig. 5c).
Fig. 5.
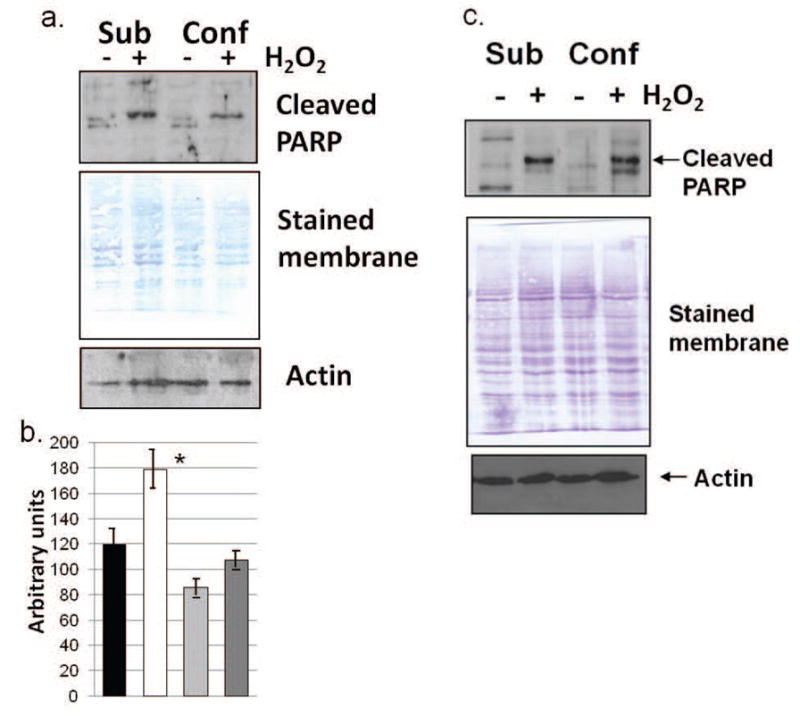
Cleaved PARP is present at higher levels in H2O2-treated subconfluent fibroblasts (BJ) than in treated confluent, contact inhibited fibroblasts. (a) Western blot analysis in H2O2-treated and control subconfluent and confluent fibroblast cultures. Equivalency of loading is shown by the Coomassie blue stained membrane and reprobing with actin. Results shown are representative of 10 independent experiments. (b) The average densitometry of cleaved PARP results from normal fibroblasts is shown (from all independent experiments and normalized to actin). Significant variance (F<0.001) was determined through use of ANOVA. (c) Cleaved PARP is present at equal levels in treated subconfluent and confluent fibrosarcoma (HT-1080) cells as shown through western blot analysis. Results shown are representative of 8 independent experiments. Equivalency of loading is shown by the Coomassie blue stained membrane and reprobing with actin. Sub = subconfluent; Conf = confluent.
Increasing the antioxidant capacity of fibroblasts through NAC-treatment lessens H2O2-stimulated JNK activation
N-acetylcysteine (NAC) increases the antioxidant capacity by increasing GSH [16]. Cells were pre-treated with 5 mM NAC for 24 h, then stressed with 300 μM H2O2 for 10 min. JNK became phosphorylated in normal fibroblast cultures upon treatment with H2O2, however, cells pre-treated with NAC showed a slight decrease in JNK activation. (Fig. 6a). Total JNK increased upon treatment with H2O2, but JNK levels were not influenced by NAC treatment (Fig. 6a, middle panel). Pre-treatment with NAC also did not significantly prevent PARP cleavage in H2O2-treated fibroblasts (lower panel). As JNK was still phosphorylated with the NAC treatment, it is apparent that this treatment was not robust enough to prevent PARP cleavage. H2O2-stimulation also increased phosphorylation of JNK in fibrosarcoma cells, however, NAC pre-treatment did not demonstrate an effect on JNK phosphorylation (Fig. 6c). Similarly, in fibrosarcoma cells, total JNK levels were not influenced by either H2O2 treatment or pre-treatment with NAC. Pre-treatment with NAC also did not prevent PARP cleavage in H2O2-treated fibrosarcoma cells. Equivalency of loading is shown by the Coomassie blue stained membranes and re-probing with actin (Fig. 6).
Fig. 6.
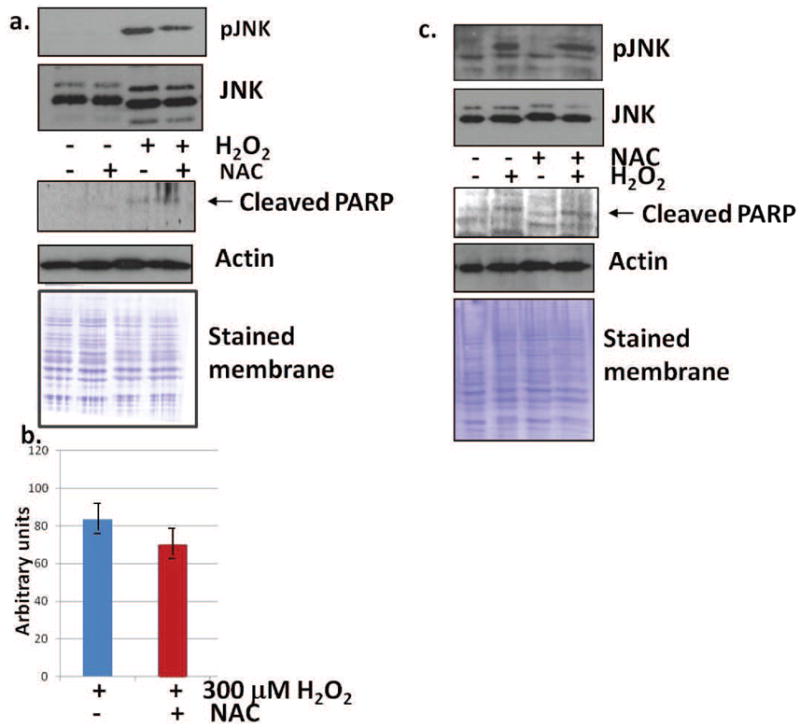
JNK activation is slightly lessened by NAC treatment in normal fibroblasts. (a) Western blot analysis demonstrated that JNK was phosphorylated with H2O2 treatment, but not with NAC treatment alone. Pre-treatment with NAC, followed by stressing with H2O2 resulted in slightly less activation of JNK. Results shown are representative of 10 independent experiments. Total levels of JNK were slightly higher with peroxide treatment. (a, middle panel) PARP cleavage is not prevented by pre-treatment with NAC (a, lower panel, representative of 4 independent experiments). Equivalency of loading is shown by the Coomassie blue stained membrane and reprobing with actin. (b) Densitometry of H2O2-treated pJNK in normal fibroblasts (BJ). (c) pJNK is equally activated by H2O2 in fibrosarcoma cells (HT-1080) pre-treated with NAC or control cultures, as shown by western blot analysis. Total JNK is also found at equal levels in control, H2O2-treated, NAC-treated, or cultures pre-treated with NAC and stressed with H2O2. Results shown are representative of 8 independent experiments. PARP cleavage is not prevented by pre-treatment with NAC (lower panel, representative of 4 independent experiments). Equivalency of loading is shown by the Coomassie blue stained membrane and reprobing with actin.
Expression of MKP-1 was not influenced by increasing the antioxidant capacity
Though expression of MKP-1 is slightly increased upon treatment with H2O2 in normal fibroblasts (Fig. 4a), treatment with NAC does not significantly increase the expression of MKP-1 (Fig. 7). Similarly, treatment with NAC 24 hr prior to H2O2-stimulation also does not alter MKP-1 expression in comparison to H2O2-stressed cells (Fig. 7), suggesting that the decrease seen in JNK activity upon pre-treatment with NAC is not due to feedback regulation by this phosphatase, but may be the effect of other phosphatases or the result of alterations in upstream activators.
Fig. 7.
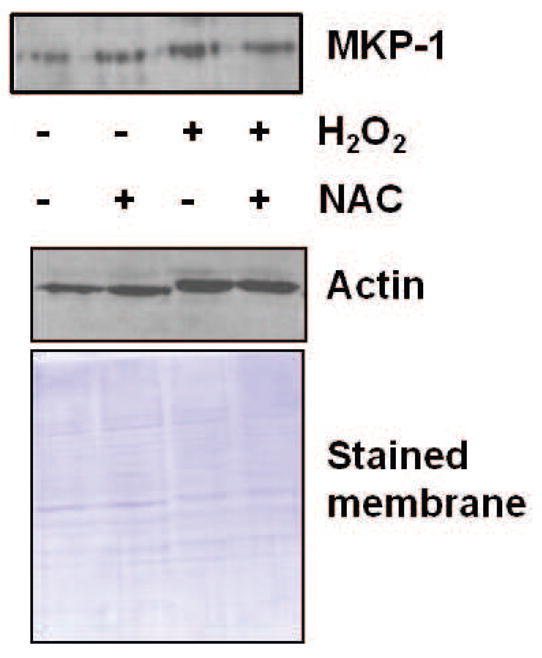
MKP-1 levels were not altered with NAC treatment in normal fibroblasts. Western blot analysis was performed to detect MKP-1 in fibroblast cultures following treatment with 300 μM H2O2, 5 mM NAC, or pre-treatment with 5 mM NAC followed by stressing with 300 μM H2O2. Results shown are representative of 12 independent experiments. Equivalency of loading is shown by the Coomassie blue stained membrane and reprobing with actin.
Limiting MAPK activation diminishes apoptosis
Contact-inhibited fibroblast cells demonstrated both decreased H2O2-stimulated activation of JNK and decreased apoptosis (Figs. 2 and 5). To determine the involvement of MAPK activation in the oxidative stress-stimulated apoptosis of subconfluent normal fibroblasts, subconfluent normal fibroblasts were transfected with FLAG, FLAG-MKP-1, or were mock transfected. Cleaved PARP was not detectable in untreated lysates, however, PARP cleavage was evident following H2O2-stimulation. Transfection with FLAG-MKP-1 resulted in a reduction in the level of cleaved PARP (Fig. 8). Though the expression of cleaved PARP was only slightly reduced by MKP-1 expression, it should be noted that FLAG-MKP-1 is a relatively unstable protein [22], and this reduction in cleaved PARP expression was seen despite very low levels of FLAG-MKP-1 expression in the cells (Fig. 8).
Fig. 8.
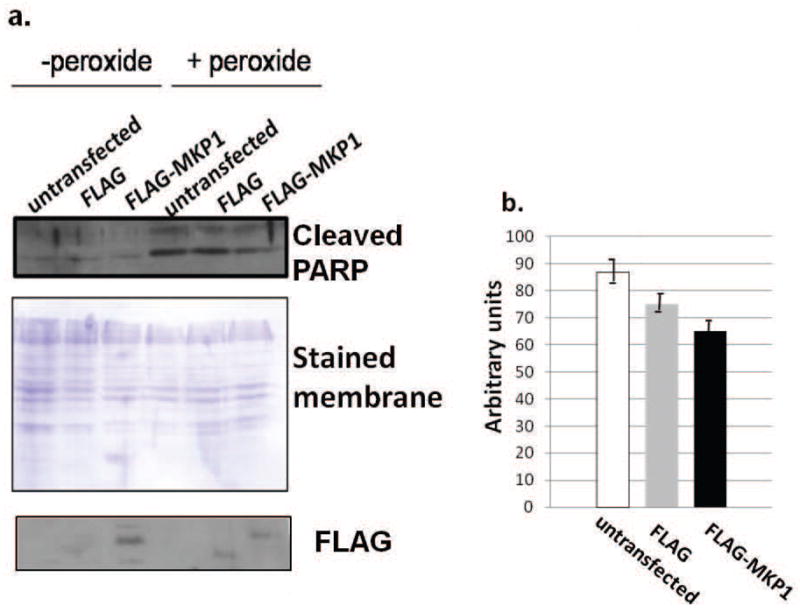
Exogenous expression of FLAG-MKP-1 limits apoptosis. (a) Fibrosarcoma cells were mock-transfected, or were transfected with FLAG or FLAG-MKP-1 expression vectors. Western blot analysis was performed to detect cleaved PARP and FLAG in control transfected cultures, or in transfected cultures treated with 300 μM H2O2. Equivalency of loading is shown by the Coomassie blue stained membrane. Results are representative of five independent experiments. (b) Average densitometry of cleaved PARP bands following treatment with H2O2.
Discussion
Oxidative stress has been linked to many age-related disorders as well as apoptosis. The production of free radicals due to dysregulation of the electron transport chain can cause extensive damage to proteins, lipids, and DNA [25]. The redox state of the cell is balanced between the production of reactive oxygen species (ROS) and antioxidant defense systems. The presence of intracellular glutathione (GSH), which scavenges free radicals, plays a protective antioxidant role. Proliferating, normal human fibroblasts have increased GSH compared to contact-inhibited cells [16]. In addition to GSH, the antioxidant capacity is also maintained by a variety of enzymes that work to detoxify free radicals, including catalase, superoxide dismutase, and glutathione peroxidase [25]. The antioxidant capacity, reflecting the combined ability of the antioxidant systems in the cell to protect against ROS, was measured in subconfluent and contact-inhibited normal human fibroblasts. Subconfluent fibroblasts maintained a much greater antioxidant capacity than contact-inhibited cells (Fig. 1). Natural changes in the redox state which occur upon contact inhibition may greatly influence the activity of proteins. Activation of ERK and p38 is curtailed in contact-inhibited cells, and was correlated with increased levels of MKPs [14, 15]. Modulation of the redox state of normal human fibroblasts with buthionine sulfoximine (BSO), an inhibitor of glutathione synthesis, was found to transiently activate ERK and induce MKP-1 [14], suggesting that redox changes upon contact inhibition may, in part, regulate the activity of MAPKs.
Modulation of MAP kinase regulation during the transition to a contact inhibited state may influence the cell’s ability to respond to a more potent exogenous stress. In support, cell density has been shown to influence the ability of cells to survive stress. Sparse cultures of endothelial cells have been found to be more susceptible than dense cultures to apoptosis induced by low serum because of decreased cell levels of FGF-2 on their cell surface [20]. Activation of FGF involves the ERK pathway [26], which may imply that density-dependent changes in ERK regulation are key to survival under this condition. Confluent fibroblasts failed to activate p38 and exhibited resistance to apoptosis following H2O2 treatment, implying a relationship between cell cycle state and anti-oxidative defense [21]. Confluence has also been shown to delay apoptosis in human DNA repair deficient cell lines [27]. Cell-cell contact may influence signaling pathways necessary for stress survival: high cell density attenuates the response of p53 to DNA damage [28] and contact between multiple myeloma and osteoclasts enhances multiple myeloma cell growth and survival [29]. In this work, the transition to a contact-inhibited state was correlated with a decrease in the level of H2O2-stimulated JNK1 phosphorylation (Fig. 2a). Concurrently, a decrease was also seen in oxidative stress-stimulated apoptosis (Fig. 5a), shown by less cleaved PARP in confluent than subconfluent H2O2-treated cells. Activation of JNK has been implicated in apoptosis in a variety of cell lines and stress conditions [30, 31]. The regulation of the JNK pathway in contact-inhibited normal fibroblasts results in less activation of JNK and subsequently less apoptosis than subconfluent, actively proliferating cultures. Previously, it was shown that levels of MKP-1 increase upon contact inhibition [14]. Such an increase in MKP-1 could curtail H2O2-stimulated JNK-1 activation in contact-inhibited cultures. However, when MKP-1 levels were quantified following stress treatment of subconfluent and contact-inhibited fibroblasts, no significant difference in MKP-1 expression was seen between subconfluent and contact-inhibited cells - expression of MKP-1 was equally enhanced by H2O2 treatment (Fig. 4a). In contrast, expression of MKP-1 was not significantly enhanced by H2O2-treatment in either subconfluent or confluent fibrosarcoma cells (Fig. 4b), suggesting that feedback regulation of MKP-1 is not triggered by oxidative stress in these cells. In addition, treatment of normal fibroblasts with the antioxidant NAC does not alter MKP-1 expression (Fig. 7). Together, these results suggest that the decreased H2O2-stimulated JNK-1 activation in contact-inhibited cells is more likely a result of decreased upstream activation of JNK-1 or possibly the result of deactivation by other phosphatases. However, the possibility that MKP-1 activity may be regulated by post-translational modifications following H2O2-treatment of these cells has not been fully explored. In addition to MKP-1, other dual specificity phosphatases, including MKP-2, have been implicated in feedback regulation of JNK-1 [3].
Feedback regulation of MAPK pathways is essential to their regulation, and such curtailment of MAPK activity may limit apoptosis. Stimulation of MAPK activity by oxidative stress is transient as such stress, in addition to causing phosphorylation of MAPKs, also tends to activate transcription of the MKPs that regulate them [2, 3, 5]. Activation of MKPs following oxidative stress may be key in limiting apoptosis. Overexpression of MKP-1 in fibrosarcoma cells slightly decreased apoptosis, as shown by the decrease in cleaved PARP in H2O2-treated cells (Fig. 8). These results may have been hindered by poor expression of MKP-1, with FLAG-MKP-1 being expressed at low levels (Fig. 8). As found in previous work, MKP-1 is an unstable protein which is difficult to expression in eukaryotic cells [22]. However, with minimal expression of MKP-1, a slight decrease in cleaved PARP was still detectable. Expression of MKPs in other cellular systems has been shown to be protective against apoptosis. Induction of MKP-1 has also been shown to be protective against H2O2-induced apoptosis of rat mesangial cells [32], repression of MKP-1 expression by anthracyclines activates antiapoptotic p44/p42 MAPK phosphorylation in breast carcinoma cells [33], and over-expression of MKP-1 was partially protective from apoptosis following ischemia-reperfusion in mice [34]. Stress-stimulated apoptosis has also been shown to be prevented by pre-treatment with antioxidants. NAC, specifically, has been used to prevent dopamine-induced melanocyte death [35] and pre-treatment of alveolar epithelial cells with NAC prevented H2O2-induced apoptosis by suppressing JNK activation [36]. Increasing the antioxidant capacity of fibroblasts through NAC-treatment was found to lessen H2O2-stimulated JNK activation (Fig. 6a). In summary, H2O2-treatment of subconfluent, normal fibroblasts leads to apoptosis due to activation of the JNK pathway. Contact inhibition of normal fibroblasts attenuates the activation of JNK, and is protective against apoptosis.
Acknowledgments
We wish to thank Mary Grace Baker, Julia Arpino, and Ruth Adekunle for technical assistance. This work was supported by grant R15GM076076 from the National Institute of General Medical Sciences. The content is solely the responsibility off the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health.
References
- 1.Robinson MJ, Cobb MH. Mitogen-activated protein kinase pathways. Curr Opin Cell Bio. 1997;9:180–186. doi: 10.1016/s0955-0674(97)80061-0. [DOI] [PubMed] [Google Scholar]
- 2.Pelech SL. Networking with protein kinases. Curr Biol. 1993;3:513–515. doi: 10.1016/0960-9822(93)90043-n. [DOI] [PubMed] [Google Scholar]
- 3.Theodosiou A, Ashworth A. MAP kinase phosphatases. Genome Biol. 2002;3:1–10. doi: 10.1186/gb-2002-3-7-reviews3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Slack DN, Seternes OM, Gabrielsen M, Keyse SM. Distinct binding determinants forERK2/p38α and JNK MAP kinases mediate catalytic activation and substrate selectivity of MAP kinase phosphatase-1. J Biol Chem. 2001;276:16491–16500. doi: 10.1074/jbc.M010966200. [DOI] [PubMed] [Google Scholar]
- 5.Keshet Y, Seger R. The MAP kinase signaling cascades: a system of hundreds of components regulates a diverse array of physiological functions. Methods Mol Biol. 2010;661:3–38. doi: 10.1007/978-1-60761-795-2_1. [DOI] [PubMed] [Google Scholar]
- 6.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 7.Pelech SL, Charest DL. MAP kinase-dependent pathways in cell cycle control. Prog Cell Cycle Res. 1995;1:33–52. doi: 10.1007/978-1-4615-1809-9_4. [DOI] [PubMed] [Google Scholar]
- 8.Zhang Z-Y, Zhou B, Xie L. Modulation of protein kinase signaling by protein phosphatases and inhibitors. Pharm Therapeutics. 2002;93:307–317. doi: 10.1016/s0163-7258(02)00199-7. [DOI] [PubMed] [Google Scholar]
- 9.Keyse SM. Protein phosphatases and the regulation of mitogen-activated protein kinase signaling. Curr Opin Cell Biol. 2000;12:186–192. doi: 10.1016/s0955-0674(99)00075-7. [DOI] [PubMed] [Google Scholar]
- 10.Liu Y, Gorospe M, Yang C, Holbrook NJ. Role of mitogen-activated protein kinase phosphatase during the cellular response to genotoxic stress. Inhibition of c-Jun N-terminal kinase activity and AP-1 dependent gene activation. J Biol Chem. 1995;270:8377–8380. doi: 10.1074/jbc.270.15.8377. [DOI] [PubMed] [Google Scholar]
- 11.Tanoue T, Nishida E. Molecular recognitions in MAP kinase cascades. Cellular Signalling. 2003;15:455–462. doi: 10.1016/s0898-6568(02)00112-2. [DOI] [PubMed] [Google Scholar]
- 12.Weston CR, Davis RJ. The JNK signal transduction pathway. Curr Opin Genetics Dev. 2002;12:14–21. doi: 10.1016/s0959-437x(01)00258-1. [DOI] [PubMed] [Google Scholar]
- 13.Holley RW. Control of growth of mammalian cells in culture. Nature. 1975;258:487–490. doi: 10.1038/258487a0. [DOI] [PubMed] [Google Scholar]
- 14.Wayne J, Sielski J, Rizvi A, Georges K, Hutter D. ERK regulation upon contact inhibition in fibroblasts. Mol Cell Biochem. 2006;286:181–189. doi: 10.1007/s11010-005-9089-z. [DOI] [PubMed] [Google Scholar]
- 15.Slisz M, Rothenberger E, Hutter D. Attenuation of p38 MAPK activity upon contact inhibition in fibroblasts. Molecular and Cellular Biochemistry. 2007;308:65–73. doi: 10.1007/s11010-007-9613-4. [DOI] [PubMed] [Google Scholar]
- 16.Hutter DE, Till BG, Greene JJ. Redox state changes in density-dependent regulation of proliferation. Exp Cell Res. 1997;232:435–438. doi: 10.1006/excr.1997.3527. [DOI] [PubMed] [Google Scholar]
- 17.Kamata H, Hirata H. Redox regulation of cellular signaling. Cell Signal. 1999;11:1–14. doi: 10.1016/s0898-6568(98)00037-0. [DOI] [PubMed] [Google Scholar]
- 18.Hutter D, Greene JJ. Influence of the cellular redox state on NF-KB-regulated gene expression. J Cell Physiol. 2000;183:45–52. doi: 10.1002/(SICI)1097-4652(200004)183:1<45::AID-JCP6>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 19.Mendelson KG, Contois LR, Tevosian SG, Davis RJ, Paulson KE. Independent regulation of JNK/p38 mitogen-activated protein kinases by metabolic oxidative stress in the liver. Proc Natl Acad Sci USA. 1996;93:12908–23913. doi: 10.1073/pnas.93.23.12908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kinoshita M, Shimokado K. Autocrine FGF-2 is responsible for the cell-density-dependent susceptibility to apoptosis of HUVEC. Arterioscler Thromb Vasc Biol. 1999;19:2323–2329. doi: 10.1161/01.atv.19.10.2323. [DOI] [PubMed] [Google Scholar]
- 21.Naderi J, Hung M, Pandey S. Oxidative stress-induced apoptosis in dividing fibroblasts involves activation of p38 MAP kinase and over-expression of Bax: resistance of quiescent cells to oxidative stress. Apoptosis. 2003;8:91–100. doi: 10.1023/a:1021657220843. [DOI] [PubMed] [Google Scholar]
- 22.Hutter D, Chen P, Barnes J, Liu Y. Catalytic activation of mitogen-activated protein (MAP) kinase phosphatase-1 by binding to p38 MAP kinase: critical role of the p38 C-terminal domain in its negative regulation. Biochem J. 2000;352:155–163. [PMC free article] [PubMed] [Google Scholar]
- 23.Reddy SP. The antioxidant response element and oxidative stress modifiers in airway diseases. Curr Mol Med. 2008;8:376–383. doi: 10.2174/156652408785160925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boulares AH, Yakovlev AG, Ivanova V, Stoica BA, Wang G, Iyer S, Smulson M. Role of Poly(ADP-ribose) Polymerase (PARP) Cleavage in Apoptosis. J Biol Chem. 1999;274:22932–22940. doi: 10.1074/jbc.274.33.22932. [DOI] [PubMed] [Google Scholar]
- 25.Romano AD, Serviddio G, de Matthaeis A, Bellanti F, Vendemiale G. Oxidative stress and aging. J Nephrol. 2010;15:S29–36. [PubMed] [Google Scholar]
- 26.Harris VK, Coticchia CM, Kagan BL, Ahmad S, Wellstein A, Riegel AT. Induction of the angiogenic modulator fibroblast growth factor-binding protein by epidermal growth factor is mediated through both MEK/ERK and p38 signal transduction pathways. J Biol Chem. 2000;275:10802–10811. doi: 10.1074/jbc.275.15.10802. [DOI] [PubMed] [Google Scholar]
- 27.Carvalho H, de Costa RM, Chigancas V, Weinlich R, Brumatti G, Amarante-Mendes GP, Sarasin A, Menck CF. Effect of cell confluence on Ultraviolet light apoptotic responses in DNA repair deficient cells. Mutat Res. 2003;544:159–166. doi: 10.1016/j.mrrev.2003.06.004. [DOI] [PubMed] [Google Scholar]
- 28.Cohen-Noyman BJ, Geiger B, Oren M. Attenuation of the p53 response to DNA damage by high cell density. Oncogene. 2004;23:2128–2137. doi: 10.1038/sj.onc.1207325. [DOI] [PubMed] [Google Scholar]
- 29.Abe M, Hiura K, Wilde J, Shioyasono A, Moriyama K, Hashimoto T, Ki S, Oshima T, Shibata H, Ozaki S, Inuoue D, Matsumoto T. Osteoclasts enhance myeloma cell growth and survival via cell-cell contact: a vicious cycle between bone destruction and myeloma expansion. Blood. 2004;104:2484–2491. doi: 10.1182/blood-2003-11-3839. [DOI] [PubMed] [Google Scholar]
- 30.Mandlekar S, Yu R, Tan TH, Kong AN. Activation of caspase-3 and c-Jun NH2-terminal kinase-1 signaling pathways in tamoxifen-induced apoptosis of human breast cancer cells. Cancer Res. 2000;60:5995–6000. [PubMed] [Google Scholar]
- 31.Franklin CC, Srikanth S, Kraft AS. Conditional expression of mitogen-activated protein kinase phosphatase-1, MKP-1, is cytoprotective against UV-induced apoptosis. Proc Natl Acad Sci USA. 1998;95:3014–3019. doi: 10.1073/pnas.95.6.3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu Q, Konta T, Nakayama K, Furusu A, Moreno-Manzano V, Lucio-Cazana J, Ishikawa Y, Fine LG, Yao J, Kitamura M. Cellular defense against H2O2-induced apoptosis via MAP kinase-MKP-1 pathway. Free Rad Biol Med. 2004;36:985–993. doi: 10.1016/j.freeradbiomed.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 33.Small GW, Somasundaram S, Moore DT, Shi YY, Orlowski RZ. Repression of mitogen-activated protein kinase (MAPK) phosphatase-1 by anthracyclines contributes to their antiapoptotic activation of p44/p42-MAPK. J Pharmacol Exp Ther. 2003;307:861–869. doi: 10.1124/jpet.103.055806. [DOI] [PubMed] [Google Scholar]
- 34.Kaiser RA, Bueno OF, Lips DJ, Doevendans PA, Jones F, Kimball TF, Molkentin JD. Targeted inhibition of p38 mitogen-activated protein kinase antagonizes cardiac injury and cell death following ischemia-reperfusion in vivo. J Biol Chem. 2004;279:15524–15530. doi: 10.1074/jbc.M313717200. [DOI] [PubMed] [Google Scholar]
- 35.Choi HR, Shin JW, Lee HK, Kim JY, Huh CH, Youn SW, Park KC. Potential redox-sensistive Akt activation by dopamine activates Bad and promotes cell death in melanocytes. Oxid Med Cell Longev. 2010;3:219–224. doi: 10.4161/oxim.3.3.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fu YQ, Fang F, Lu ZY, Kuang FW, Xu F. N-acetylcysteine protects alveolar epithelial cells from hydrogen peroxide-induced apoptosis through scavenging reactive oxygen species and suppressing c-Jun N-terminal kinase. Exp Lung Res. 2010;36:352–361. doi: 10.3109/01902141003678582. [DOI] [PubMed] [Google Scholar]