

Abstract
Chronic inflammation has been strongly associated with tumor progression, but the underlying mechanisms remain elusive. Here we demonstrate that E3 ligase Itch and deubiquitinase Cyld form a complex via the interaction through ‘WW-PPXY’ motifs. The Itch-Cyld complex sequentially cleaved K63-linked ubiquitin chains and catalyzed K48-linked ubiquitination on the kinase Tak1 to terminate inflammatory tumor necrosis factor signaling. Reconstitution of wild-type Cyld but not mutant Cyld(Y485A), which cannot associate with Itch, blocked the sustained Tak1 activation and proinflammatory cytokine production by Cyld−/− bone marrow-derived macrophages. Itch or Cyld deficiency resulted in chronic production of tumor-promoting cytokines by the tumor-associated macrophages and aggressive growth of lung carcinoma. Thus, we have uncovered an Itch-Cyld mediated regulatory mechanism in innate inflammatory cells.
INTRODUCTION
Accumulating evidence indicates that tumor progression is governed not only by genetic changes in cancer cells, but also by epigenetic and environmental factors. Chronic infection and subsequent inflammation are considered critical environmental and epigenetic factors contributing to tumor progression1, 2. Primary examples are gastric cancer and Helicobacter pylori infections, hepatocellular carcinoma and viral hepatitis, colitis-associated cancer, and pancreatic cancer associated with chronic pancreatitis1. Similarly, chronic obstructive pulmonary disease (COPD), cigarette smoking, exposure to asbestos and silica are all associated with lung inflammation and lung carcinoma3–5. Inflammatory cells found in the tumor microenvironment, particularly tumor-associated macrophages (TAMs), constitute an important interface between tumor cells and the immune system6. Production of pro-inflammatory cytokines such as tumor necrosis factor (TNF), interleukin 6 (IL-6) and IL-1β, by TAMs not only trigger pro-survival signals in tumor cells, but also support their growth and metastasis6, 7. However, the molecular mechanisms that regulate inflammation and tumor progression remain unclear.
Post-translational modifications mediated by ubiquitin conjugation have emerged as a key regulatory mechanism in the immune cells8, 9. The ubiquitin signal is interpreted based on the number of attached ubiquitin chains and the topology of their linkage. A polyubiquitin chain is formed when one of the seven lysines within ubiquitin is linked to the C-terminal glycine of another ubiquitin. Although ubiquitin contains seven lysine residues, linkage generally occurs via either K48 or K63. K48-linked polyubiquitination predominantly targets proteins for proteasomal degradation, whereas K63-linked polyubiquitination results in non-proteasomal modifications, such as subcellular localization or protein-protein interactions8. We and others have demonstrated HECT type E3 ligase Itch as a critical regulator of inflammation10, 11. On a mouse C57BL/6 background, Itch deficiency results in the development of a late onset lymphoproliferation disorder and chronic pulmonary inflammation12, 13. Itch possesses four WW domains that recognize the Pro-rich PPXY (PY) consensus sequence in their substrate targets14. Although it is clear that Itch regulates inflammation, no systematic study has linked Itch to tumorigenesis.
Similar to phosphorylation, protein ubiquitination is reversible, and removal of ubiquitin molecules is mediated by de-ubiquitinating (DUB) enzymes, such as Cyld15. Cyld is encoded by a NF-κB-inducible gene16, inhibits NF-κB and the kinase JNK by removing K63-linked polyubiquitin chains from signaling pathway molecules such as Bcl-3, TRAF2, TRAF6 and Nemo (also known as IKK-γ)17–21. Cyld through its deubiquitinating function has been shown to regulate various types of human cancers, including lung, hepatocellular, colon and cervical cancers15. Cyld was also shown to negatively regulate tumor-promoting cytokine secretion by inflammatory cells22. Despite these important observations, the molecular mechanisms regulating Cyld function and its role in inflammation remain largely unknown. Here, we demonstrate that Itch interacts with Cyld and cooperatively regulates Tak1 and inflammation.
RESULTS
Augmented tumor growth in Itch−/− and Cyld−/− mice
We have demonstrated earlier that Itch deficiency results in elevated lung inflammation10, 11. Similarly, Cyld−/− mice exhibit inflammatory infiltrates in numerous tissues including lung, liver, spleen and the salivary glands22. Cyld deficiency resulted in increased susceptibility to spontaneous and chemically induced colitis and colon tumorigenesis22, 23. Cyld was also shown to regulate lung inflammation24, 25 and Cyld expression is frequently downregulated in non-small cell lung carcinoma (NSCLC) cell lines26. Over-expression of the full-length but not the catalytically inactive mutant of Cyld in NSCLC cell lines significantly reduces cell growth, suggesting that Cyld acts as tumor suppressor gene for the progression of NSCLC and that its deubiquitin function is important for this mechanism26. To determine the contribution of Itch and Cyld in inflammation and metastasis of lung carcinoma, we inoculated 1 × 106 Lewis Lung Carcinoma (LLC) cells into Cyld−/−, Itch−/− and control C57BL/6 mice intravenously via tail vein. Both Itch−/− and Cyld−/− mice exhibited higher mortality rate compared to wild-type mice (Fig. 1a). Histological analysis revealed aggressive tumor growth in the lungs of Itch−/− and Cyld−/− mice (Fig. 1b). Similarly, remarkable differences were seen in tumor multiplicity (Fig. 1c) and tumor size (Fig. 1d). To investigate the effect of Itch and Cyld deficiency on metastasis, we inoculated LLC cells subcutaneously (s.c.) and spontaneous lung metastasis was analyzed. A marked increase in the metastasis of LLC tumors was observed in Itch−/− and Cyld−/− mice (Supplementary Fig. 1). These data suggest that Itch and Cyld deficiency in the hosts result in elevated growth and metastasis of lung carcinoma.
Figure 1.

Itch−/− and Cyld−/− mice develop elevated growth and metastasis of Lewis Lung Carcinoma. (a) % survival of WT (n=12), Cyld−/− (n=13) and Itch−/−(n=11) mice inoculated with 1×106 Lewis Lung Carcinoma (LLC) cells via tail vein. (b) Representative H&E stained histological sections of the lungs of wild type, Itch−/− and Cyld−/− mice on day 15 after inoculation of LLC cells (1×106 cells). (c) Lung tumor multiplicity and (d) Tumor size. (e) Cytokine mRNA levels in CD11b+ macrophages from lung tumors of WT, Itch−/− and Cyld−/− mice inoculated with LLC cells (1×106) via tail vein. (f) Cytokine abundance in naïve Itch−/− and Cyld−/− lung tissues. *P<0.01(paired t-test). Data are representative of three independent experiments.
Since proinflammatory cytokines secreted by TAMs have been associated with enhanced tumor growth and metastasis6, we analyzed cytokine expression by tumor infiltrated CD11b+ cells. Enhanced expression of TNF, IL-6 and IL-1β was observed in both Itch−/− and Cyld−/− cells (Fig. 1e). Similarly, we have observed elevated expression of these cytokines from the whole tumor nodules isolated from the lungs of Itch−/− and Cyld−/− mice (data not shown). No significant difference in cytokine expression was observed in the lung tissues of naïve Itch−/− and Cyld−/− mice that were not inoculated with LLC cells (Fig. 1f), suggesting that Itch and Cyld negatively regulate TAM activity.
Itch interacts with Cyld via the WW-PPXY motifs
We previously reported that Itch regulated TNF-induced NF-κB signaling by modulating cFLIPL turnover27. Cyld deubiquitin activity was shown earlier to interfere with the ubiquitination of TRAF molecules and Tak1, which are involved directly in the TNF-associated NF-κB signaling pathway15. Since Itch and Cyld can regulate similar pathways, we tested whether these enzymes can cooperate to terminate NF-κB signaling and inflammatory response during tumor growth and metastasis. To determine whether Itch and Cyld can directly interact with each other, we performed sequence analysis and found a highly conserved Cyld ‘PPXY’ motif (Fig. 2a) by which it could interact with Itch ‘WW’ domains. We tested this interaction by transiently transfecting 293T cells with Flag-Itch and Myc-Cyld. Cell lysates were immunoprecipitated using control mouse IgG or antibodies against Flag or Myc. Antibody against Myc immunoprecipitated Flag-Itch and vice versa, suggesting that Itch physically interacts with Cyld (Fig. 2b). Further, we generated a fusion protein of glutathione S-transferase (GST) and Itch (GST-Itch) and performed pull-down assay using lysates of 293T cells transfected with Myc-Cyld, GST-Itch precipitated Myc-Cyld, but not GST alone, confirming the association (Supplementary Fig. 2). We further tested whether the interaction is direct by using GST-Itch and His-Cyld (expressed in Escherichia coli). GST-Itch precipitated His-Cyld in the pull-down assay confirming a direct interaction between Itch and Cyld (Fig. 2c).
Figure 2.

Itch and Cyld form a complex via interaction through PPXY motif. (a) Sequence alignment of PPXY motif of Cyld which is conserved across species. Y485 critical for interaction with Itch is indicated (*). (b) 293T cells transiently co-transfected with expression vectors for Flag-Itch and Myc-Cyld. The cell lysate was immunoprecipitated using mouse IgG, antibodies against Flag and Myc. The immunoprecipitates were blotted using antibodies against Flag and Myc. (c) Immunoblot of His-Cyld precipitated using GST or GST-Itch purified from E. coli. The input controls for His-Cyld, GST and GST-Itch are shown below (d) BMDMs were stimulated with TNF for thirty minutes. The cell lysates were immunoprecipitated using mouse IgG, rabbit IgG, antibodies against Itch and Cyld. The immunoprecipitates were analyzed by immunoblot with antibodies against Itch and Cyld. (e) 293T cells were co-transfected with expression vectors for HX-Cyld(Y485A) and Flag-Itch. The cells lysate was immunoprecipitated using antibody against Flag and immunoblotted using antibody against HX. Data are representative of three (b, c and e) and two (d) experiments.
To determine if this interaction occurs endogenously in primary cells, we stimulated bone marrow derived macrophages (BMDMs) with TNF for 30 min followed by immunoprecipitation experiments using antibodies against Itch and Cyld. Itch co-immunoprecipitated with Cyld (Fig. 2d). Itch and Cyld interaction in BMDMs seems to be stimulation dependent as we did not detect the association in unstimulated cells (data not shown). To test whether Itch-Cyld interaction occurs via the ‘PPXY’ motif, we generated Y to A mutant Cyld (Cyld(Y485A)) by site-directed mutagenesis. The Cyld(Y485A) mutation disrupted Itch-Cyld interaction (Fig. 2e). Taken together these data collectively indicate that Itch forms a complex with Cyld via its interaction through ‘PPXY’ motif.
Itch targets Tak1 for K48-linked ubiquitination
Next, we sought to understand the role of Itch-Cyld complex in the regulation of inflammatory response. Inflammatory stimuli driven signaling converge at the IκB kinase (IKK) complex, a trimeric holoenzyme comprised of catalytic subunits IKKα, IKKβ, and the regulatory subunit Nemo28. Tak1 is a key intermediate that transmits signals from receptor complexes to IKK. Tak1 associates with TNF-receptor associated factors TRAF2 or TRAF6 following stimulation with TNF or Toll-like receptor ligands and IL-1, respectively, to initiate a signaling cascade. Both TRAF2 and TRAF6 are RING domain ubiquitin ligases that catalyze K63-linked polyubiquitin chains, which results in Tak1 autophosporylation29. Activated Tak1 phosphorylates IKKβ within the activation loop, leading to the activation of IKK30. IKK in turn phosphorylates IκBα, resulting in NF-κB nuclear translocation and transcription of inflammatory target genes28. Tak1 activation is transient and is rapidly inactivated, which is important for preventing prolonged inflammatory responses31–33. Cyld was previously shown to cleave K63-linked polyubiquitin chain on Tak123. Because Itch deficiency resulted in persistent IKK and Jnk kinase activation34, similar to Cyld deficiency17–21, we reasoned that Itch may participate in terminating Tak1 activation by K48-linked polyubiquitination. To test this notion, we co-expressed Flag-Tak1 with Myc-Itch and either Hemaglutinin (HA) tagged Ub(WT), Ub(K48) [in which all the lysine residues, except K48, are mutated], or Ub(K63) [in which all the lysine residues, except K63, are mutated]. Slow-migrating high molecular weight polyubiquitinated forms of Tak1 was observed when Itch and Tak1 were co-expressed with Ub(WT) (Fig. 3a, lane1). Interestingly, Itch catalyzed Tak1 polyubiquitination when co-expressed with Ub(K48) (lane3), but not with Ub(K63) (lane5) suggesting that Itch targets Tak1 for K48-linked but not K63-linked ubiquitination. To confirm the specificity of Itch as the E3 ligase, we generated an Itch(C830A) mutant in which the conserved cysteine is mutated to alanine35. Expression of the Itch(C830A) mutant with either Ub(WT) or Ub(K48) did not result in Tak1 ubiquitination (Fig. 3a, lanes 2 and 4). To further confirm that Itch targets Tak1 for K48-linked polyubiquitination, we utilized Ub(K48R) in which only K48 was mutated to arginine. Co-expression of Ub(K48R) and Itch failed to polyubiquitinate Tak1 (Fig. 3b). Next, we tested whether another functionally related E3 ligase, Cbl-b8 ubiquitinates Tak1. Co-expression of Cbl-b with Tak1 did not result in Tak1 ubiquitination (Supplementary Fig. 3), suggesting that Itch is the specific ligase for Tak1.
Figure 3.

Itch and Cyld sequentially cleave K63-linked ubiquitination and catalyze K48-linked ubiquitination to deactivate Tak1. (a) 293T cells co-transfected with Flag-Tak1 along with Myc-Itch, HX-Itch (C830A), HA tagged Ub(WT), Ub(K63) and Ub(K48) as indicated. The cell lysate was immunoprecipitated using antibody against Flag and immunoblotted using antibody against HA. (b) 293T cells co-transfected with Flag-Tak1 along with Myc-Itch, HA-Ub(WT), HA-Ub(K48) and Ub(K48R) as indicated. The cell lysate was immunoprecipitated using antibody against Flag and immunoblotted using antibody against HA. (c) 293T cells co-transfected with Flag-Tak1 along with Myc-TRAF2, HX-TRAF2Δ, HA-Ub(K63), HX-Cyld, HX-Cyld(C601A), HX-Cyld(Y485A), Myc-Itch, HA-Ub(K48) in various combinations as indicated. The cell lysate was immunoprecipitated using antibody against Flag and immunoblotted using antibody against HA. (d) 293T cells were transfected as in (b) utilizing Ub(K48R) and Ub(K63R). (e) HX-Tak1, HX-TRAF2 and Flag-Ub(K63), HA-Ub(K48), Myc-Itch, and HX-Cyld were co-expressed in 293T cells. The cell lysate was immunoprecipitated using antibody against Tak1 and immunoblotted with antibody against Flag to detect K63-linked ubiquitination. The membranes were reprobed with antibody against HA to detect K48-linked ubiquitination. Data are representative of three or more independent experiments.
Itch-Cyld complex edits Tak1 ubiquitination
Because Cyld cleaves K63-linked ubiquitination of Tak123, we hypothesized that the ‘Itch-Cyld’ complex sequentially cleaves K63-linked ubiquitin chain and catalyzes K48-linked ubiquitination to deactivate Tak1 and terminate NF-κB signaling. Co-expression of TRAF2 with Tak1 and Ub(K63) resulted in Tak1 polyubiquitination (Fig. 3c, lane1). As expected the RING deletion mutant of TRAF2 that lacks the ligase activity36 failed to ubiquitinate Tak1 (Fig. 3c, lane2). Inclusion of Cyld(WT) (Fig. 3c, lane3) but not Cyld(C601A) mutant (Fig. 3c, lane4) that lacks the DUB activity37 resulted in diminished Tak1 ubiquitination suggesting that Cyld deubiquitinated Tak1. Similarly, Cyld(Y485A) mutant was defective in deubiquitinating Tak1 (Fig. 3c, lane5). Itch failed to ubiquitinate Tak1 in the presence of Cyld, TRAF2, and Ub(K63) (Fig. 3c, lane6), but in a similar combination co-expression of Ub(K48) resulted in Tak1 ubiquitination (Fig. 3c, lane7). This result suggests that following cleavage of K63-linked polyubiquitin chain, Itch catalyzes K48-linked polyubiquitination. The possibility of cross reactivity of antibodies against Xpress (HX) and Flag in these experiments are ruled out as we obtained single specific bands (Supplementary Fig. 4).
To further confirm these results we utilized Ub(K48R) (in which all lysines, except K48, are intact) and Ub(K63R) (in which all lysines, except K63, are intact). Expression of TRAF2 with Ub(K48R) (Fig. 3d, lane 2) but not Ub(K63R) (Fig. 3d, lane 1) resulted in Tak1 polyubiquitination. As expected RING deletion mutant of TRAF236 was defective in catalyzing ubiquitin chain on Tak1 (Fig. 3, lane 3). Cyld(WT) (Fig. 3d, lane 4) but not Cyld(C601A) mutant 37 deubiquitinated Tak1 (Fig. 3d, lane 5). Similarly, Cyld(Y485A) mutant was defective in deubiquitination (Fig. 3d, lane 6). Itch was unable to ubiquitinate Tak1 (Fig. 3d, lane 7) in the presence of Ub(K63R) but in a similar combination co-expression of Ub(K48R) resulted in Tak1 polyubiquitination (Fig. 3d, lane8). Similar results were obtained when TRAF6 was used instead of TRAF2 (Supplementary Fig. 5). To further demonstrate that Cyld-mediated cleavage of K63-linked ubiquitin chain formed by TRAF2 is a prerequisite for Itch-mediated K48-linked ubiquitination, we used Flag-Ub(K63) and HA-Ub(K48). Itch failed to form K48-linked ubiquitin chain on Tak1 without Cyld-mediated removal of K63-linked ubiquitin chain (Fig. 3e). These results suggest that Itch and Cyld cooperatively ‘switch’ K63-linked to K48-linked ubiquitination.
We also tested whether additional deubiquitinases, A20 and Cezanne38, 39, which deubiquitinate the signaling molecule RIP1, likewise regulate Tak1 ubiquitination. We co-expressed Tak1, Ub(K63), and TRAF2 along with either A20 or Cezanne. Both A20 and Cezanne failed to deubiquitinate Tak1 suggesting the substrate specificity of Cyld (Supplementary Fig. 6). Next, we analyzed whether Itch-mediated K48-linked polyubiquitination results in Tak1 degradation. Co-expression of HA-Tak1 with increasing Flag-Itch but not Itch(C830A) mutant resulted in reduced Tak1 abundance, suggesting that Itch regulates Tak1 turnover (Fig. 4a). Further, MG132 a proteasome inhibitor blocked Itch-mediated degradation of Tak1 in a similar experiment (Fig. 4b). This suggests that Itch-mediated K48-linked ubiquitination promotes proteasomal degradation of Tak1.
Figure 4.
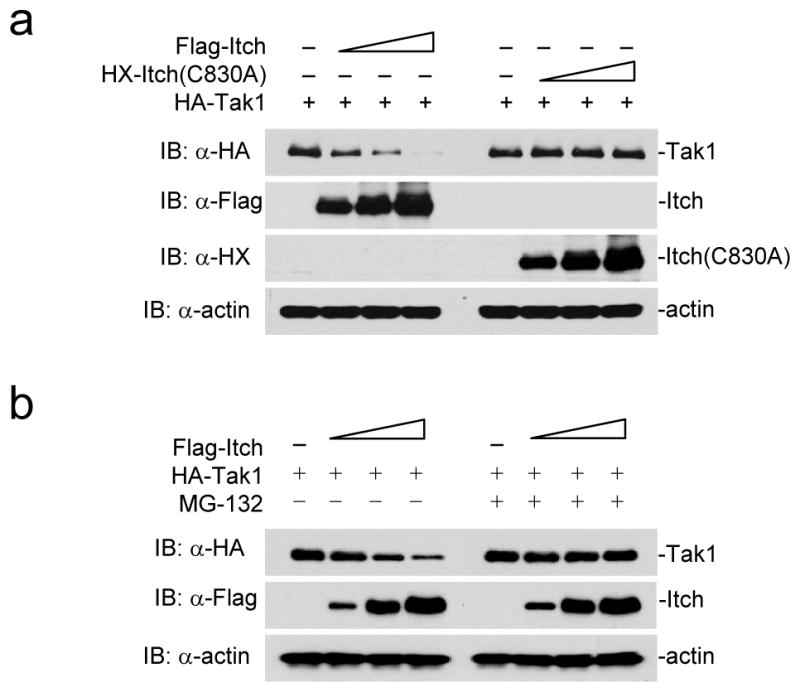
Itch-mediated polyubiquitination results in Tak1 degradation. (a) 293T cells were co-transfected with HA-Tak1 (1μg) and increasing concentrations of Flag-Itch or Itch(C830A). The cellular Tak1 level was analyzed by immunoblotting the lysate using antibody against HA. The level of expression of Itch and Itch-C830A was analyzed by reprobing the membranes using antibodies against Flag and HX. (b) 293T cells were transfected with HA-Tak1 (1μg) and increasing concentrations of Flag-Itch, the cells were cultured in the presence or absence of MG132. The cellular Tak1 level was analyzed by immunoblotting the lysate using antibody against HA. Data are representative of two experiments.
Tak1 activation in Itch−/− and Cyld−/− BMDMs
Subsequently, we investigated the effect of Itch and Cyld deficiency on Tak1 activation in BMDMs following treatment with TNF. The cell lysate was immunoprecipitated using antibody against TRAF2 and immunoblotted with antibody against phospho-Tak1 to test TRAF2-associated Tak1 activation. Tak1 phosphorylation peaked within five minutes following stimulation in wild-type macrophages and the phosphorylation was reduced to minimal within thirty minutes, however, we have observed sustained Tak1 phosphorylation in Itch−/− and Cyld−/− BMDMs (Fig. 5a). We reprobed the membranes with Tak1-specific antibody to analyze total TRAF2-associated Tak1. Similar to Tak1 phosphorylation, association of Tak1 with TRAF2 was transient in wild-type macrophages; however, Itch and Cyld deficiency prolonged the association. To analyze the effect of Itch and Cyld deficiency on total cellular Tak1 abundance, we immunoblotted the cell lysate with antibody against Tak1. We observed only a marginal decrease in the total Tak1 protein in wild-type BMDMs stimulated with TNF. This finding suggests that Itch-Cyld complex degrades the Tak1 that is associated with TRAF complex but not the entire pool of Tak1 within the cell during an inflammatory response.
Figure 5.

Sustained Tak1 activation results in chronic production of inflammatory cytokines by Itch−/− and Cyld−/− BMDMs. (a) Itch−/− and Cyld−/− BMDMs were stimulated with TNF and lysed at the indicated time points. The lysate was immunoprecipitated with antibody against TRAF2 and immunoblotted with antibody against p-Tak1. The membranes were reprobed with antibodies against Tak1 and TRAF2. The total cell lysate was blotted with antibodies against Tak1, TRAF2, Cyld, Itch and actin. (b) BMDMs were stimulated with TNF, total RNA was isolated at the indicated time points and expression of IL-6, TNF and IL-1β was analyzed using real-time PCR. (c) IL-6 and IL-1β concentration were assayed in the culture supernatant by ELISA. Data are representative of three experiments (b–c mean and s.d. of triplicate wells).
Because Tak1 activation is directly linked to pro-inflammatory cytokine secretion, we analyzed the expression of IL-6, TNF and IL-1β in BMDMs after stimulation with TNF. Expression of proinflammatory cytokine mRNA was sustained until 12 hours in Itch−/− and Cyld−/− BMDMs (Fig. 5b). We also analyzed the cytokine concentrations in the culture supernatant by ELISA. Itch and Cyld deficiency resulted in elevated secretion of proinflammatory cytokines (Fig. 5c). This result provides evidence that cleavage of K63-linked polyubiquitination is not sufficient to deactivate Tak1 and downstream effector function, rather sequential Itch-mediated K48 linked ubiquitination is essential. Similarly, sustained Tak1 phosphorylation and elevated cytokine mRNA expression were observed in Itch−/− and Cyld−/− BMDMs stimulated with lipopolysaccharide (LPS) (Supplementary Fig. 7).
To test whether inhibiting Tak1 activation attenuates cytokine production, we used (5Z)-7-Oxozeanol, a selective inhibitor of Tak140. BMDMs were pre-incubated for 30 minutes prior to TNF stimulation with indicated concentrations of (5Z)-7-Oxozeaenol, which decreased cytokine mRNA abundance in a dose-dependent manner (Fig. 6a). Similar results were obtained when BMDMs were stimulated with LPS in the presence of (5Z)-7-Oxozeaenol (Supplementary Fig. 8). Also, pre-incubation of BMDMs with (5Z)-7-Oxozeaenol inhibited sustained Tak1 phosphorylation observed in Itch−/− and Cyld−/− BMDMs (Fig. 6b). Next, we tested whether ectopic expression of a catalytically inactive dominant-negative dnTak1 (Tak1(K63W))41 inhibits elevated cytokine production by Itch−/− and Cyld−/− BMDMs. Expression of dnTak1 in primary BMDMs inhibited cytokine production by Itch−/− and Cyld−/− BMDMs (Fig. 6c–d). These data collectively suggest that sustained Tak1 activation in Itch−/− and Cyld−/− BMDMs results in chronic production of proinflammatory cytokines.
Figure 6.

Inhibiting Tak1 activation rescues elevated inflammatory cytokine production by Itch−/− and Cyld−/− BMDMs (a) BMDMs were pre-incubated with indicated amounts of (5Z)-7-Oxozeaenol(Ox) and stimulated with TNF for 6 hours, expression of IL-6, TNF, and IL-1β was analyzed by real-time PCR. (b) BMDMs were pretreated with 100 nM of Ox for 30 minutes and then stimulated with TNF for the indicated time points. The cell lysates were immunoprecipitated using antibody against TRAF2 and immunoblotted using antibody against p-Tak1. The total lysate was immunoblotted using antibodies against TRAF2 and actin. (c) WT, Itch−/− and Cyld−/− BMDMs were transduced with a lentiviral vector encoding dominant negative Tak1 (dnTak1). The efficiency of ectopic expression was analyzed by immunoblotting the total cell lysate using antibody against V5. (d) WT, Itch−/− and Cyld−/− BMDMs transduced with dnTak1 were stimulated with TNF and expression of IL-6, TNF and IL-1β was analyzed by real-time PCR. Data are representative of three experiments. (a, d mean and s.d. of triplicate wells).
Itch and Cyld enzyme activity regulates Tak1
To determine the functional relevance of Tak1 regulation by Itch and Cyld, we reconstituted Itch−/− MEFs with Itch(WT) and the ligase deficient Itch(C830A) mutant. As expected, reconstitution of Itch−/− MEFs with Itch(WT) but not Itch(C830A) mutant normalized Tak1 activation (Fig. 7a). Similarly, IL-6 production by Itch−/− MEFs reconstituted with Itch(WT) but not Itch(C830A) mutant was comparable to wild-type MEFs (Fig. 7b). This result is consistent with the finding that Itch(C830A) mutant failed to ubiquitinate Tak1 (Fig. 3a). We also reconstituted Cyld−/− MEFs with Cyld(WT), Cyld(C601A) that lacks the DUB activity37 and Cyld(Y485A) mutant that fails to interact with Itch. Reconstitution of Cyld−/− MEFs with Cyld(WT) restored transient Tak1 activation (Fig. 7c–d) and normalized IL-6 production. However, Cyld(C601A) and Cyld(Y485A) mutants were impaired in the termination of Tak1 activation. Collectively these data indicate that DUB function of Cyld and its interaction with Itch is essential for the termination of Tak1 and inflammatory cytokine production.
Figure 7.

Ligase activity of Itch, DUB activity of Cyld, and Cyld-Itch interaction is necessary for termination of TNF induced Tak1 activation. (a) Itch−/− MEFs were reconstituted with either V5-Itch(WT) or V5-Itch(C830A) mutant using a lentiviral transduction system. The cells lysed at indicated time points post TNF stimulation. The lysate was immunoprecipitated using against TRAF2 and immunoblotted with antibody against p-Tak1. The same membrane was reprobed with antibody against Tak1. The cell lysate from WT MEFs stimulated with TNF was used as control. (b) Itch−/− MEFs transduced with V5-Itch(WT) or V5-Itch(C830A) mutant as in (a) were stimulated with TNF and expression of IL-6 was analyzed by ELISA in the culture supernatant. (c) Cyld−/− MEFs were reconstituted with V5-Cyld(WT), V5-Cyld(C601A) or V5-Cyld(Y485A) mutant. The cells lysed at indicated time points post TNF stimulation. The lysate was immunoprecipitated using antibody against TRAF2 and immunoblotted with antibody against p-Tak1. The same membrane was reprobed with antibody against Tak1. (d) Cyld−/− MEFs transduced with V5-Cyld(WT), V5-Cyld(Y485A) or V5-Cyld(C601A) mutant as in (c) stimulated with TNF and expression of IL-6 was analyzed by ELISA in the culture supernatant. Data are representative of three (b, d) and two (a, c) experiments. (b,d mean and s.d. of triplicate wells).
To further confirm the role of Itch and Cyld interaction for terminating Tak1 activation in macrophages, we reconstituted Cyld−/− BMDMs with Cyld(WT) or Cyld(Y485A) mutant that fails to interact with Itch. Expression of Cyld(WT) but not Cyld(Y485A) mutant restored transient Tak1 activation (Fig. 8a). Similarly, wild-type Cyld almost completely reverted elevated cytokine production by Cyld−/− BMDMs; however, expression of Cyld(Y485A) mutant had only minimum effect (Fig. 8b–c). These data clearly suggest that Itch and Cyld cooperatively regulate cytokine production by the inflammatory cells.
Figure 8.

Reconstitution of Cyld−/− BMDMs with WT but not Cyld(Y485A) mutant rescues defects in termination of Tak1 activation and chronic production of inflammatory cytokines. (a) V5-Cyld(WT) or V5-Cyld(Y485A) mutant was ectopically expressed in Cyld−/− BMDMs using a lentiviral transduction system. The cells lysed at indicated time points post TNF stimulation. The lysate was immunoprecipitated using antibody against TRAF2 and immunoblotted with antibody against p-Tak1. The same membrane was reprobed with antibody against Tak1. The cell lysate from WT BMDMs stimulated with TNF was used as control in these experiments. (b) WT and Cyld−/− transduced with V5- Cyld(WT) or V5-Cyld(Y485A) mutant were stimulated with TNF and expression of IL-6, TNF and IL-1β was analyzed by real-time PCR. (c) IL-6 and IL-1β concentration were assayed in the culture supernatant by ELISA. Data are representative of at least three independent experiments (b–c mean and s.d. of triplicate wells).
DISCUSSION
In this study, we demonstrate that Itch forms a ubiquitin-editing complex with Cyld via the interaction through WW-PPXY motifs. Itch-Cyld complex promotes the transition of K63-linked to K48-linked ubiquitination by sequentially cleaving K63-linked ubiquitin chain and by catalyzing K48-linked ubiquitination on Tak1. During the early phase of TNF- or TLR-induced inflammatory response RING type E3 ligases TRAF2 and TRAF6 promote K63-linked ubiquitination within the kinase domain29, promoting phosphorylation of Tak1 that leads to IKK-NF-κB activation and transcription of proinflammatory cytokine gene expression42. We propose that the combined activity of Itch and Cyld shifts Tak1 ubiquitination from K63-linked toward K48-linked chains that triggers the proteasomal degradation of Tak1, thus terminating the inflammatory response. Itch-Cyld association was detected only following stimulation of BMDMs with TNF suggesting the requirement of extracellular signals. At present the molecular mechanisms that regulate Itch-Cyld complex formation remain unclear. One possibility is that Cyld expression is upregulated by TNF and IL-1β, via IKK-NF-κB pathway16, 24, 25 triggers its association with Itch. Alternatively, Itch-Cyld association could be regulated by NF-κB-dependent phosphorylation mediated mechanisms. Such a phenomenon has recently been demonstrated for the recruitment of Itch to A20 complex by TAXIBP134, 43.
The molecular basis for the chronic multi-organ inflammatory disorder observed in Itch−/− mice has been attributed to biased TH2 differentiation due to the defect in the degradation of JunB13. Our previous studies have shown that Itch also regulates T cell tolerance, as Itch−/− T cells are resistant to anergy induction44 and TGF-β mediated suppression11. Our present results indicate a critical role for Itch in the regulation innate immune cells via targeting Tak1 for proteasomal degradation. These findings along with recent reports that Itch is a component of A20, TAXIBP1 and RNF11 complex34, 43 raise the possibility that Itch has more prominent role as a negative regulator of inflammatory signaling in the innate immune cells.
Itch seems to be a specific ligase that targets Tak1 for K48-linked ubiquitination as Cbl-b a functionally related E3 ligase8 which also regulates NF-κB pathway45 but failed to ubiquitinate Tak1. Similarly, several other DUBs including A20, Cezanne, USP4, and USP21 are shown to negatively regulate NF-κB pathway39. In our assay system we did not detect DUB activity of A20 or Cezanne towards Tak1, suggesting the specificity of Cyld. However, we do not completely rule out the involvement of additional DUBs or E3 ligases in the regulation of Tak1 and downstream signaling.
An additional observation from our studies is that Itch−/− and Cyld−/− TAMs produced elevated tumor promoting inflammatory cytokines. Consistently, Itch−/− and Cyld−/− mice exhibited aggressive growth and metastasis of transplanted LLC tumors. However, it is not clear whether the enhanced tumor growth observed is exclusively due to the chronic production of tumor promoting cytokines by TAMs. Aberrant T cell response in Itch−/− and Cyld−/− mice8, 10, 11, 13 may also contribute to tumor growth. Further detailed analysis is essential for a clear and better understanding of these mechanisms that could lead to innovative therapeutic targeting.
Consistent with our findings, cytokines produced by activated innate immune cells, particularly TAMs, have been associated with inflammation and cancer6. Clinical studies have shown that increased numbers of TAMs are correlated with angiogenesis, metastasis and poor prognosis6. Tumor-promoting role of TAMs is linked to NF-κB-mediated production of pro-inflammatory cytokines, including TNF and IL-67 supporting the notion that excessive, chronically produced pro-inflammatory cytokines contribute to tumor progression. Our findings have identified an unanticipated co-ordination between Itch and Cyld that restricts prolonged inflammatory signaling and secretion of tumor promoting inflammatory cytokines by the macrophages.
Our studies also show that Tak1 selective inhibitor, (5Z)-7-Oxozeaenol inhibits tumor promoting cytokine production by BMDMs. Since NF-κB has emerged as a key internal factor that promotes inflammation and cancer1, 2, several approaches have been designed to block NF-κB, including inhibitors of IKK and proteasome inhibitors to inhibit IκB degradation46. Although proteasome inhibitors like bortezomib block NF-κB in a variety of cells and are being used in clinical trials against multiple myeloma, the results of the phase I/II clinical trials indicate the drug has limited activity against advanced NSCLC47, 48. In addition, complications have been reported after global inhibition of NF-κB using genetic and pharmacological approaches49. This highlights the requirement for more specific inhibitors to dampen inflammation and tumor progression. Our results provide a mechanistic insight into regulation of Tak1 for termination of inflammatory cytokine production which can be exploited therapeutically to target inflammatory cells in the tumor microenvironment. By targeting inflammatory cells, which are genetically normal and stable compared to genetically unstable carcinoma cells, development of drug resistance could be minimized. In addition, a combination of conventional cytotoxic cancer therapy along with inhibitors that block inflammatory responses could potentially enhance current therapeutic strategies.
METHODS
Cell culture and transfection
293T, 293FT (Invitrogen R700-07) and Lewis Lung Carcinoma cells (LLC, ATCC CRL-1642) were cultured in DMEM supplemented with 10% FBS. All of the transient cell transfections were performed using Lipofectamine 2000 (Invitrogen) following manufacturer’s instructions. MEFs were prepared from 12.5–14.5 day embryos. The genotyping of the Itch−/− and Cyld−/− MEFs was performed by PCR.
Animals
Cyld−/− and Itch−/− mice were described before12, 18. Control C57BL/6 mice were purchased from Charles River Laboratory. All the mice were housed in microisolator cages in the barrier facility of Karmanos Cancer Institute. Animal experiments were performed in accordance with the guidelines of the Institutional Animal Care and Use Committee of Wayne State University/Karmanos Cancer Institute.
Plasmids and Constructs
Full length TRAF6 was amplified from human cDNA clone (MHS1010-7296061, Open Biosystems) and cloned into the vector pEF4/HisC (Invitrogen) between BamHI-EcoRI to create HX-TRAF6. Full length human Tak1 and mouse Tak1 were amplified from human and mouse cDNA clones (MHS1010-58135 and MMM1013-64018, Open Biosystems) and cloned into the vector pCMV-Tag2B between BamHI-EcoRI to create both human and mouse Flag-Tak1. HA-Ub(WT), HA-Ub(K63), HA-Ub(K48), GST-Itch, and Itch-C830A were described before35. His-Cyld was created from myc-Cyld by subcloning into pET30a between BamHI-NotI. HX-Cyld(C601A) was created from HA-Cyld(C601A) (15507, addgene) by subcloning into pEF4/HisC between BamHI-NotI. HX-Cyld(Y485A) was created from Myc-Cyld by site-directed mutagenesis and subcloning into pEF4/HisC between BamHI-NotI. HX-TRAF2 was created from HA-Flag-TRAF2 (20229, Addgene) by subcloning into pEF4/HisC between BamHI-NotI. Myc tagged TRAF2 mutant lacking the Ring domain, amino acid residues 87–501 (Myc-TRAF2Δ), was created by subcloning into pcDNA3.1/myc-His (−) A (Invitrogen) between BamHI-HindIII. Myc-A20 was created from pEGFP-C1-A20 (22141, Addgene) by subcloning into pcDNA3.1/myc-His (−) A between XhoI-EcoRI. HX-Cezanne was created from Flag-HA-OTUD7B (22550, Addgene) by subcloning into pEF4/HisC between EcoRI-NotI. All clones were sequence verified.
Antibodies and Reagents
Following antibodies were used in this study: antibody against c-Myc (sc-40, Santa Cruz), antibody against Flag (F1804, F7425, Sigma), antibody against Itch (611198, BD), antibody against Cyld (4495, Cell Signaling), antibody against Tak1 (sc-7162, Santa Cruz), antibody against phospho-Tak1 (4531, Cell Signaling), antibody against TRAF2 (sc-876, Santa Cruz), antibody against TRAF6 (sc-7221, Santa Cruz), antibody against actin (A5441, Sigma), antibody against Ubiquitin (sc-8017, Santa Cruz), antibody against Xpress(HX) (R910-25, Invitrogen), antibody against HA (sc-805, Santa Cruz), antibody against V5 (R960-25, Invitrogen), normal Rabbit IgG (sc-2027) and normal mouse IgG (sc-2025). Tak1 inhibitor (5Z)-7-Oxozeaenol was purchased from Tocris Bioscience (Missouri, USA) and MG132 was from Calbiochem.
BMDMs
BMDMs were prepared from femurs and tibias of age matched WT, Cyld−/− and Itch−/− mice and cultured in DMEM supplemented with 15% FBS, 2mM L-glutamine, and 10 ng/ml GM-CSF (AF-315-03, Peprotech). BMDMs were stained with antibody against CD11b and subjected to flow cytometry to confirm the purity of macrophages (>90%). BMDMs were cultured for seven days (6 days with GM-CSF and 1 day without GM-CSF) prior to stimulation with 50 ng/ml TNF (RMTNFAI, Thermo Scientific) or 10 ng/ml LPS (E. coli J5, 437620, Calbiochem). In experiments that were performed along with pretreatment with Tak1 inhibitor (5Z)-7-Oxozeaenol, cells were pretreated for 30 min with varying concentrations of inhibitor as indicated before stimulation.
Lentiviral transduction
Expression clones were created using the Gateway cloning technology (Invitrogen) following the manufacturer’s instructions. Dominant negative expression clone of mouse Tak1 was constructed by first PCR subcloning Tak1K63W (BamHI-EcoRI) into pENTR-3C entry vector (Invitrogen) and then LR recombined to pLenti6.2/N-Lumio/V5-DEST (Invitrogen). Expression clones for Cyld (BamHI-BamHI), Cyld(Y485A) (BamHI-BamHI), Cyld(C601A) (BamHI-NotI), Itch (BamHI-NotI) and Itch(Y485A) (BamHI-NotI) were similarly prepared by first PCR subcloning into pENTR-3C entry vector (Invitrogen) and then LR recombined to pLenti6.2/N-Lumio/V5-DEST (Invitrogen).
Lentiviral transduction of MEFs and BMDMs was performed using the ViraPower lentiviral expression system (Invitrogen). Briefly, 293FT cells were used to package and produce the viruses. BMDMs and MEFs were incubated with the virus-containing supernatants at 37°C overnight in a 5% CO2 incubator. Successfully transduced cells were selected by further incubation for 48 h in medium containing 2 μg/ml Blasticidin before TNF treatment.
Lung metastasis assays
Sub-confluent LLC cells were trypsinized, washed three times with PBS, and filtered through a cell strainer to obtain single-cell suspension. Cells of >90% viability (Trypan Blue staining) were used. Seven weeks old mice were injected with 1×106 cells via tail vein. Mice were sacrificed at indicated days and lungs were removed for histological examination while some mice were kept until death and survival data were obtained. Lung tumor multiplicity count, the number of incidences of tumors in lungs per mouse, was scored under the microscope after the lungs were paraformaldehyde fixed, paraffin embedded, sectioned and stained by hematoxylin and eosin staining. For spontaneous lung metastasis, 2×106 cells were subcutaneously injected into the left flank of mice and tumor growth was monitored till the primary tumors reached a size of 2000 mm3 and after which mice were sacrificed to determine tumor multiplicity count.
Supplementary Material
Acknowledgments
We thank William C. Hahn (Dana-Farber Cancer Institute, Boston, MA) for Myc-Cyld, Jonathan Ashwell (NCI, Bethesda, MD) for HA tagged TRAF2Δ, Avraham Raz for pET30a, Yi-Chi M. Kong and Ramkumar Mathur(Columbia University, NY), for helpful discussions. This work was supported by National Cancer Institute (NCI) grant 1RC1CA146576-01 to K.V.
Footnotes
Contributions
N.A., M.Z., and I.S. did the experiments and analyzed data; L.P. assisted with in vivo tumor studies; W.W. assisted in manuscript preparation; C.R., R.F. provided critical reagents and mouse model; R.M. provided critical mouse model and assisted in manuscript preparation; K.V. designed the experiments, analyzed data and wrote the manuscript.
Competing financial interests
The authors declare no competing financial interests.
References
- 1.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 3.Takahashi H, Ogata H, Nishigaki R, Broide DH, Karin M. Tobacco smoke promotes lung tumorigenesis by triggering IKKbeta- and JNK1-dependent inflammation. Cancer Cell. 17:89–97. doi: 10.1016/j.ccr.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moghaddam SJ, et al. Promotion of lung carcinogenesis by chronic obstructive pulmonary disease-like airway inflammation in a K-ras-induced mouse model. Am J Respir Cell Mol Biol. 2009;40:443–453. doi: 10.1165/rcmb.2008-0198OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Van den Heuvel MM, Burgers SA, van Zandwijk N. Immunotherapy in non-small-cell lung carcinoma: from inflammation to vaccination. Clin Lung Cancer. 2009;10:99–105. doi: 10.3816/CLC.2009.n.012. [DOI] [PubMed] [Google Scholar]
- 6.Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141:39–51. doi: 10.1016/j.cell.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin WW, Karin M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J Clin Invest. 2007;117:1175–1183. doi: 10.1172/JCI31537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Venuprasad K. Cbl-b and itch: key regulators of peripheral T-cell tolerance. Cancer Res. 2010;70:3009–3012. doi: 10.1158/0008-5472.CAN-09-4076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Malynn BA, Ma A. Ubiquitin makes its mark on immune regulation. Immunity. 2010;33:843–852. doi: 10.1016/j.immuni.2010.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Venuprasad K, et al. Convergence of Itch-induced ubiquitination with MEKK1-JNK signaling in Th2 tolerance and airway inflammation. J Clin Invest. 2006;116:1117–1126. doi: 10.1172/JCI26858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Venuprasad K, et al. The E3 ubiquitin ligase Itch regulates expression of transcription factor Foxp3 and airway inflammation by enhancing the function of transcription factor TIEG1. Nat Immunol. 2008;9:245–253. doi: 10.1038/niXXXX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perry WL, et al. The itchy locus encodes a novel ubiquitin protein ligase that is disrupted in a18H mice. Nat Genet. 1998;18:143–146. doi: 10.1038/ng0298-143. [DOI] [PubMed] [Google Scholar]
- 13.Fang D, et al. Dysregulation of T lymphocyte function in itchy mice: a role for Itch in TH2 differentiation. Nat Immunol. 2002;3:281–287. doi: 10.1038/ni763. [DOI] [PubMed] [Google Scholar]
- 14.Melino G, et al. Itch: a HECT-type E3 ligase regulating immunity, skin and cancer. Cell Death Differ. 2008;15:1103–1112. doi: 10.1038/cdd.2008.60. [DOI] [PubMed] [Google Scholar]
- 15.Massoumi R. Ubiquitin chain cleavage: CYLD at work. Trends Biochem Sci. 2010;35:392–399. doi: 10.1016/j.tibs.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 16.Jono H, et al. NF-kappaB is essential for induction of CYLD, the negative regulator of NF-kappaB: evidence for a novel inducible autoregulatory feedback pathway. J Biol Chem. 2004;279:36171–36174. doi: 10.1074/jbc.M406638200. [DOI] [PubMed] [Google Scholar]
- 17.Reiley W, Zhang M, Sun SC. Negative regulation of JNK signaling by the tumor suppressor CYLD. J Biol Chem. 2004;279:55161–55167. doi: 10.1074/jbc.M411049200. [DOI] [PubMed] [Google Scholar]
- 18.Massoumi R, Chmielarska K, Hennecke K, Pfeifer A, Fassler R. Cyld inhibits tumor cell proliferation by blocking Bcl-3-dependent NF-kappaB signaling. Cell. 2006;125:665–677. doi: 10.1016/j.cell.2006.03.041. [DOI] [PubMed] [Google Scholar]
- 19.Brummelkamp TR, Nijman SM, Dirac AM, Bernards R. Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF-kappaB. Nature. 2003;424:797–801. doi: 10.1038/nature01811. [DOI] [PubMed] [Google Scholar]
- 20.Kovalenko A, et al. The tumour suppressor CYLD negatively regulates NF-kappaB signalling by deubiquitination. Nature. 2003;424:801–805. doi: 10.1038/nature01802. [DOI] [PubMed] [Google Scholar]
- 21.Trompouki E, et al. CYLD is a deubiquitinating enzyme that negatively regulates NF-kappaB activation by TNFR family members. Nature. 2003;424:793–796. doi: 10.1038/nature01803. [DOI] [PubMed] [Google Scholar]
- 22.Zhang J, et al. Impaired regulation of NF-kappaB and increased susceptibility to colitis-associated tumorigenesis in CYLD-deficient mice. J Clin Invest. 2006;116:3042–3049. doi: 10.1172/JCI28746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reiley WW, et al. Deubiquitinating enzyme CYLD negatively regulates the ubiquitin-dependent kinase Tak1 and prevents abnormal T cell responses. J Exp Med. 2007;204:1475–1485. doi: 10.1084/jem.20062694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lim JH, et al. Tumor suppressor CYLD regulates acute lung injury in lethal Streptococcus pneumoniae infections. Immunity. 2007;27:349–360. doi: 10.1016/j.immuni.2007.07.011. [DOI] [PubMed] [Google Scholar]
- 25.Lim JH, et al. Tumor suppressor CYLD acts as a negative regulator for non-typeable Haemophilus influenza-induced inflammation in the middle ear and lung of mice. PLoS One. 2007;2:e1032. doi: 10.1371/journal.pone.0001032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhong S, Fields CR, Su N, Pan YX, Robertson KD. Pharmacologic inhibition of epigenetic modifications, coupled with gene expression profiling, reveals novel targets of aberrant DNA methylation and histone deacetylation in lung cancer. Oncogene. 2007;26:2621–2634. doi: 10.1038/sj.onc.1210041. [DOI] [PubMed] [Google Scholar]
- 27.Chang L, et al. The E3 ubiquitin ligase itch couples JNK activation to TNFalpha-induced cell death by inducing c-FLIP(L) turnover. Cell. 2006;124:601–613. doi: 10.1016/j.cell.2006.01.021. [DOI] [PubMed] [Google Scholar]
- 28.Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 29.Fan Y, et al. Lysine 63-linked polyubiquitination of TAK1 at lysine 158 is required for tumor necrosis factor alpha- and interleukin-1beta-induced IKK/NF-kappaB and JNK/AP-1 activation. J Biol Chem. 2010;285:5347–5360. doi: 10.1074/jbc.M109.076976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang C, et al. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature. 2001;412:346–351. doi: 10.1038/35085597. [DOI] [PubMed] [Google Scholar]
- 31.Broglie P, Matsumoto K, Akira S, Brautigan DL, Ninomiya-Tsuji J. Transforming growth factor beta-activated kinase 1 (TAK1) kinase adaptor, TAK1-binding protein 2, plays dual roles in TAK1 signaling by recruiting both an activator and an inhibitor of TAK1 kinase in tumor necrosis factor signaling pathway. J Biol Chem. 285:2333–2339. doi: 10.1074/jbc.M109.090522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kajino T, et al. Protein phosphatase 6 down-regulates TAK1 kinase activation in the IL-1 signaling pathway. J Biol Chem. 2006;281:39891–39896. doi: 10.1074/jbc.M608155200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takaesu G, et al. Interleukin-1 (IL-1) receptor-associated kinase leads to activation of TAK1 by inducing TAB2 translocation in the IL-1 signaling pathway. Mol Cell Biol. 2001;21:2475–2484. doi: 10.1128/MCB.21.7.2475-2484.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shembade N, et al. The E3 ligase Itch negatively regulates inflammatory signaling pathways by controlling the function of the ubiquitin-editing enzyme A20. Nat Immunol. 2008;9:254–262. doi: 10.1038/ni1563. [DOI] [PubMed] [Google Scholar]
- 35.Peng DJ, et al. Noncanonical K27-Linked Polyubiquitination of TIEG1 Regulates Foxp3 Expression and Tumor Growth. J Immunol. 2011 doi: 10.4049/jimmunol.1003801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Conze DB, et al. Posttranscriptional downregulation of c-IAP2 by the ubiquitin protein ligase c-IAP1 in vivo. Mol Cell Biol. 2005;25:3348–3356. doi: 10.1128/MCB.25.8.3348-3356.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stegmeier F, et al. The tumor suppressor CYLD regulates entry into mitosis. Proc Natl Acad Sci U S A. 2007;104:8869–8874. doi: 10.1073/pnas.0703268104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wertz IE, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature. 2004;430:694–699. doi: 10.1038/nature02794. [DOI] [PubMed] [Google Scholar]
- 39.Harhaj EW, Dixit VM. Deubiquitinases in the regulation of NF-kappaB signaling. Cell Res. 2011;21:22–39. doi: 10.1038/cr.2010.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ninomiya-Tsuji J, et al. A resorcylic acid lactone, 5Z-7-oxozeaenol, prevents inflammation by inhibiting the catalytic activity of TAK1 MAPK kinase kinase. J Biol Chem. 2003;278:18485–18490. doi: 10.1074/jbc.M207453200. [DOI] [PubMed] [Google Scholar]
- 41.Holtmann H, et al. The MAPK kinase kinase TAK1 plays a central role in coupling the interleukin-1 receptor to both transcriptional and RNA-targeted mechanisms of gene regulation. J Biol Chem. 2001;276:3508–3516. doi: 10.1074/jbc.M004376200. [DOI] [PubMed] [Google Scholar]
- 42.Bhoj VG, Chen ZJ. Ubiquitylation in innate and adaptive immunity. Nature. 2009;458:430–437. doi: 10.1038/nature07959. [DOI] [PubMed] [Google Scholar]
- 43.Shembade N, Pujari R, Harhaj NS, Abbott DW, Harhaj EW. The kinase IKKalpha inhibits activation of the transcription factor NF-kappaB by phosphorylating the regulatory molecule TAX1BP1. Nat Immunol. 2011 doi: 10.1038/ni.2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Heissmeyer V, et al. Calcineurin imposes T cell unresponsiveness through targeted proteolysis of signaling proteins. Nat Immunol. 2004;5:255–265. doi: 10.1038/ni1047. [DOI] [PubMed] [Google Scholar]
- 45.Qiao G, et al. T-cell receptor-induced NF-kappaB activation is negatively regulated by E3 ubiquitin ligase Cbl-b. Mol Cell Biol. 2008;28:2470–2480. doi: 10.1128/MCB.01505-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Karin M, Yamamoto Y, Wang QM. The IKK NF-kappa B system: a treasure trove for drug development. Nat Rev Drug Discov. 2004;3:17–26. doi: 10.1038/nrd1279. [DOI] [PubMed] [Google Scholar]
- 47.Fanucchi MP, et al. Randomized phase II study of bortezomib alone and bortezomib in combination with docetaxel in previously treated advanced non-small-cell lung cancer. J Clin Oncol. 2006;24:5025–5033. doi: 10.1200/JCO.2006.06.1853. [DOI] [PubMed] [Google Scholar]
- 48.Davies AM, et al. Bortezomib plus gemcitabine/carboplatin as first-line treatment of advanced non-small cell lung cancer: a phase II Southwest Oncology Group Study (S0339) J Thorac Oncol. 2009;4:87–92. doi: 10.1097/JTO.0b013e3181915052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Greten FR, et al. NF-kappaB is a negative regulator of IL-1beta secretion as revealed by genetic and pharmacological inhibition of IKKbeta. Cell. 2007;130:918–931. doi: 10.1016/j.cell.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.