

Abstract
Hsp104 in yeast and ClpB in bacteria are homologous, hexameric AAA+ proteins and Hsp100 chaperones that function in the stress response as ring-translocases that drive protein disaggregation and reactivation. Both Hsp104 and ClpB contain a distinctive coiled-coil middle domain (MD) inserted in the first AAA+ domain, which distinguishes them from other AAA+ proteins and Hsp100 family members. Here, we focus on recent developments concerning the location and function of the MD in these hexameric molecular machines, which remains an outstanding question. While the atomic structure of the hexameric assembly of Hsp104 and ClpB remains uncertain, recent advances have illuminated that the MD is critical for the intrinsic disaggregase activity of the hexamer and mediates key functional interactions with the Hsp70 chaperone system (Hsp70 and Hsp40) that empower protein disaggregation.
Introduction
Hsp104 and ClpB are homologous protein disaggregases, which are classified in the Hsp100 family of proteins [1–9]. This family, in turn, is a member of the AAA+ (ATPases Associated with various cellular Activities) super-family [10–12]. Hsp104, which is found in S. cerevisiae, has two main functions. First, in collaboration with Hsp70 and Hsp40, Hsp104 confers thermo- and chemical-tolerance to yeast by resolubilizing stress-induced protein aggregates (Fig. 1a) [13–18]. These aggregates are typically disordered or amorphous in structure [19, 20]. Additionally, Hsp104 can directly remodel amyloid and this activity governs prion inheritance in yeast (Fig. 1b) [21–33]. Prions, which are proteins that assume an infectious amyloid fold, are structurally distinct from disordered aggregates in that they form ordered assemblies with a characteristic ‘cross-β’ structure [20, 34–39]. Unlike their mammalian counterparts, yeast prions confer selective advantages, which are only made possible by the Hsp104-remodeling activities that facilitate stable prion inheritance through successive generations [22, 27, 40–42]. Curiously, Hsp104 is absent from metazoan lineages [43]. Thus, it has been suggested that the ability of Hsp104 to remodel amyloid conformers as well as toxic preamyloid oligomers might even be harnessed, engineered and potentiated for therapeutics against numerous neurodegenerative amyloidoses [6, 39, 43–45]. Despite sharing over 50% identity with Hsp104, the bacterial protein ClpB does not possess the same dual functionality as Hsp104. Like Hsp104, ClpB is able to disaggregate amorphous substrates in response to environmental stresses that induce widespread protein aggregation [46–50]. However, unlike Hsp104, ClpB appears to be ineffective at remodeling amyloid conformers [21, 51, 52].
Figure 1. In vitro disaggregase activity of Hsp104 and ClpB.
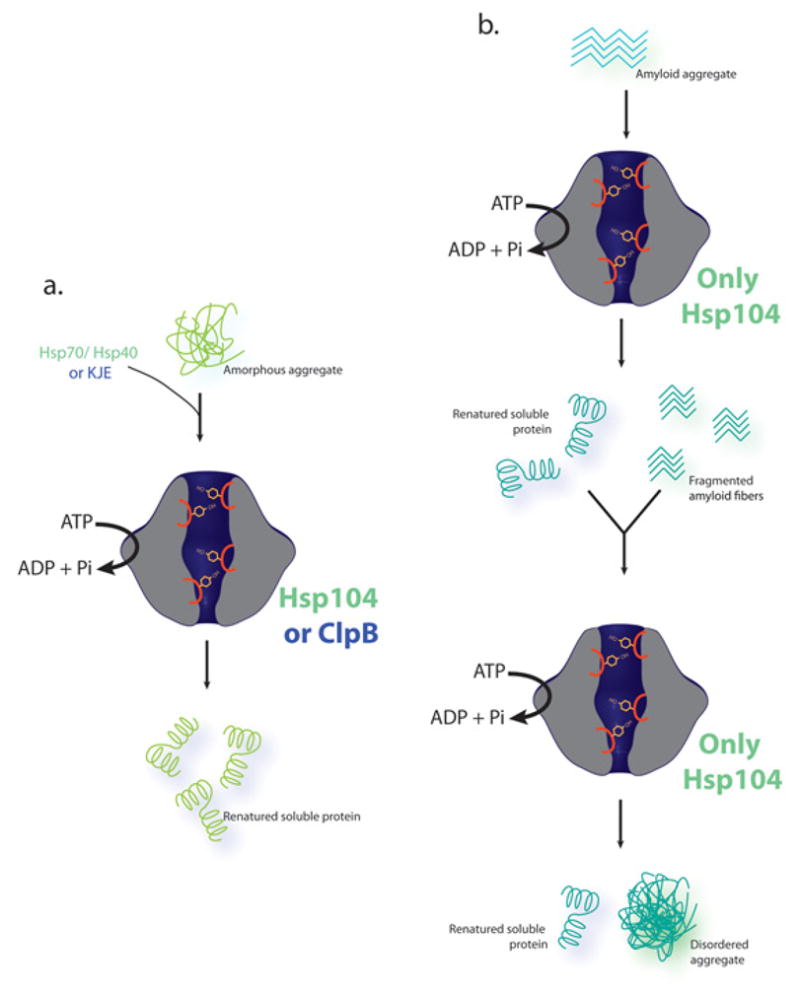
(a) Hsp104 remodels amorphous aggregates via collaboration with Hsp70 and Hsp40, while ClpB remodels these types of aggregates via collaboration with DnaK, DnaJ, and GrpE (KJE). The product of disaggregation of amorphous aggregates is natively folded protein. Hsp104 or ClpB hexamer is shown in gray with the front half cutaway to reveal the axial channel running down the length of the structure. Four pore loops with conserved tyrosines residues are shown in orange. These pore loops are important for substrate binding and threading through the axial channel. (b) Only Hsp104 is able to remodel amyloid aggregates and in vitro this can proceed without the aid of Hsp70 and Hsp40. Products of amyloid disaggregation are soluble natively folded protein and fragmented amyloids. However, for some amyloids (e.g. Sup35 prions) remodeling can continue to generate disordered-type aggregates that Hsp104 cannot remodel and which lack the seeding activity of amyloid [23].
Significant efforts have been made toward gaining a structural understanding of Hsp104 and ClpB. Like many AAA+ proteins, Hsp104 and ClpB are functional as ring-shaped hexamers [1, 53, 54], which are thought to drive protein disaggregation by directly translocating substrates through their central channel (Fig. 1a, b) [55–60]. However, there is still no general consensus about the gross domain organization within these hexameric molecular machines [53, 61–66]. Thus, how the hexamer couples conformational change to generate the mechanical force necessary to drive protein disaggregation continues to remain uncertain. A point of particular contention is the location and orientation of a unique coiled-coil insertion, termed the Middle Domain (MD), within the hexameric assembly. Despite a lack of detailed structural information, a number of studies have recently revealed key mechanistic insights about how ClpB and Hsp104 functionally interact with their respective Hsp70 chaperone system (Hsp70 and Hsp40), which is also activated during the stress response [18, 67–70]. Here, the elusive MD plays a critical role that mediates the functional interaction between Hsp104/ClpB and the Hsp70 chaperone system [67–69]. In this review, we will first outline the debate concerning the quaternary structural organization of Hsp104 and ClpB and then explore the implications that the MD mediates functional interactions with Hsp70.
Two Structural Models of Hsp104 and ClpB hexamers: Whither the Middle Domain?
Each Hsp104 or ClpB monomer contains an N-terminal domain, two AAA+ nucleotide-binding domains (NBD1 and NBD2), and a predicted coiled-coil middle domain (MD) that is inserted toward the C-terminal end of NBD1 (Fig. 2a) [1, 2, 71–73]. Additionally, Hsp104 contains a short C-terminal extension, absent from ClpB, that is required for Hsp104 hexamerization [74]. The tertiary structure of the individual domains was largely resolved in 2003 when a 3.0Å crystal structure of ClpB (TClpB) from the thermophilic eubacterium, Thermus thermophilus, was solved by Tsai and colleagues (Fig. 2b) [63]. These studies revealed that both NBDs adopt a canonical AAA+ fold [63]. The structure of NBD1 agreed with a previously solved structure of this isolated domain [75]. The N-terminal domain, which is the least conserved domain in the disaggregase family, was also structurally similar to the isolated E. coli ClpB N-terminal domain [76]. The MD was revealed as a broken anti-parallel coiled-coil (Fig. 2b, Fig. 3b) [63]. For the purpose of this review, we have provided a unified nomenclature to describe positions along the MD in ClpB, TClpB, and Hsp104 (Fig. 3a, b). The MD is less well conserved than NBD1 or NBD2, with Hsp104 and ClpB sharing only ~36% identity. Unfortunately, full-length TClpB did not crystallize in its functional hexameric structure, but rather in a trimeric spiral with protomer-protomer interactions that might, at least in part, reflect crystal contacts rather than native protomer interfaces. In a dynamic, multi-domain molecular machine like Hsp104/ClpB, understanding the relative domain positioning and protomer organization is absolutely critical to understanding the mechanism of action.
Figure 2. Predicted structures and hexameric models of Hsp104 and ClpB.

(a) Domain organization of one monomer of Hsp104 and ClpB. N-terminal domain (N) shown in purple, Nucleotide binding domain 1 (NBD1) shown in cyan, Middle domain (MD) in yellow, Nucleotide binding domain 2 (NBD2) in dark blue. Only Hsp104 has the short C-terminal extension (C) shown in green. Sequence numbering for ClpB is shown on top and for Hsp104 is shown on the bottom. (b) TClpB crystal structure. Domain coloring corresponds with part (a). A 180° rotation about the vertical axis is shown on the right. (c) The Tsai model for the hexameric quaternary structure of TClpB. The Tsai model, which was initially based on Cryo-EM envelopes generated with TClpB is shown on left. (d) The Saibil model, which used Hsp104 to generate Cryo-EM density, is shown in the middle. (e) The 6.93Å crystal structure of hexameric, full length ClpC is shown on the right. The adaptor protein MecA was omitted for clarity. A side view is shown on top and a view down the axial channel from the N-terminus is shown on the bottom. One subunit is colored as described in part (a). The other five subunits are in gray.
Figure 3. Middle domain nomenclature.
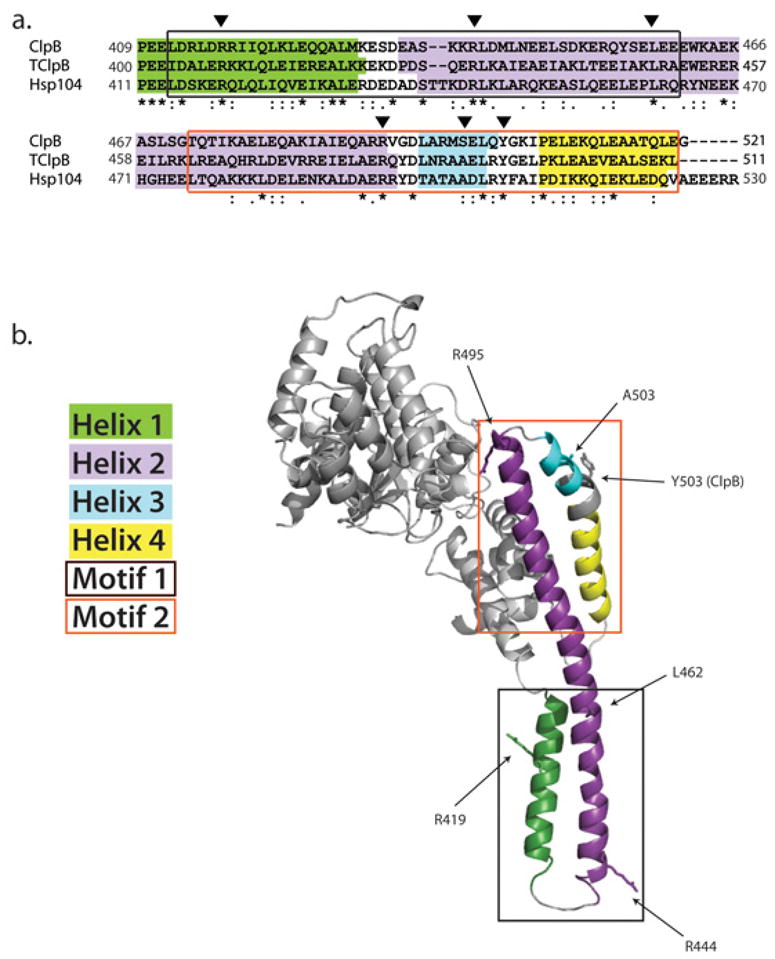
(a) An alignment of the MD from E. coli ClpB, T. thermophilus ClpB, and S. cerevisiae Hsp104. Helix 1 is colored in green, helix 2 is colored in purple, helix 3 is colored in light blue, and helix four is colored in yellow. Motif 1 (also called wing 2) is boxed in black while motif 2 (also called wing 1) is boxed in orange. Arrowheads denote key residues discussed in the text. (b) Close up of the MD in the TClpB crystal structure. Each helix and motif is colored as indicated in part (a). NBD2 is omitted for clarity. Arrows point to side chains (shown as sticks) of key residues discussed in the text.
The overall quaternary structure of Hsp104 and ClpB has been investigated primarily by Cryo Electron Microscopy (Cryo-EM), which has led to recent debate. It is generally agreed that Hsp104 and ClpB are native hexamers and that oligomerization is promoted by increased protein concentration [53, 77–79], low salt [77, 80], and the presence of ADP or ATP [53, 54, 79–81]. Curiously, however, while nucleotide binding to NBD1 is critical for ClpB hexamerization [80], this situation is reversed in Hsp104 where nucleotide binding to NBD2 is key [53, 78, 79]. The underlying reason for this switch between NBDs is unknown and continues to remain puzzling and unaddressed. NBD1 contributes the majority of basal ATPase activity in Hsp104 [53, 78, 79], whereas both NBDs contribute to basal ATPase activity in ClpB [80]. The ATPase activities of both NBDs are required for the full repertoire of protein-remodeling activities and are modulated by allosteric communication within and between NBD1 and NBD2 [73, 77, 79, 80, 82–84]. Gross domain position and the protomer-protomer interfacial packing of Hsp104 and ClpB hexamers still remain uncertain. Of particular interest is the position of the coiled-coil MD, which is necessary for disaggregase activity and is unique to the Hsp100 chaperones that function primarily in disaggregation [63, 80, 85]. In the TClpB crystal structure, this domain was jutting obliquely from the axis of the other domains [63] (Fig. 2b). Thus, in the original cryo-EM reconstructions of TClpB in the presence of AMP-PNP (a non-hydrolyzable ATP analog) the MD was assigned to protrusions that appeared to emanate from one tier of the hexamer [63] (Fig. 2c, Tsai model).
In subsequent studies, to determine any conformational reorganizations that take place through the ATPase cycle, Cryo-EM envelopes of TClpB in the ADP and apo state were reconstructed as well as the envelope of the Double Walker B TClpB mutant (E271A:E668A) in the presence of ATP [61]. This mutant binds but does not hydrolyze ATP at both NBDs and has increased affinity for substrate [61, 86]. In all states, a two-tiered hexamer with an axial channel running through the center was clearly visible [61]. However, the N-terminal domain was not visible as electron density [61]. In the AMP-PNP-bound state, the TClpB envelope shows clear, well defined protrusions on the outside of the hexamer, which, when the individual domains of the TClpB monomeric crystal structure were rigid body fit into the density, overlapped partially with predicted MD density [61, 63]. It was suggested that the exterior position of the MD might enable it to act as a ‘crowbar’ to pry apart large aggregates [63]. Glover and Lindquist had originally suggested that Hsp104 might possess such ‘crowbar activity’, but did not ascribe this activity to any particular domain [16]. All the other nucleotide states of TClpB (ATP, ADP, and apo) do not have such large protrusion of density that could correlate with a MD projection. It was suggested that this might be due to the inherent mobility of the coiled-coil MD [61]. Indeed, the main difference between the different nucleotide states were the length of these radially extending protrusions [61]. By contrast, the positions of the AAA+ domains remained almost identical in the various nucleotide states [61]. Consequently, these reconstructions do not clarify the mechanochemical coupling events that drive substrate translocation through the central channel.
This structural model of the hexamer was challenged by cryo-EM reconstructions of Hsp104 by Saibil and colleagues [64–66]. In these studies, cryo-EM reconstructions were generated of Hsp104 lacking its N-terminal domain, ΔN-Hsp104, and an NBD2 sensor-1 Hsp104 mutant, Hsp104N728A, which has slowed hydrolysis in NBD2 and is able to catalyze disaggregation of disordered aggregates in the absence of Hsp70 and Hsp40, but is unable to remodel amyloid [77, 82]. Contrary to the TClpB reconstruction, Hsp104N728A had a structured N-terminal domain and presented as a three-tiered hexamer with no oblique MD protrusion [64–66]. The central cavity of Hsp104N728A was also much larger than that observed with TClpB and the modeled protomer-protomer packing was unlike that of typical AAA+ structures [64–66]. It was hypothesized that the enlarged cavity might serve as an adaptation necessary to remodel large aggregated structures [64–66]. A TClpB-homology model of Hsp104 was rigid-body fit into the electron density with each domain connected by a flexible linker (Fig. 2d, Saibil model) [64–66]. The resulting fits placed the MD intercalated within NBD1 and NBD2, rather than projecting out into solution (Fig. 2d) [64–66]. This physical proximity of the MD to both NBDs, which is also partially supported by fluorescence proximity studies of ClpB [87], might help explain how the MD mediates communication pathways between NBD1 and NBD2 [73].
The cryo-EM reconstructions of Hsp104N728A with ATPγS (a slowly hydrolysable ATP analog), ATP and ADP were also generated [65, 66]. Hsp104N728A displayed large nucleotide-dependent domain reorganizations, with the ATPγS state having the most expanded central cavity [65, 66]. These domain movements displaced the substrate-binding tyrosine loops in the central channel and triggered a peristaltic motion that provides a structural basis for N- to C-terminal substrate translocation [65, 66]. Asymmetric reconstructions of Hsp104N728A with ATPγS and ATP provided unprecedented insight into disaggregase activity [65, 66]. These reconstructions suggested a sequential mechanism of ATP hydrolysis in NBD1 coupled to clockwise handover of substrate in the NBD1 ring and a coordinated handover between NBD1 and NBD2 [65, 66]. Thus, the first detailed structural picture of the mechanochemistry that underpins protein disaggregation emerged [4, 65, 66].
It remains unclear why the Cryo-EM reconstructions of TClpB and Hsp104N728A would be so disparate. Could the differences simply reflect a fundamental difference in hexamer architecture for the prokaryotic and eukaryotic enzymes? Or could the different reconstructions reflect the different protein preparations for the Cryo-EM? For example, glutaraldehyde fixation was employed for the TClpB studies [61, 63], whereas fixative was omitted for the Hsp104 studies [64, 65]. The story takes another twist when Tsai and colleagues presented Cryo-EM reconstructions of a double Walker B Hsp104 mutant (E285A:E687A) in the presence of ATP and ΔN-Hsp104 in the presence of ATPγS with and without glutaraldehyde fixation [62]. Both sets of envelopes appeared very similar, indicating that fixation might not be an issue [62]. Intriguingly, and in contrast to their prior TClpB reconstructions, there was a striking absence of additional mass density on the outside of the ΔN-Hsp104 or Hsp104E285A:E687A hexamer [62]. Indeed, the Cryo-EM envelopes of the different Hsp104 variants constructed by the different groups are remarkably similar, particularly for ΔN-Hsp104 plus ATPγS (especially when either reconstruction is subjected to a 180° vertical rotation) [62, 64, 65]. This similarity among Hsp104 envelopes might even suggest that TClpB hexamers possess a subtly distinct architecture. After all, Hsp104 is equipped to remodel amyloid, whereas ClpB is not [21, 51, 52], so perhaps subtle structural differences might be anticipated. However, despite the absence of lateral projections emanating from the Hsp104 hexamer, the authors placed the MD outside of the hexamer walls in their atomic structure fitting as with TClpB [62]. Thus, it is still unclear whether the MD of Hsp104 and ClpB is located inside or outside of the hexamer. It should also be noted that no reconstructions have been presented with full-length wild-type Hsp104.
In an effort to visualize the MD during Cryo-EM, an Hsp104 chimera was generated with T4 lysozyme inserted into the MD [62]. Because the lysozyme was visible as density on the outside of the hexamer, it was suggested that the MD must also be located on the exterior [62]. However, density corresponding to the MD itself on the outside of the hexamer could still not be readily visualized even with this highly artificial construct [62]. It is extremely probable that this large-scale (19kDa) insertion disrupts the native quaternary structure of Hsp104, especially since the insertion was located in a helical region of the MD (between residues N467 and E468 of helix 2 (Fig. 3a, b)) and not in a predicted loop region. Indeed, although this chimera possesses some disaggregation activity against non-native substrates in vitro, it should be noted that its ATPase activity was elevated, hexamerization was perturbed, disaggregase activity was dysregulated and the functionality was not assessed in vivo [62, 69]. Thus, it is not clear whether the two models can really be distinguished with this insertion variant despite it displaying partial functionality in some settings. Rather, the ability of the Hsp104 to accommodate such a large insertion and retain some activity indicates an extraordinary plasticity of the hexamer.
Additionally, the structure of Hsp104 and ClpB has been investigated using biochemical techniques other than Cryo-EM. Engineered disulfide cross-links in ClpB that covalently link NBD1 to motif 2 of the MD (Fig. 3a, b) have shown that these two domains are closely associated and that movement of the MD is crucial for disaggregation [63, 67]. Unfortunately, these cross-links do not differentiate between the two models because the MD is closely associated with NBD1 in both proposed structures [61–66]. Importantly, the MD is inaccessible to three monoclonal antibodies that recognize native MD epitopes [73] in Hsp104 hexamers, but is exposed in Hsp104 monomers [65]. These data suggest that the MD becomes shielded upon hexamerization but remains accessible and solvated when Hsp104 is monomeric [64–66]. This observation might explain why monomeric TClpB crystallized with the MD jutting away from the axis of the NBDs. However, in contrast to these results, Lee et al. inserted a short StrepII tag (WSHPQFEK) into the MD (between residues N467 and E468 of helix 2 (Fig. 3a, b)) of Hsp104 and employed dot blots to determine whether the tag was exposed in monomeric and hexameric StrepII-tagged Hsp104 [62]. These studies suggested that the StrepII tag was accessible in monomeric and hexameric forms of Hsp104 [62]. Unfortunately, however, it is uncertain whether this epitope tag is truly innocuous or partially disruptive. Moreover, these data are difficult to assess because it is not clear whether monomeric and hexameric Hsp104 were spotted on the same dot blot [62]. Thus, one cannot be certain from the presented data whether the signal intensities for monomeric and hexameric conformers are directly comparable [62].
Another major difference between the hexameric models is the positioning of the putative arginine fingers [61–66]. Typically, an arginine finger is an arginine residue that coordinates the ©-phosphate of a bound nucleotide and is a recurring characteristic of AAA+ family members [10]. In many AAA+ proteins the arginine residue is located distal to the nucleotide-binding site and is provided by the adjacent subunit of the oligomer [10, 88, 89]. Arginine fingers contribute to ATP hydrolysis through stabilization of the transition state [10]. When the MD is positioned on the exterior of the hexamer the arginine-finger residues are positioned in a canonical position reaching into the nucleotide-binding site of an adjacent protomer [10, 61–63]. By contrast, when the MD is intercalated between NBDs the arginine fingers are in a non-canonical position [64, 65]. However, it should be noted that the arginine finger is not conserved in all members of the AAA+ family, and it is not clear if the position of this motif in the oligomeric structure is stringently conserved among all the different clades of the family. For example, in some crystal structures, the predicted arginine finger of HslU, an Hsp100 and AAA+ family member with only one NBD, is roughly 8Å from the nucleotide binding site [90]. In this regard, it is interesting to note that the missense mutation R444M in the distal loop region between helices 1 and 2 in the MD (Fig. 3a, b) reduces ATPase activity of Hsp104 and impairs thermotolerance in a dominant-negative manner and disrupts amyloid remodeling functionality [64]. This deficiency may suggest a close contact between the distal loop of the MD and the NBD2 [64]. Thus, it is possible that other conserved arginines might fulfill the role of the arginine finger in Hsp104.
Finally, a crystal structure of another Hsp100 family member from Bacillus subtilis, ClpC, was recently solved and has weighed in on the debate [91] (Fig. 2e). Like ClpB and Hsp104, ClpC is a AAA+ disaggregase involved in modulation of stress response [92] and protein quality control [93]. Typically, ClpC passes disaggregated substrates to the chambered protease, ClpP, for degradation [91–94]. While ClpC is natively a hexamer, it requires an adaptor protein, MecA, to oligomerize and form an active enzyme [95, 96]. By employing a variant with four loop deletions and two ATPase-obliterating mutations, a 6.93Å crystal structure of hexameric ClpC in complex with the adaptor protein MecA was obtained. [91]. Interestingly, the packing within the NBD1 ring corresponded well with that proposed for TClpB [61, 63, 91] (Fig. 4). Additionally, the MD of ClpC, which is approximately half the length of the MD of ClpB [96], was also jutting out away from NBDs and was distinctly located on the outside of the hexamer [91]. These data provide independent support for the TClpB hexameric model. However, the coiled-coil domain of ClpC is considerably shorter than that of Hsp104 or ClpB and must interact with MecA so that ClpC can hexamerize [91]. Thus, the MD of ClpC plays a very different role to the MD of ClpB or Hsp104 where it is dispensable for hexamerization [69, 80].
Figure 4. Overlay of ClpB and ClpC monomers.

ClpB and ClpC monomers were aligned by their AAA+ domains using Pymol. The resulting RMSD was 3.4. Domains for ClpB are colored as in Figure 2 with the N domain in purple, NBD1 in cyan, MD in yellow, and NBD2 in dark blue. All domains in ClpC are colored in orange. Note the drastically different position of the ClpB N-terminal domain (marked with an arrow) and the different angle of the ClpC MD (marked with an asterisk).
In closing this section, we suggest that further biochemical characterization and alternative techniques are urgently needed to discern which hexameric model is correct or whether a revised model is required for Hsp104 and ClpB. It is probable that both models are partially correct because Hsp104 and ClpB are large, oligomeric machines that possess significant domain plasticity [62, 63, 69]. Potentially, the MD might be able to occupy both structural conformations depending on the stage of the disaggregase cycle. Two factors are likely to have greatly increased the difficulty in obtaining high-resolution structural information on the Hsp104 or ClpB hexamer. First, the hexamer is highly dynamic and monomers are exchanged on the minute timescale [97, 98]. Second, conformational heterogeneity is likely generated by individual subunits or domains adopting different conformations within the hexamer [65, 66]. Future studies employing a full gamut of complementary techniques will be essential to refine our understanding of the hexameric structure of Hsp104 and ClpB.
The function of the MD
Despite the ambiguity in MD location, several studies have probed MD function. For instance, in ClpB, deletion of the MD causes a loss of thermotolerance function [80]. Furthermore, partial truncations in ClpB MD cause a decreased ATPase activity, hexamerization defects, and impaired disaggregation [99]. Even point mutations along the MD have been implicated in altered ATPase activity, loss of thermotolerance activity, and/or destabilization of the hexamer [64, 67, 85, 100]. The dynamic mobility of the MD is also critical for Hsp104/ClpB function, as crosslinks that hinder MD flexibility reduce or ablate disaggregation activity of ClpB [63, 67]. Additionally, the MD appears to be involved in facilitating NBD1 and NBD2 communication [73]: when motif 2 is covalently attached to NBD1, ATPase activity in NBD2 increases by ~30-fold [87]. Clearly, the structural integrity and mobility of the MD must be maintained for full disaggregase functionality.
At first glance, the MD appears to differentiate Hsp100 proteins that possess disaggregase activity from other Hsp100s. The disaggregases ClpBand ClpC [98, 101, 102] in bacteria, Hsp104 and Hsp78 [103–106] in yeast, as well as Hsp101 [107–110] in plants, all contain a coiled-coil insertion toward the C-terminal end of NBD1. ClpE is another bacterial homologue that contains a coiled-coil insertion[111, 112]. Genetic studies have also implicated that ClpE is part of the cellular disaggregation machinery, but this has yet to be demonstrated with pure protein biochemistry[113]. While there are many examples of Hsp100 proteins that do not contain a MD and also do not possess disaggregase activity[114], the correlation between MD and disaggregase function is not quite so straightforward.. For instance, ClpV, another bacterial Hsp100 protein, also contains a predicted MD insertion between two AAA+ domains but does not possess any disaggregation activity against substrates in vivo [115]. Additionally, ClpA, another Hsp100 and AAA+ protein with two NBDs per monomer found in E. coli (but not in yeast), does display disaggregase activity against disordered aggregates but does not harbor a coiled-coil MD [82, 116]. Indeed, in collaboration with its adaptor protein, ClpS, ClpA disaggregates substrates and delivers them to ClpP for degradation [116]. ClpA disaggregates substrates without any need for the Hsp70 chaperone system [114, 116], indicating that Hsp70 is not absolutely required for disaggregation per se. Importantly, however, the Hsp70 chaperone system inhibits protein disaggregation by ClpA [116]. In this way, DnaK prevents degradation of aggregated substrates by ClpAP in E. coli and permits their disaggregation and reactivation by ClpB [116]. Since the MD is a major distinguishing feature of ClpB compared to ClpA, these studies provided the first hint that the MD might contribute to collaboration with Hsp70
Hsp104 and ClpB require collaboration with Hsp70 and Hsp40 chaperones for successful disaggregation of amorphous aggregates, both in vitro and in vivo [13, 16, 47, 49, 117, 118]. Hsp70 and Hsp40 can also ameliorate the amyloid-remodeling activity of Hsp104 [24, 28, 44, 51, 119]. In yeast, members of the Hsp70 chaperone family (e.g. Ssa1, Ssb1) and Hsp40 family (e.g. Ydj1 and Sis1) have been identified as being able to collaborate with Hsp104 [16, 24]. In bacteria, the homologous DnaK and DnaJ (along with the nucleotide exchange factor GrpE) (KJE) collaborate with ClpB [46, 48, 49]. The exact mechanisms by which Hsp70 and Hsp40 collaborate with Hsp104 are largely unresolved. However, two key functions are commonly ascribed. First, Hsp70 is proposed to act upstream of Hsp104 or ClpB by interacting with the aggregate and shuttling substrate into the axial channel of Hsp104 or ClpB (Fig. 1a) [16, 58, 67, 98, 120–122]. Second, Hsp70 is proposed to act downstream of Hsp104 and promotes refolding of unfolded polypeptides once they emerge from axial channel [16, 49, 82, 86].
The Hsp70 chaperone system, however, may fulfill a third role by modulating the ATPase activity of Hsp104/ClpB in a manner that promotes the successful disaggregation of disordered aggregates [2, 82, 114]. Indeed, mixing of ClpB with KJE results in a synergistic increase in global ATPase activity [114]. Moreover, the requirement for Hsp70 to disaggregate certain disordered aggregates can be bypassed altogether by addition of specific mixtures of ATP:ATPγS (3:1 for Hsp104 and 1:1 for ClpB), or by introducing specific Walker A (e.g. K620T in Hsp104), Walker B (e.g. E279Q or E678Q in ClpB) or sensor-1 (e.g. N728A in Hsp104) mutations into either NBD1 or NBD2 [82–84, 114, 123]. Thus, part of the function of Hsp70 might be to set Hsp104 ATPase activity in a mode in which predominantly one NBD is rapidly hydrolyzing ATP, which can be sufficient to promote the disaggregation of disordered aggregates [65, 82, 114, 123]. However, this mode of ATPase activity precludes amyloid remodeling by Hsp104 [82]. Further studies are needed to define the precise mechanisms by which Hsp70 promotes protein disaggregation by Hsp104. Moreover, it has long remained unclear how the functional interaction between Hsp104 and Hsp70 is mediated and what domains of either protein are required. Recent studies, however, ascribe a critical role for the MD in promoting communication with the Hsp70 system.
The role of the MD in Hsp70 Chaperone Collaboration
Interestingly, it has long been clear that Hsp104 is unable to collaborate with the bacterial Hsp70 chaperone system [16]. Likewise, ClpB is unable to collaborate with the yeast Hsp70 chaperone system [106]. This situation is often referred to as the ‘species-specific’ interaction between Hsp104 and Hsp70 [2, 68, 69]. However, the term ‘species specific’ should not be taken too literally. Hsp104 is able to collaborate effectively with the mammalian Hsp70 chaperone system [44, 84, 124] and multiple eukaryotic Hsp104 homologues can complement the thermotolerance function of Hsp104 in yeast (but interestingly not always the prion propagation function of Hsp104) [125–128], whereas ClpB cannot [51, 70]. Rather, it seems that eukaryotic Hsp100s are unable to collaborate with prokaryotic Hsp70, and likewise prokaryotic Hsp100s cannot collaborate with eukaryotic Hsp70. Within prokaryotes, TClpB cannot collaborate with DnaK from E. coli [129]. Moreover, Ssa1 and DnaJ can collaborate with Hsp104, whereas DnaK and Ydj1 cannot [16]. Thus, the key determinant of this specificity appears to be the Hsp70 chaperone rather than the Hsp40.
Yet, defining whether or how Hsp70 or Hsp40 might interact directly with Hsp104 has proven remarkably difficult. Initial evidence suggested that Hsp104 coimmunoprecipitates with Ydj1, but not Sis1, from yeast extracts [16]. Using pure proteins, a weak physical association has been observed between hexameric ClpB and DnaK [129]. In another study, DnaJ was found not to interact with ClpB, whereas the substrate-binding domain of DnaK was required for an interaction with ClpB that was disrupted by deletion of the N-terminal domain of ClpB and was partially disrupted by deletion of portions of the coiled-coil MD of ClpB [130]. These data indicated that DnaK may interact with ClpB at the N-terminal domain and the MD [130]. However, it is difficult to know whether these interactions reflect specific functional interactions between ClpB and DnaK that are required for protein disaggregation, or whether they reflect the chaperone activity of DnaK that would recognize and bind transiently unfolded portions of ClpB.
The first functional evidence for the interaction between the MD and the Hsp70 chaperone system was uncovered by characterization of specific helix 3 MD mutants (e.g. Y503D; Fig. 3a, b) [67] (Table 1)ClpBY503D was unable to collaborate with KJE in protein disaggregation [67]. However, ClpBY503D formed hexamers, had elevated ATPase activity and translocated unfolded polypeptides [67]. The specific defect in ClpBY503D was pinpointed to an inability of KJE to shuttle aggregated substrates to the NBD1 channel loop [67]. Thus, it was concluded that the MD functions to couple the interaction with Hsp70 to translocation of aggregated substrates [67]. Yet, a direct interaction between helix 3 of the MD and DnaK was not observed [67]. Rather, helix 3 of the MD displays conformation flexibility that is necessary to regulate a direct interaction between helix 3 of the MD and NBD1 [67]. Mutations in helix 3 (e.g. Y503D) cause this interaction to become dysregulated and consequently ATPase activity becomes elevated to such an extent that substrate handover from KJE to the ClpB channel can no longer be coordinated [67]. Thus, these studies revealed that helix 3 of the MD indirectly controls the functional interaction with Hsp70 through a direct interaction with NBD1.
Table 1.
DeSantis & Shorter.
Selected MD point mutations. Activity relative to wild-type (WT) Hsp104 is shown in green symbols and activity relative to WT ClpB is shown in blue symbols. ++ indicates that mutant displayed greater activity than WT, + indicates that the mutant displayed comparable activity to WT, − indicates that mutant displayed less activity than WT, while Ø indicates that no activity was observed for the mutant tested. Blank cells indicate that the mutant was not tested for this activity. Please note that this is not an exhaustive table of tested MD mutations. For a more exhaustive listing of characterized MD point mutations, see Barends and colleagues review [4].
| Mutation | Reference # | Forms Hexamers? | ATPase activity? | Able to Translocate? | Disaggregate Amorphous Aggregates in vitro? | Disaggregate Amyloid Aggregates in vitro? | Functional in Thermotolerance? | Functional in [PSI+] Propagation? |
|---|---|---|---|---|---|---|---|---|
| ClpBY503D | 67 | Yes |
|
|
|
n/a |
|
n/a |
| Hsp104R419M | 64 | Yes |
|
|
|
|||
| Hsp104M444M | 64 | Yes |
|
|
|
|||
| Hsp104R495M | 64 | Yes |
|
|
|
|||
| Hsp104L462R | 134, |
|
|
|||||
| Hsp104A503V | 85,134–136 |
|
|
|
 No activity observed
No activity observed
 As active as WT Hsp104 or CIpB
As active as WT Hsp104 or CIpB
 Less active than WT 104 or CIpB
Less active than WT 104 or CIpB
 More active than WT 104 or CIpB
More active than WT 104 or CIpB
![]()
![]()
The established connection between the MD and Hsp70 [67] stimulated efforts to utilize chimeras of Hsp104 and ClpB to determine which domain(s) of Hsp104 would enable ClpB to function in yeast (Fig. 5) [51, 68, 131]. Thus, each domain (N, NBD1, MD and NBD2) was treated as a module and chimeras were constructed that had various combinations of modules from either ClpB or Hsp104 [51, 68, 131] (Fig. 5). For example, the chimera 444B contains the N-terminal domain, NBD1 and MD of Hsp104 plus NBD2 of ClpB. In an initial study, 444B and _44B (444B with the N-terminal domain deleted) could function in thermotolerance and prion propagation, indicating that either the N-terminal domain, NBD1 or the MD of ClpB might preclude activity in yeast [51]. Subsequent studies revealed that the MD plays a critical role [68]. Thus, BB4B was able to partially complement Hsp104 in thermotolerance (Fig. 5) [68], but was inactive in propagation of the yeast prion [PSI+] [51]. Moreover, BB4B and a similar construct could now collaborate with eukaryotic Hsp70 to promote the disaggregation of disordered aggregates in vitro (Fig. 5) [68, 69]. Thus, the MD of Hsp104 enables ClpB to functionally interact with eukaryotic Hsp70.
Figure 5. Summary of ClpB and Hsp104 chimera activity.

Cartoon representation of the domain chimeras utilized in references [51, 68, 69]. Green cylinders represent Hsp104 domains and blue cylinders represent ClpB domains. Activity relative to wild-type (WT) Hsp104 is shown in green symbols and activity relative to WT ClpB is shown in blue symbols. ++ indicates that the chimera displayed greater activity than WT, + indicates that the chimera displayed comparable activity to WT, − indicates that chimera displayed less activity than WT, while Ø indicates that no activity was observed for the chimera tested. Blank cells indicate that chimera was not tested for this activity. [PSI+] propagation, [RNQ+] propagation, ATPase activity, increase in ATPase activity upon binding α-casein, the ability to bind α-casein, conferred thermotolerance in E. coli, conferred thermotolerance in S. cerevisiae, and association with DnaK as measured by a bacterial 2-hybrid system were tested. Additionally, DnaK, DnaJ, and GrpE and Hsp70/40 mediated disaggregation of Green fluorescent protein (GFP), β-galactosidase (β-Gal), and malate dehydrogenase (MDH) were tested.
Curiously, however, the converse domain transplant was not nearly as successful (Fig. 5). Thus, 44B4 could not collaborate with KJE in vitro or in E. coli (Fig. 5) [68]. Another study found that 44B4 had ATPase activity, formed hexamers and could weakly collaborate with KJE in disaggregation in vitro [131]. Thus, simply transplanting the MD of ClpB into Hsp104 does not enable Hsp104 to collaborate with DnaK in the same way that ClpB can [68]. Rather, additional sequences appeared to be required. The addition of the NBD1 of ClpB helped restore activity to wild-type ClpB levels [68]. Thus, 4BB4 was active in vitro with KJE and in E. coli [68]. Curiously, 44BB has not been studied. Collectively, these data assert that the MD of Hsp104 is extremely important for a functional interaction with eukaryotic Hsp70 in the disaggregation of disordered aggregates [68, 69]. However, the situation with ClpB is more complex as the MD of ClpB does not enable or only minimally enables Hsp104 to interact functionally with KJE [68, 69]. Surprisingly, no mechanistic explanation for this disparity has been advanced [68, 69]. Moreover, although these studies [68, 69] strengthen the functional interaction between the MD and Hsp70, there is still no compelling evidence for a direct interaction between the MD and Hsp70 that is required for protein disaggregation. A hint is provided by a bacterial two-hydrid system in which the MD of ClpB appears to confer (in some constructs) the ability to interact physically with DnaK, although it remains possible that this interaction might still be indirect [68]. Even in this experiment, 44B4 displayed a signal that was only slightly higher than background, indicating that the MD of ClpB is insufficient to confer an interaction with DnaK [68]. Here too, there is still the concern that apparent specificity in the bacterial two-hydrid system might stem from the chaperone activity of DnaK rather than from a direct, functional interaction that is required for disaggregase activity. Further studies in this system, which pinpoint the exact residues in ClpB and DnaK that are necessary for the interaction might be informative.
To determine the precise region within the MD that is required for collaboration with DnaK or Hsp70 another series of ClpB chimeras were generated [68]. Thus, the MD of these chimeras was comprised of various combinations of helices 1, 2, 3 or 4 from the Hsp104 or ClpB primary sequence (Fig. 5). These studies indicate that helix 2 and 3 of the ClpB MD are important for a functional interaction with DnaK, whereas helix 1, 2 and 3 of the Hsp104 MD are important for an interaction with eukaryotic Hsp70 [68]. However, a single missense mutation in helix 2 that disrupted the functional interaction with DnaK could not be identified [68]. By contrast, the Y503D mutation in helix 3 of the ClpB MD ablates any collaboration with DnaK, even though this region does not interact with DnaK directly [67]. This latter finding raises the possibility that all the chimeric proteins that fail to interact with DnaK in protein disaggregation [68, 69] might only fail to do so because, in the chimera, the communication between NBD1 and helix 3 of the MD that is necessary for collaboration with KJE is disrupted in a manner akin to ClpBY503D. That is, perhaps due to unanticipated, long-range structural effects, the chimera is unable to support the appropriate communication between NDB1 and helix 3 of the MD, which is paramount for collaboration with KJE. Thus, chimera dysfunction might not reflect loss of a direct interaction between DnaK and the MD per se. Rather, chimeras might be unable to collaborate with KJE because of an indirect, long-range structural effect that disrupts communication between NBD1 and helix 3 of the MD. Further studies are needed to resolve these issues and to pinpoint the exact sites where DnaK and ClpB might interact directly.
Hsp70-independent functions of the MD
Interestingly, amyloid remodeling by Hsp104 does not absolutely require Hsp70 and Hsp40 in vitro [21, 23, 24, 44, 132–134], which suggests that the disaggregation of ordered aggregates may proceed by a different reaction mechanism than amorphous aggregate remodeling. This independence from Hsp70 also provides an opportunity to assess the function of the MD toward the intrinsic disaggregase activity of Hsp104. The MD plays an important role in this intrinsic amyloid-remodeling activity of Hsp104, as individual missense mutations in conserved MD arginines (R419, R444 or R495) ablate the ability of Hsp104 to remodel infectious amyloid forms of Sup35 [64] (Table 1). Thus, the MD plays a critical role in disaggregase activity even in the absence of Hsp70 [64].
Consistent with these in vitro observations, several mutations in the MD of Hsp104 impair prion propagation in vivo [135–137]. Intriguingly, some of these mutations impair propagation of the yeast prion [PSI+], which is comprised of infectious amyloid forms of Sup35, but have no effect on thermotolerance. MD mutants that fall in this class include L462R and A503V (Fig. 3a, b) [85, 135–137] (Table 1). The mechanistic explanation for the ability of these specific MD mutants to promote the disaggregation of stress-induced disordered aggregates while simultaneously being unable to promote the amyloid-remodeling events required for prion propagation is unknown. Nonetheless, these data hint that Hsp104 might exploit subtly different mechanisms to remodel amyloid versus disordered aggregates, and that the MD plays a key role in these different mechanisms.
Concluding remarks
In closing, we note that despite several important advances described above, many aspects of the role of the MD in Hsp104 and ClpB activity remain elusive. First, the location of the MD within the hexameric assembly remains a point of debate [61–66, 91]. Second, although the MD appears to control the functional collaboration with the Hsp70 chaperone system and even confers some ability to enable chimeric disaggregases to traverse the S. cerevisiae - E. coli species barrier (in one direction at least), precisely how this is achieved and whether the MD interacts with Hsp70 directly is still unclear [51, 67–69]. Finally, single missense mutations in the MD of Hsp104 can have dominant gain-of-function mutations with highly unexpected biological consequences [85]. Perhaps one of the most surprising examples is provided by A503V [85, 135, 136]. This mutant confers a growth defect at 37°C but surprisingly is more active than wild-type Hsp104 in thermotolerance [85]. Furthermore, Hsp104A503V cannot support propagation of the yeast prion [PSI+] [85, 135]. Yet, remarkably, Hsp104A503V is considerably more effective than wild-type Hsp104 in antagonizing polyglutamine aggregation and toxicity in yeast models of Huntington’s disease [136]. This surprising therapeutic gain-of-function suggests that MD mutations should be intensively explored in our efforts to identify novel Hsp104 variants with enhanced disaggregase activity against specific proteins that misfold, aggregate and confer toxicity in a diverse collection of increasingly prevalent, devastating and presently untreatable neurodegenerative disorders [6, 39, 43].
Highlights.
Discussion review of hexameric models of Hsp104 and ClpB
Discussion of function of middle domain of Hsp104 and ClpB in Hsp70 collaboration.
Discussion of Hsp70-independent functions of middle domain of Hsp104 and ClpB.
Acknowledgments
We thank Elizabeth Sweeny, Meredith Jackrel, and Laura Castellano for critical reading of the manuscript. These studies were supported by a Chemistry-Biology Interface Fellowship from the NIH training grant (2T32GM071339-06A1) (to M.E.D.); an NIH Director’s New Innovator Award (1DP2OD002177-01), an NINDS R21 (5R21NS067354-02), an Ellison Medical Foundation New Scholar in Aging Award, and a Bill and Melinda Gates Foundation Grand Challenges Explorations Award (to J.S.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Schirmer EC, Glover JR, Singer MA, Lindquist S. HSP100/Clp proteins: a common mechanism explains diverse functions. Trends in Biochemical Sciences. 1996;21:289–296. [PubMed] [Google Scholar]
- 2.Doyle SM, Wickner S. Hsp104 and ClpB: protein disaggregating machines. Trends Biochem Sci. 2009;34:40–48. doi: 10.1016/j.tibs.2008.09.010. [DOI] [PubMed] [Google Scholar]
- 3.Glover JR, Lum R. Remodeling of protein aggregates by Hsp104. Protein Pept Lett. 2009;16:587–597. doi: 10.2174/092986609788490087. [DOI] [PubMed] [Google Scholar]
- 4.Barends TR, Werbeck ND, Reinstein J. Disaggregases in 4 dimensions. Curr Opin Struct Biol. 2010;20:46–53. doi: 10.1016/j.sbi.2009.12.014. [DOI] [PubMed] [Google Scholar]
- 5.Haslberger T, Bukau B, Mogk A. Towards a unifying mechanism for ClpB/Hsp104-mediated protein disaggregation and prion propagation. Biochem Cell Biol. 2010;88:63–75. doi: 10.1139/o09-118. [DOI] [PubMed] [Google Scholar]
- 6.Vashist S, Cushman M, Shorter J. Applying Hsp104 to protein-misfolding disorders. Biochem Cell Biol. 2010;88:1–13. doi: 10.1139/o09-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grimminger-Marquardt V, Lashuel HA. Structure and function of the molecular chaperone Hsp104 from yeast. Biopolymers. 2010;93:252–276. doi: 10.1002/bip.21301. [DOI] [PubMed] [Google Scholar]
- 8.Bosl B, Grimminger V, Walter S. The molecular chaperone Hsp104--a molecular machine for protein disaggregation. J Struct Biol. 2006;156:139–148. doi: 10.1016/j.jsb.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 9.Lindquist S, Patino MM, Chernoff YO, Kowal AS, Singer MA, Liebman SW, Lee KH, Blake T. The role of Hsp104 in stress tolerance and [PSI+] propagation in Saccharomyces cerevisiae. Cold Spring Harb Symp Quant Biol. 1995;60:451–460. doi: 10.1101/sqb.1995.060.01.050. [DOI] [PubMed] [Google Scholar]
- 10.Hanson PI, Whiteheart SW. AAA+ proteins: have engine, will work. Nat Rev Mol Cell Biol. 2005;6:519–529. doi: 10.1038/nrm1684. [DOI] [PubMed] [Google Scholar]
- 11.Erzberger JP, Berger JM. Evolutionary relationships and structural mechanisms of AAA+ proteins. Annu Rev Biophys Biomol Struct. 2006;35:93–114. doi: 10.1146/annurev.biophys.35.040405.101933. [DOI] [PubMed] [Google Scholar]
- 12.Neuwald AF, Aravind L, Spouge JL, Koonin EV. AAA+: A class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res. 1999;9:27–43. [PubMed] [Google Scholar]
- 13.Parsell DA, Kowal AS, Singer MA, Lindquist S. Protein disaggregation mediated by heat-shock protein Hspl04. Nature. 1994;372:475–478. doi: 10.1038/372475a0. [DOI] [PubMed] [Google Scholar]
- 14.Sanchez Y, Taulin J, Borkovich KA, Lindquist S. Hsp104 is required for tolerance to many forms of stress. EMBO Journal. 1992;11:2357–2364. doi: 10.1002/j.1460-2075.1992.tb05295.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sanchez Y, Lindquist SL. HSP104 required for induced thermotolerance. Science. 1990;248:1112–1115. doi: 10.1126/science.2188365. [DOI] [PubMed] [Google Scholar]
- 16.Glover JR, Lindquist S. Hsp104, Hsp70, and Hsp40: A Novel Chaperone System that Rescues Previously Aggregated Proteins. Cell. 1998;94:73–82. doi: 10.1016/s0092-8674(00)81223-4. [DOI] [PubMed] [Google Scholar]
- 17.Lindquist S, Kim G. Heat-shock protein 104 expression is sufficient for thermotolerance in yeast. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:5301–5306. doi: 10.1073/pnas.93.11.5301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Parsell DA, Taulien J, Lindquist S. The role of heat-shock proteins in thermotolerance. Philos Trans R Soc Lond B Biol Sci. 1993;339:279–285. doi: 10.1098/rstb.1993.0026. discussion 285–276. [DOI] [PubMed] [Google Scholar]
- 19.Fink AL. Protein aggregation: folding aggregates, inclusion bodies and amyloid. Folding and Design. 1998;3:R9–R23. doi: 10.1016/S1359-0278(98)00002-9. [DOI] [PubMed] [Google Scholar]
- 20.Wang L, Schubert D, Sawaya MR, Eisenberg D, Riek R. Multidimensional Structure–Activity Relationship of a Protein in Its Aggregated States. Angewandte Chemie International Edition. 2010;49:3904–3908. doi: 10.1002/anie.201000068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shorter J, Lindquist S. Hsp104 Catalyzes Formation and Elimination of Self-Replicating Sup35 Prion Conformers. Science. 2004;304:1793–1797. doi: 10.1126/science.1098007. [DOI] [PubMed] [Google Scholar]
- 22.Shorter J, Lindquist S. Prions as adaptive conduits of memory and inheritance. Nat Rev Genet. 2005;6:435–450. doi: 10.1038/nrg1616. [DOI] [PubMed] [Google Scholar]
- 23.Shorter J, Lindquist S. Destruction or Potentiation of Different Prions Catalyzed by Similar Hsp104 Remodeling Activities. 2006;23:425–438. doi: 10.1016/j.molcel.2006.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shorter J, Lindquist S. Hsp104, Hsp70 and Hsp40 interplay regulates formation, growth and elimination of Sup35 prions. The EMBO journal. 2008;27:2712–2724. doi: 10.1038/emboj.2008.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chernoff YO, Lindquist SL, Ono B, Inge-Vechtomov SG, Liebman SW. Role of the Chaperone Protein Hsp 104 in Propagation of the Yeast Prion- Like Factor [psi+] Science. 1995;268:880–884. doi: 10.1126/science.7754373. [DOI] [PubMed] [Google Scholar]
- 26.Moriyama H, Edskes HK, Wickner RB. [URE3] Prion Propagation in Saccharomyces cerevisiae: Requirement for Chaperone Hsp104 and Curing by Overexpressed Chaperone Ydj1p. Mol Cell Biol. 2000;20:8916–8922. doi: 10.1128/mcb.20.23.8916-8922.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alberti S, Halfmann R, King O, Kapila A, Lindquist S. A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell. 2009;137:146–158. doi: 10.1016/j.cell.2009.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sweeny EA, Shorter J. Prion proteostasis: Hsp104 meets its supporting cast. Prion. 2008;2:135–140. doi: 10.4161/pri.2.4.7952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sondheimer N, Lindquist S. Rnq1: an epigenetic modifier of protein function in yeast. Mol Cell. 2000;5:163–172. doi: 10.1016/s1097-2765(00)80412-8. [DOI] [PubMed] [Google Scholar]
- 30.Du Z, Park KW, Yu H, Fan Q, Li L. Newly identified prion linked to the chromatin-remodeling factor Swi1 in Saccharomyces cerevisiae. Nature genetics. 2008;40:460–465. doi: 10.1038/ng.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Patel BK, Gavin-Smyth J, Liebman SW. The yeast global transcriptional co-repressor protein Cyc8 can propagate as a prion. Nature cell biology. 2009;11:344–349. doi: 10.1038/ncb1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.DiSalvo S, Derdowski A, Pezza JA, Serio TR. Dominant prion mutants induce curing through pathways that promote chaperone-mediated disaggregation. Nat Struct Mol Biol. 2011;18:486–492. doi: 10.1038/nsmb.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Satpute-Krishnan P, Langseth SX, Serio TR. Hsp104-dependent remodeling of prion complexes mediates protein-only inheritance. PLoS Biol. 2007;5:e24. doi: 10.1371/journal.pbio.0050024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sunde M, Serpell LC, Bartlam M, Fraser PE, Pepys MB, Blake CCF. Common core structure of amyloid fibrils by synchrotron X-ray diffraction. Journal of Molecular Biology. 1997;273:729–739. doi: 10.1006/jmbi.1997.1348. [DOI] [PubMed] [Google Scholar]
- 35.Nelson R, Sawaya MR, Balbirnie M, Madsen AO, Riekel C, Grothe R, Eisenberg D. Structure of the cross-[beta] spine of amyloid-like fibrils. Nature. 2005;435:773–778. doi: 10.1038/nature03680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tessier PM, Lindquist S. Unraveling infectious structures, strain variants and species barriers for the yeast prion [PSI+] Nat Struct Mol Biol. 2009;16:598–605. doi: 10.1038/nsmb.1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aguzzi A, O’Connor T. Protein aggregation diseases: pathogenicity and therapeutic perspectives. Nat Rev Drug Discov. 2010;9:237–248. doi: 10.1038/nrd3050. [DOI] [PubMed] [Google Scholar]
- 38.Aguzzi A, Rajendran L. The transcellular spread of cytosolic amyloids, prions, and prionoids. Neuron. 2009;64:783–790. doi: 10.1016/j.neuron.2009.12.016. [DOI] [PubMed] [Google Scholar]
- 39.Cushman M, Johnson BS, King OD, Gitler AD, Shorter J. Prion-like disorders: blurring the divide between transmissibility and infectivity. J Cell Sci. 2010;123:1191–1201. doi: 10.1242/jcs.051672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Halfmann R, Lindquist S. Epigenetics in the extreme: prions and the inheritance of environmentally acquired traits. Science. 2010;330:629–632. doi: 10.1126/science.1191081. [DOI] [PubMed] [Google Scholar]
- 41.Shorter J. Emergence and natural selection of drug-resistant prions. Mol Biosyst. 2010;6:1115–1130. doi: 10.1039/c004550k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.True HL, Lindquist SL. A yeast prion provides a mechanism for genetic variation and phenotypic diversity. Nature. 2000;407:477–483. doi: 10.1038/35035005. [DOI] [PubMed] [Google Scholar]
- 43.Shorter J. Hsp104: a weapon to combat diverse neurodegenerative disorders. Neurosignals. 2008;16:63–74. doi: 10.1159/000109760. [DOI] [PubMed] [Google Scholar]
- 44.Lo Bianco C, Shorter J, Ragulier E, Lashuel H, Iwatsubo T, Lindquist S, Aebischer P. Hsp104 antagonizes alpha-synuclein aggregation and reduces dopaminergic degeneration in a rat model of Parkinson disease. The Journal of Clinical Investigation. 2008;118:3087–3097. doi: 10.1172/JCI35781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vacher C, Garcia-Oroz L, Rubinsztein DC. Overexpression of yeast hsp104 reduces polyglutamine aggregation and prolongs survival of a transgenic mouse model of Huntington’s disease. Human molecular genetics. 2005;14:3425–3433. doi: 10.1093/hmg/ddi372. [DOI] [PubMed] [Google Scholar]
- 46.Motohashi K, Watanabe Y, Yohda M, Yoshida M. Heat-inactivated proteins are rescued by the DnaK.J-GrpE set and ClpB chaperones. Proc Natl Acad Sci USA. 1999;96:7184–7189. doi: 10.1073/pnas.96.13.7184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mogk A, Tomoyasu T, Goloubinoff P, Rudiger S, Roder D, Langen H, Bukau B. Identification of thermolabile E. coli proteins: prevention and reversion of aggregation by DnaK and ClpB. EMBO J. 1999;18:6934–6949. doi: 10.1093/emboj/18.24.6934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zolkiewski M. ClpB cooperates with DnaK, DnaJ, and GrpE in suppressing protein aggregation. A novel multi-chaperone system from Escherichia coli. J Biol Chem. 1999;274:28083–28086. doi: 10.1074/jbc.274.40.28083. [DOI] [PubMed] [Google Scholar]
- 49.Goloubinoff P, Mogk A, Zvi APB, Tomoyasu T, Bukau B. Sequential mechanism of solubilization and refolding of stable protein aggregates by a bichaperone network. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:13732–13737. doi: 10.1073/pnas.96.24.13732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Squires CL, Pedersen S, Ross BM, Squires C. ClpB is the Escherichia coli heat shock protein F84.1. J Bacteriol. 1991;173:4254–4262. doi: 10.1128/jb.173.14.4254-4262.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tipton KA, Verges KJ, Weissman JS. In Vivo Monitoring of the Prion Replication Cycle Reveals a Critical Role for Sis1 in Delivering Substrates to Hsp104. Cell. 2008;32:584–591. doi: 10.1016/j.molcel.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hinault MP, Cuendet AF, Mattoo RU, Mensi M, Dietler G, Lashuel HA, Goloubinoff P. Stable alpha-synuclein oligomers strongly inhibit chaperone activity of the Hsp70 system by weak interactions with J-domain co-chaperones. J Biol Chem. 2010;285:38173–38182. doi: 10.1074/jbc.M110.127753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Parsell DA, Kowal AS, Lindquist S. Saccharomyces cerevisiae Hsp104 protein. Purification and characterization of ATP-induced structural changes. J Biol Chem. 1994;269:4480–4487. [PubMed] [Google Scholar]
- 54.Zolkiewski M, Kessel M, Ginsburg A, Maurizi MR. Nucleotide-dependent oligomerization of C1pB from Escherichia coli. Protein Science. 1999;8:1899–1903. doi: 10.1110/ps.8.9.1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lum R, Niggemann M, Glover JR. Peptide and Protein Binding in the Axial Channel of Hsp104: INSIGHTS INTO THE MECHANISM OF PROTEIN UNFOLDING. J Biol Chem. 2008;283:30139–30150. doi: 10.1074/jbc.M804849200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lum R, Tkach JM, Vierling E, Glover JR. Evidence for an unfolding/threading mechanism for protein disaggregation by Saccharomyces cerevisiae Hsp104. J Biol Chem. 2004;279:29139–29146. doi: 10.1074/jbc.M403777200. [DOI] [PubMed] [Google Scholar]
- 57.Schlieker C, Weibezahn J, Patzelt H, Tessarz P, Strub C, Zeth K, Erbse A, Schneider-Mergener J, Chin JW, Schultz PG, Bukau B, Mogk A. Substrate recognition by the AAA+ chaperone ClpB. Nat Struct Mol Biol. 2004;11:607–615. doi: 10.1038/nsmb787. [DOI] [PubMed] [Google Scholar]
- 58.Weibezahn J, Tessarz P, Schlieker C, Zahn R, Maglica Z, Lee S, Zentgraf H, Weber-Ban EU, Dougan DA, Tsai FTF, Mogk A, Bukau B. Thermotolerance Requires Refolding of Aggregated Proteins by Substrate Translocation through the Central Pore of ClpB. Cell. 2004;119:653–665. doi: 10.1016/j.cell.2004.11.027. [DOI] [PubMed] [Google Scholar]
- 59.Shorter J, Lindquist S. Navigating the ClpB channel to solution. Nat Struct Mol Biol. 2005;12:4–6. doi: 10.1038/nsmb0105-4. [DOI] [PubMed] [Google Scholar]
- 60.Horwich AL. Chaperoned protein disaggregation--the ClpB ring uses its central channel. Cell. 2004;119:579–581. doi: 10.1016/j.cell.2004.11.018. [DOI] [PubMed] [Google Scholar]
- 61.Lee S, Choi JM, Tsai FTF. Visualizing the ATPase Cycle in a Protein Disaggregating Machine: Structural Basis for Substrate Binding by ClpB. Molecular Cell. 2007;25:261–271. doi: 10.1016/j.molcel.2007.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee S, Sielaff B, Lee J, Tsai FTF. CryoEM structure of Hsp104 and its mechanistic implication for protein disaggregation. Proceedings of the National Academy of Sciences. 2010;107:8135–8140. doi: 10.1073/pnas.1003572107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lee S, Sowa ME, Watanabe Y-h, Sigler PB, Chiu W, Yoshida M, Tsai FTF. The Structure of ClpB: A Molecular Chaperone that Rescues Proteins from an Aggregated State. Cell. 2003;115:229–240. doi: 10.1016/s0092-8674(03)00807-9. [DOI] [PubMed] [Google Scholar]
- 64.Wendler P, Shorter J, Plisson C, Cashikar AG, Lindquist S, Saibil HR. Atypical AAA+ Subunit Packing Creates an Expanded Cavity for Disaggregation by the Protein-Remodeling Factor Hsp104. Cell. 2007;131:1366–1377. doi: 10.1016/j.cell.2007.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wendler P, Shorter J, Snead D, Plisson C, Clare DK, Lindquist S, Saibil HR. Motor Mechanism for Protein Threading through Hsp104. Molecular Cell. 2009;34:81–92. doi: 10.1016/j.molcel.2009.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wendler P, Saibil HR. Cryo electron microscopy structures of Hsp100 proteins: crowbars in or out? Biochem Cell Biol. 2010;88:89–96. doi: 10.1139/o09-164. [DOI] [PubMed] [Google Scholar]
- 67.Haslberger T, Weibezahn J, Zahn R, Lee S, Tsai FTF, Bukau B, Mogk A. M Domains Couple the ClpB Threading Motor with the DnaK Chaperone Activity. Molecular Cell. 2007;25:247–260. doi: 10.1016/j.molcel.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 68.Miot M, Reidy M, Doyle SM, Hoskins JR, Johnston DM, Genest O, Vitery MC, Masison DC, Wickner S. Species-specific collaboration of heat shock proteins (Hsp) 70 and 100 in thermotolerance and protein disaggregation. Proceedings of the National Academy of Sciences. 2011;108:6915–6920. doi: 10.1073/pnas.1102828108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sielaff B, Tsai FTF. The M-Domain Controls Hsp104 Protein Remodeling Activity in an Hsp70/Hsp40-Dependent Manner. Journal of Molecular Biology. 2010;402:30–37. doi: 10.1016/j.jmb.2010.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Parsell DA, Lindquist S. The function of heat-shock proteins in stress tolerance: degradation and reactivation of damaged proteins. Annu Rev Genet. 1993;27:437–496. doi: 10.1146/annurev.ge.27.120193.002253. [DOI] [PubMed] [Google Scholar]
- 71.Parsell DA, Sanchez Y, Stitzel JD, Lindquist S. Hspl04 is a highly conserved protein with two essential nucleotide-binding sites. Nature. 1991;353:270–273. doi: 10.1038/353270a0. [DOI] [PubMed] [Google Scholar]
- 72.Barnett ME, Zolkiewska A, Zolkiewski M. Structure and Activity of ClpB from Escherichia coli. Journal of Biological Chemistry. 2000;275:37565–37571. doi: 10.1074/jbc.M005211200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cashikar AG, Schirmer EC, Hattendorf DA, Glover JR, Ramakrishnan MS, Ware DM, Lindquist SL. Defining a Pathway of Communication from the C-Terminal Peptide Binding Domain to the N-Terminal ATPase Domain in a AAA Protein. Mol Cell. 2002;9:751–760. doi: 10.1016/s1097-2765(02)00499-9. [DOI] [PubMed] [Google Scholar]
- 74.Mackay RG, Helsen CW, Tkach JM, Glover JR. The C-terminal Extension of Saccharomyces cerevisiae Hsp104 Plays a Role in Oligomer Assembly. Biochemistry. 2008;47:1918–1927. doi: 10.1021/bi701714s. [DOI] [PubMed] [Google Scholar]
- 75.Li J, Sha B. Crystal structure of E. coli Hsp100 ClpB nucleotide-binding domain 1 (NBD1) and mechanistic studies on ClpB ATPase activity. J Mol Biol. 2002;318:1127–1137. doi: 10.1016/S0022-2836(02)00188-2. [DOI] [PubMed] [Google Scholar]
- 76.Li J, Sha B. Crystal Structure of the E. coli Hsp100 ClpB N-Terminal Domain. Structure (Camb) 2003;11:323–328. doi: 10.1016/s0969-2126(03)00030-3. [DOI] [PubMed] [Google Scholar]
- 77.Hattendorf DA, Lindquist SL. Analysis of the AAA sensor-2 motif in the C-terminal ATPase domain of Hsp104 with a site-specific fluorescent probe of nucleotide binding. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:2732–2737. doi: 10.1073/pnas.261693199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schirmer EC, Queitsch C, Kowal AS, Parsell DA, Lindquist S. The ATPase activity of Hsp104, effects of environmental conditions and mutations. J Biol Chem. 1998;273:15546–15552. doi: 10.1074/jbc.273.25.15546. [DOI] [PubMed] [Google Scholar]
- 79.Schirmer EC, Ware DM, Queitsch C, Kowal AS, Lindquist SL. Subunit interactions influence the biochemical and biological properties of Hsp104. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:914–919. doi: 10.1073/pnas.031568098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mogk A, Schlieker C, Strub C, Rist W, Weibezahn J, Bukau B. Roles of individual domains and conserved motifs of the AAA+ protein ClpB in oligomerization, ATP-hydrolysis and chaperone activity. J Biol Chem. 2003:M209686200. doi: 10.1074/jbc.M209686200. [DOI] [PubMed] [Google Scholar]
- 81.Tkach JM, Glover JR. Amino Acid Substitutions in the C-terminal AAA+ Module of Hsp104 Prevent Substrate Recognition by Disrupting Oligomerization and Cause High Temperature Inactivation. Journal of Biological Chemistry. 2004;279:35692–35701. doi: 10.1074/jbc.M400782200. [DOI] [PubMed] [Google Scholar]
- 82.Doyle SM, Shorter J, Zolkiewski M, Hoskins JR, Lindquist S, Wickner S. Asymmetric deceleration of ClpB or Hsp104 ATPase activity unleashes protein-remodeling activity. Nat Struct Mol Biol. 2007;14:114–122. doi: 10.1038/nsmb1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Franzmann TM, Czekalla A, Walter SG. Regulatory Circuits of the AAA+ Disaggregase Hsp104. J Biol Chem. 2011;286:17992–18001. doi: 10.1074/jbc.M110.216176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Schaupp A, Marcinowski M, Grimminger V, Bosl B, Walter S. Processing of proteins by the molecular chaperone Hsp104. J Mol Biol. 2007;370:674–686. doi: 10.1016/j.jmb.2007.04.070. [DOI] [PubMed] [Google Scholar]
- 85.Schirmer EC, Homann OR, Kowal AS, Lindquist S. Dominant Gain-of-Function Mutations in Hsp104p Reveal Crucial Roles for the Middle Region. Mol Biol Cell. 2004;15:2061–2072. doi: 10.1091/mbc.E02-08-0502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Weibezahn J, Schlieker C, Bukau B, Mogk A. Characterization of a trap mutant of the AAA+ chaperone ClpB. J Biol Chem. 2003;278:32608–32617. doi: 10.1074/jbc.M303653200. [DOI] [PubMed] [Google Scholar]
- 87.Watanabe Y-h, Takano M, Yoshida M. ATP Binding to Nucleotide Binding Domain (NBD)1 of the ClpB Chaperone Induces Motion of the Long Coiled-coil, Stabilizes the Hexamer, and Activates NBD2. Journal of Biological Chemistry. 2005;280:24562–24567. doi: 10.1074/jbc.M414623200. [DOI] [PubMed] [Google Scholar]
- 88.Ogura T, Whiteheart SW, Wilkinson AJ. Conserved arginine residues implicated in ATP hydrolysis, nucleotide-sensing, and inter-subunit interactions in AAA and AAA+ ATPases. Journal of Structural Biology. 2004;146:106–112. doi: 10.1016/j.jsb.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 89.Lupas AN, Martin J. AAA proteins. Current Opinion in Structural Biology. 2002;12:746–753. doi: 10.1016/s0959-440x(02)00388-3. [DOI] [PubMed] [Google Scholar]
- 90.Bochtler M, Hartmann C, Song HK, Bourenkov GP, Bartunik HD, Huber R. The structures of HsIU and the ATP-dependent protease HsIU-HsIV. Nature. 2000;403:800–805. doi: 10.1038/35001629. [DOI] [PubMed] [Google Scholar]
- 91.Wang F, Mei Z, Qi Y, Yan C, Hu Q, Wang J, Shi Y. Structure and mechanism of the hexameric MecA-ClpC molecular machine. Nature. 2011;471:331–335. doi: 10.1038/nature09780. [DOI] [PubMed] [Google Scholar]
- 92.Kirstein J, Zuhlke D, Gerth U, Turgay K, Hecker M. A tyrosine kinase and its activator control the activity of the CtsR heat shock repressor in B. subtilis. Embo J. 2005;24:3435–3445. doi: 10.1038/sj.emboj.7600780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kruger E, Witt E, Ohlmeier S, Hanschke R, Hecker M. The clp proteases of Bacillus subtilis are directly involved in degradation of misfolded proteins. J Bacteriol. 2000;182:3259–3265. doi: 10.1128/jb.182.11.3259-3265.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kirstein J, Moliere N, Dougan DA, Turgay K. Adapting the machine: adaptor proteins for Hsp100/Clp and AAA+ proteases. Nat Rev Microbiol. 2009;7:589–599. doi: 10.1038/nrmicro2185. [DOI] [PubMed] [Google Scholar]
- 95.Schlothauer T, Mogk A, Dougan DA, Bukau B, Turgay K. MecA, an adaptor protein necessary for ClpC chaperone activity. Proceedings of the National Academy of Sciences. 2003;100:2306–2311. doi: 10.1073/pnas.0535717100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kirstein J, Schlothauer T, Dougan DA, Lilie H, Tischendorf G, Mogk A, Bukau B, Turgay K. Adaptor protein controlled oligomerization activates the AAA+ protein ClpC. Embo J. 2006;25:1481–1491. doi: 10.1038/sj.emboj.7601042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Werbeck ND, Schlee S, Reinstein J. Coupling and Dynamics of Subunits in the Hexameric AAA+ Chaperone ClpB. J Mol Biol. 2008;378:178–190. doi: 10.1016/j.jmb.2008.02.026. [DOI] [PubMed] [Google Scholar]
- 98.Haslberger T, Zdanowicz A, Brand I, Kirstein J, Turgay K, Mogk A, Bukau B. Protein disaggregation by the AAA+ chaperone ClpB involves partial threading of looped polypeptide segments. Nat Struct Mol Biol. 2008;15:641–650. doi: 10.1038/nsmb.1425. [DOI] [PubMed] [Google Scholar]
- 99.Kedzierska S, Akoev V, Barnett ME, Zolkiewski M. Structure and Function of the Middle Domain of ClpB from Escherichia coli. Biochemistry. 2003;42:14242–14248. doi: 10.1021/bi035573d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Watanabe Yh, Nakazaki Y, Suno R, Yoshida M. Stability of the two wings of the coiled-coil domain of ClpB chaperone is critical for its disaggregation activity. Biochemical Journal. 2009;421:71–77. doi: 10.1042/BJ20082238. [DOI] [PubMed] [Google Scholar]
- 101.Kruger E, Volker U, Hecker M. Stress induction of clpC in Bacillus subtilis and its involvement in stress tolerance. J Bacteriol. 1994;176:3360–3367. doi: 10.1128/jb.176.11.3360-3367.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Turgay K, Hamoen LW, Venema G, Dubnau D. Biochemical characterization of a molecular switch involving the heat shock protein ClpC, which controls the activity of ComK, the competence transcription factor of Bacillus subtilis. Genes & Development. 1997;11:119–128. doi: 10.1101/gad.11.1.119. [DOI] [PubMed] [Google Scholar]
- 103.Leidhold C, Janowsky Bv, Becker D, Bender T, Voos W. Structure and function of Hsp78, the mitochondrial ClpB homolog. Journal of Structural Biology. 2006;156:149–164. doi: 10.1016/j.jsb.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 104.Lewandowska A, Gierszewska M, Marszalek J, Liberek K. Hsp78 chaperone functions in restoration of mitochondrial network following heat stress. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 2006;1763:141–151. doi: 10.1016/j.bbamcr.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 105.Röttgers K, Zufall N, Guiard B, Voos W. The ClpB Homolog Hsp78 Is Required for the Efficient Degradation of Proteins in the Mitochondrial Matrix. Journal of Biological Chemistry. 2002;277:45829–45837. doi: 10.1074/jbc.M207152200. [DOI] [PubMed] [Google Scholar]
- 106.Krzewska J, Langer T, Liberek K. Mitochondrial Hsp78, a member of the Clp/Hsp100 family in Saccharomyces cerevisiae, cooperates with Hsp70 in protein refolding. FEBS Lett. 2001;489:92–96. doi: 10.1016/s0014-5793(00)02423-6. [DOI] [PubMed] [Google Scholar]
- 107.Nieto-Sotelo J, Kannan KB, Martínez LM, Segal C. Characterization of a maize heat-shock protein 101 gene, HSP101, encoding a ClpB/Hsp100 protein homologue. Gene. 1999;230:187–195. doi: 10.1016/s0378-1119(99)00060-8. [DOI] [PubMed] [Google Scholar]
- 108.Queitsch C, Hong SW, Vierling E, Lindquist S. Heat Shock Protein 101 Plays a Crucial Role in Thermotolerance in Arabidopsis. Plant Cell. 2000;12:479–492. doi: 10.1105/tpc.12.4.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lee Y, Nagao RT, Key JL. A Soybean 101-kD Heat Shock Protein Complements a Yeast HSP104 Deletion Mutant in Acquiring Thermotolerance. The Plant Cell Online. 1994;6:1889–1897. doi: 10.1105/tpc.6.12.1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gallie DR, Fortner D, Peng J, Puthoff D. ATP-dependent Hexameric Assembly of the Heat Shock Protein Hsp101 Involves Multiple Interaction Domains and a Functional C-proximal Nucleotide-binding Domain. Journal of Biological Chemistry. 2002;277:39617–39626. doi: 10.1074/jbc.M204998200. [DOI] [PubMed] [Google Scholar]
- 111.Derré I, Rapoport G, Devine K, Rose M, Msadek T. ClpE, a novel type of HSP100 ATPase, is part of the CtsR heat shock regulon of Bacillus subtilis. Molecular Microbiology. 1999;32:581–593. doi: 10.1046/j.1365-2958.1999.01374.x. [DOI] [PubMed] [Google Scholar]
- 112.Ingmer H, Vogensen FK, Hammer K, Kilstrup M. Disruption and Analysis of the clpB, clpC, and clpE Genes in Lactococcus lactis: ClpE, a New Clp Family in Gram-Positive Bacteria. J Bacteriol. 1999;181:2075–2083. doi: 10.1128/jb.181.7.2075-2083.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Miethke M, Hecker M, Gerth U. Involvement of Bacillus subtilis ClpE in CtsR Degradation and Protein Quality Control. J Bacteriol. 2006;188:4610–4619. doi: 10.1128/JB.00287-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Doyle SM, Hoskins JR, Wickner S. Collaboration between the ClpB AAA+ remodeling protein and the DnaK chaperone system. Proceedings of the National Academy of Sciences. 2007;104:11138–11144. doi: 10.1073/pnas.0703980104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Schlieker C, Zentgraf H, Dersch P, Mogk A. ClpV, a unique Hsp100/Clp member of pathogenic proteobacteria. Biological Chemistry. 2005;386:1115–1127. doi: 10.1515/BC.2005.128. [DOI] [PubMed] [Google Scholar]
- 116.Dougan DA, Reid BG, Horwich AL, Bukau B. ClpS, a substrate modulator of the ClpAP machine. Mol Cell. 2002;9:673–683. doi: 10.1016/s1097-2765(02)00485-9. [DOI] [PubMed] [Google Scholar]
- 117.Sanchez Y, Parsell DA, Taulien J, Vogel JL, Craig EA, Lindquist S. Genetic evidence for a functional relationship between Hsp104 and Hsp70. J Bacteriol. 1993;175:6484–6491. doi: 10.1128/jb.175.20.6484-6491.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Vogel JL, Parsell DA, Lindquist S. Heat-shock proteins Hsp104 and Hsp70 reactivate mRNA splicing after heat inactivation. Curr Biol. 1995;5:306–317. doi: 10.1016/s0960-9822(95)00061-3. [DOI] [PubMed] [Google Scholar]
- 119.Higurashi T, Hines JK, Sahi C, Aron R, Craig EA. Specificity of the J-protein Sis1 in the propagation of 3 yeast prions. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:16596–16601. doi: 10.1073/pnas.0808934105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Tessarz P, Mogk A, Bukau B. Substrate threading through the central pore of the Hsp104 chaperone as a common mechanism for protein disaggregation and prion propagation. Molecular Microbiology. 2008;68:87–97. doi: 10.1111/j.1365-2958.2008.06135.x. [DOI] [PubMed] [Google Scholar]
- 121.Zietkiewicz S, Krzewska J, Liberek K. Successive and Synergistic Action of the Hsp70 and Hsp100 Chaperones in Protein Disaggregation. Journal of Biological Chemistry. 2004;279:44376–44383. doi: 10.1074/jbc.M402405200. [DOI] [PubMed] [Google Scholar]
- 122.Ziętkiewicz S, Lewandowska A, Stocki P, Liberek K. Hsp70 Chaperone Machine Remodels Protein Aggregates at the Initial Step of Hsp70-Hsp100-dependent Disaggregation. Journal of Biological Chemistry. 2006;281:7022–7029. doi: 10.1074/jbc.M507893200. [DOI] [PubMed] [Google Scholar]
- 123.Hoskins JR, Doyle SM, Wickner S. Coupling ATP utilization to protein remodeling by ClpB, a hexameric AAA+ protein. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:22233–22238. doi: 10.1073/pnas.0911937106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Mosser DD, Ho S, Glover JR. Saccharomyces cerevisiae Hsp104 enhances the chaperone capacity of human cells and inhibits heat stress-induced proapoptotic signaling. Biochemistry. 2004;43:8107–8115. doi: 10.1021/bi0493766. [DOI] [PubMed] [Google Scholar]
- 125.Zenthon JF, Ness F, Cox B, Tuite MF. The [PSI+] prion of Saccharomyces cerevisiae can be propagated by an Hsp104 orthologue from Candida albicans. Eukaryot Cell. 2006;5:217–225. doi: 10.1128/EC.5.2.217-225.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Schirmer EC, Lindquist S, Vierling E. An Arabidopsis heat shock protein complements a thermotolerance defect in yeast. Plant Cell. 1994;6:1899–1909. doi: 10.1105/tpc.6.12.1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Senechal P, Arseneault G, Leroux A, Lindquist S, Rokeach LA. The Schizosaccharomyces pombe Hsp104 disaggregase is unable to propagate the [PSI] prion. PLoS One. 2009;4:e6939. doi: 10.1371/journal.pone.0006939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lee YR, Nagao RT, Key JL. A soybean 101-kD heat shock protein complements a yeast HSP104 deletion mutant in acquiring thermotolerance. Plant Cell. 1994;6:1889–1897. doi: 10.1105/tpc.6.12.1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Schlee S, Beinker P, Akhrymuk A, Reinstein J. A Chaperone Network for the Resolubilization of Protein Aggregates: Direct Interaction of ClpB and DnaK. Journal of Molecular Biology. 2004;336:275–285. doi: 10.1016/j.jmb.2003.12.013. [DOI] [PubMed] [Google Scholar]
- 130.Kedzierska S, Chesnokova LS, Witt SN, Zolkiewski M. Interactions within the ClpB/DnaK bi-chaperone system from Escherichia coli. Archives of Biochemistry and Biophysics. 2005;444:61–65. doi: 10.1016/j.abb.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 131.Sielaff B, Tsai FTF. The M-Domain Controls Hsp104 Protein Remodeling Activity in an Hsp70/Hsp40-Dependent Manner. Journal of Molecular Biology. 2010;402 doi: 10.1016/j.jmb.2010.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Liu YH, Han YL, Song J, Wang Y, Jing YY, Shi Q, Tian C, Wang ZY, Li CP, Han J, Dong XP. Heat shock protein 104 inhibited the fibrillization of prion peptide 106–126 and disassembled prion peptide 106–126 fibrils in vitro. Int J Biochem Cell Biol. 2011;43:768–774. doi: 10.1016/j.biocel.2011.01.022. [DOI] [PubMed] [Google Scholar]
- 133.Narayanan S, Walter S, Reif B. Yeast prion-protein, sup35, fibril formation proceeds by addition and substraction of oligomers. Chembiochem. 2006;7:757–765. doi: 10.1002/cbic.200500382. [DOI] [PubMed] [Google Scholar]
- 134.Savistchenko J, Krzewska J, Fay N, Melki R. Molecular chaperones and the assembly of the prion Ure2p in vitro. J Biol Chem. 2008;283:15732–15739. doi: 10.1074/jbc.M800728200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Takahashi A, Hara H, Kurahashi H, Nakamura Y. A systematic evaluation of the function of the protein-remodeling factor Hsp104 in [PSI+] prion propagation in S. cerevisiae by comprehensive chromosomal mutations. Prion. 2007;1:69–77. doi: 10.4161/pri.1.1.4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Gokhale KC, Newnam GP, Sherman MY, Chernoff YO. Modulation of prion-dependent polyglutamine aggregation and toxicity by chaperone proteins in the yeast model. J Biol Chem. 2005;280:22809–22818. doi: 10.1074/jbc.M500390200. [DOI] [PubMed] [Google Scholar]
- 137.Kurahashi H, Nakamura Y. Channel mutations in Hsp104 hexamer distinctively affect thermotolerance and prion-specific propagation. Mol Microbiol. 2007;63:1669–1683. doi: 10.1111/j.1365-2958.2007.05629.x. [DOI] [PubMed] [Google Scholar]