

Abstract
Background
In pediatric neuroblastoma (NBL), high anaplastic lymphoma kinase (ALK) levels appear to be correlated with an unfavorable prognosis, regardless of ALK mutation status. This suggests a therapeutic role for ALK inhibitors in NBL patients. We examined the correlation between levels of ALK, phosphorylated ALK (pALK) and downstream signaling proteins and response to ALK inhibition in a large panel of both ALK mutated and wild type (WT) NBL cell lines.
Methods
We measured protein levels by western blot and ALK inhibitor sensitivity (TAE684) by viability assays in 19 NBL cell lines of which 6 had a point mutation and 4 an amplification of the ALK gene.
Results
ALK 220 kDa (p = 0.01) and ALK 140 kDa (p = 0.03) protein levels were higher in ALK mutant than WT cell lines. Response to ALK inhibition was significantly correlated with ALK protein levels (p < 0.01). ALK mutant cell lines (n = 4) were 14,9 fold (p < 0,01) more sensitive to ALK inhibition than eight WT cell lines.
Conclusion
NBL cell lines often express ALK at high levels and are responsive to ALK inhibitors. Mutated cell lines express ALK at higher levels, which may define their superior response to ALK inhibition.
Electronic supplementary material
The online version of this article (doi:10.1007/s13402-011-0048-2) contains supplementary material, which is available to authorized users.
Keywords: ALK, Kinase inhibitor, Neuroblastoma, PHOX2B, TAE684
Introduction
In neuroblastoma (NBL), few genetic alterations have been described including MYCN amplification (20–25%), PHOX2B mutation (6.4% of familial NBL) and CCND1 amplification (2.4%). Recently, mutations have been found in the anaplastic lymphoma kinase (ALK) gene in NBL patients [1–5]. The discovery of new gene aberrations may improve risk stratifications and could lead to new therapies for NBL patients.
ALK is a tyrosine kinase receptor mainly expressed in the neonatal brain, but expression levels sharply decline after birth [6, 7]. In general, ALK expression is restricted to neural tissues. Expression of ALK in cell lines is mainly seen in neuro-ectodermal cell lines, such as neuroblastoma cell lines [8, 9]. The ALK receptor is activated through autophosphorylation upon ligand binding. Signaling of phosphorylated ALK (pALK) protein occurs through SHC3, AKT and MAPK pathways [2, 3, 10]. Through these pathways ALK influences both proliferation and differentiation. At the protein level, two main isoforms can be identified: the 220 kDa full length receptor and the truncated 140 kDa protein that is the result of extracellular cleavage. Kinase activity of both isoforms has been described although in nociceptive neurons only the 220 kDa was observed. [11] ALK gene translocations, and mainly the t(2;5), have been described in anaplastic large cell lymphoma, and result in the fusion protein NPM-ALK. These fusion proteins induce the downstream pathways AKT, JAK-STAT and MAPK, which become constitutively active [12–14].
In 2008, ALK point mutations were described in 3–11% of sporadic NBL and found to be one of the most important mutations in hereditary NBL (33–40% of the families) [4, 5]. In 20–35% of the NBL cell lines a point mutation of the ALK gene was identified [2–5, 15]. Amplification of the ALK gene has also been described in 1.2–4.4% of NBL patients and 12% of NBL cell lines [1, 4, 5, 16]. Mutations in the ALK gene have been correlated with higher proliferation and increased expression of pALK and downstream targets. Aberrations of the ALK gene have been correlated with inferior prognosis, although results have been inconclusive [1–5, 17, 18]. In NBL cell lines, higher pALK is associated with resistance to apoptosis and enhanced DNA synthesis and mitosis [2–4, 19]. Recently, Passoni et al. (2009) described NBL patients with high ALK levels without a mutation of the ALK gene. They showed that high ALK levels irrespective of mutation status were strongly correlated with prognosis [18]. This correlation between high ALK levels and unfavorable prognosis was confirmed by de Brouwer et al. [20].
In addition, ALK inhibitors may be of therapeutic value in NBL patients [1–4, 17, 18]. Since the survival rates for high risk NBL are still unsatisfactory despite intensive multimodal treatment, the potential of including ALK inhibitor treatment in the therapeutic strategy is promising [21]. ALK mutation status and ALK protein levels have been implied to increase in vitro sensitivity to ALK inhibitors [3, 18, 22]. Furthermore, ALK inhibitor treatment was shown to result in decreased proliferation and decreased protein levels of pALK and downstream targets (pAKT, pERK1, pERK2 and pSTAT3) in ALK mutated NBL cell lines [3, 22]. The silencing of high ALK expression with siRNA’s seemed to have similar effects [2, 4, 16, 18]. The results for ALK wild type and amplified neuroblastoma cell lines have been contradictory. Clarification of the biological mechanism that results in sensitivity to ALK inhibition is important to correctly identify patients that might respond to ALK inhibitor treatment [23].
Here, we further examined the correlation between ALK, pALK and downstream signaling protein levels and response to ALK inhibitor treatment in a large panel of both ALK mutated (MUT) and wild type (WT) NBL cell lines.
Methods
Cell lines
A panel of 19 NBL cell lines (AMC-106c, SK-N-FI, GI-ME-N, IMR-32, KCNR, Lan-5, SK-N-AS, N206, NGP-C4, NMB, SJNB-1, SJNB-6, SJNB-8, SJNB-10, SJNB-12, SK-N-BE, TR-14, UGH-NP, SK-N-SH) was cultured in DMEM (Invitrogen, Breda, The Netherlands) containing 10% heat inactivated fetal calf serum (Integro, Zaandam, The Netherlands), 0.05% fungizone (Invitrogen), 0.1 U/ml penicillin (Invitrogen), 0.1 μg/ml streptomycine (Invitrogen), 1% 100× glutamax (Invitrogen) and 1% 100× Non-essential amino acids (MEM, Invitrogen). Two derivatives of the SK-N-SH cell lines, SHEP-2/tet2 and SHEP-21N/tet2/N were cultered in RPMI medium (Invitrogen), containing 10% heat inactivated fetal calf serum, 0.05% fungizone, 0.1 U/ml penicillin, 0.1 ug/ml streptomycine, 0.15% NaBic (Invitrogen) and 1% 1 M HEPES (Invitrogen). Cells were maintained at 37°C under 5% CO2.
DNA and RNA isolation
Total DNA and RNA was extracted using TRIzol reagent (Invitrogen) according to manufacturer’s protocol. The quality of the extracted RNA was assessed on an Agilent 2100 Bioanalyzer (Agilent, Santa Clara, CA, USA) and the quality of the extracted DNA was checked by gel electrophoresis.
Sequencing
PCR primers for the genomic region, corresponding to ALK exon 20 and exon 22 to 25 were designed. (ALK 20 F:GATTTGCCCAGACTCAGCTC, ALK20 R: TACACTGCACCCCTCTCCTC, ALK22_F: TTCTCAGCTCACAGCCTCCT, ALK22_R: AAACCTCTCCAGGTTCTTTGG, ALK23_F: GATTTGCCCAGACTCAGCTC, ALK23_R: CACTCTTGCTCCTTCCATCC, ALK24_F: GGAAGCCAGCATTTCAGATT, ALK24_R: AGCACACAGATCAGCGACAG, ALK25_F: AATCCTAGTGATGGCCGTTG, ALK25_R: CCACACCCCATTCTTGAGG). PCR was performed in 96-well formats in 15 μl reaction volumes containing 7.6 μl H2O, 3.0 μl 5X colorless Gotaq flexi buffer, 0.9 μl 25 mM MgCl2, 1 μl each of forward and reverse primer, 0.3 μl deoxynucleotide triphosphates and 0.2 μl 5unit/μl Gotaq (Promega, Leiden, The Netherlands). To this 50 ng of DNA was added. PCR conditions were as follows: 35 cycles of 95°C for 30 s, 58˚ for 45 s and 72°C for 45 s and ending with 72°C for 10 min. Cycle sequencing was performed using the BigDye Terminator v3.1 sequencing kit (Applied Biosystems, Foster City, USA) according to the manufacturers’ protocol. Sequencing products were analyzed on the 3130XL Genetic analyzer (Applied Biosystems). Sequence traces were analyzed using Mutation Surveyor software (SoftGenetics LLC, State College PA, USA).
Multiplex ligation-dependent probe amplification
The neuroblastoma Salsa 251-B mix (MRC Holland, Amsterdam, The Netherlands) was used with standard conditions and fragment separation was performed on an ABI3100 (Applied Biosystems). Data analysis was performed with Genemarker software (SoftGenetics) using internal control probe normalization. Amplification was defined as 5 gene copies or more (ratio sample to normal control =1, amplification =/>2.5). Copy number gains larger than 20 copies were not quantifiable, but these will be mentioned as having an amplification.
Gene expression profiles
For gene expression arrays high quality RNA (RIN > 8.5) was selected. This RNA was reverse transcribed using T7-linked oligo-dT primers to create cDNA and this was used as a template to synthesize biotinylated cRNA. Labeled cRNA was then fragmented and hybridized to HU133plus2.0 arrays (Affymetrix, Santa Clara, CA, USA). The gene expression data is deposited as GSE22771 (NCBI GEO) and will be made publicly available upon publication. No microarrays were performed for SJNB-12, SJNB-6, N206, UHG-NP. Two probe sets showing similar expression covered the ALK gene (r = 0.948) (Supplementary Figure 1). The mean of these two probes has been used for analysis in this article. For PHOX2B only one probe set was available (207009_at) and for S100A6 the 217728_at probe set was used.
Western blot
Cells were harvested and put in lysis buffer containing protease inhibitors and aproptinin (Sigma-Aldrich, Moerdijk, The Netherlands). Phosphatase inhibitors were added for studying phosphorylated proteins. Western blots were carried out with 25 or 50 μg of denatured protein using standard blotting procedures. The primary antibodies used are α-pALK Y1604 (Epitomics, Burlingame, USA), α-ALK (Thermo Fisher Scientific,Fremont, USA), α-AKT, α-pAKT Ser473, α-p44/42 MAPK, α-phospho-p44/42 MAPK, α-GAPDH and the secondary antibodies used were α-mouse and α-rabbit HRP linked. Antibodies were purchased from Cell Signaling (Bioke, Leiden, The Netherlands) unless otherwise stated. Imaging and quantification of the blots was performed with GeneSnap and GeneTools software (Syngene, Cambridge, UK). For comparison of samples between various blots, a reference sample was included in each experiment. Ratios of the protein of interest and GAPDH were normalized to this reference sample.
Cell viability assay
NBL cells were seeded in flat-bottomed 96 well plates (Greiner, Alphen a/d Rijn, The Netherlands) at 5,000–50,000 cells per well in a volume of 100 μl DMEM. After 24 h TAE684 (Axon Medchem, Groningen, The Netherlands) was added for 72 h, hereafter MTS/PMS ((3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2 H-tetrazolium inner salt), Promega, Leiden, The Netherlands and phenazine methosulfate, Sigma-Aldrich, Zwijndrecht, The Netherlands) solution was added to each well followed by a 3 h incubation at 37°C. The absorbance was read at 490 nm and corrected for the 720 nm read out. We calculated the median lethal concentration (LC50).
Statistics
The data from the expression array was VSN normalized in the R environment. Spearman correlations were calculated in SPSS 15.0. A linear regression model was used to test the influence of MYCN amplification on these correlations. Differences between groups were calculated with the Mann–Whitney U test. Graphs were made in GraphPad Prism version 4 (GraphPad Software, Inc, La Jolla, USA). ALK amplified samples showed similar sensitivity to ALK inhibition and similar ALK levels as WT samples. They were therefore co-analyzed, unless otherwise stated.
Results
Characterization of cell lines
To study ALK levels and ALK inhibitor response in both ALK mutant and wild type (WT) cell lines, ALK mutation status was determined. Point mutations of the ALK gene were found in six of the 19 NBL cell lines (31.6%). We identified the F1174L mutation in four cell lines and the R1275Q mutation in two cell lines (Table 1). Amplifications (>/=5 copies) of the ALK gene were identified by multiple ligand probe amplification and ALK was amplified in four of the 19 cell lines (21.1%). These amplifications were always part of a larger region of copy number gain including the NMYC gene. NMYC was amplified in 14 of the 19 NBL cell lines (73.7%).
Table 1.
Copy number status of the MYCN and ALK gene, as well as mutation status of the ALK gene in NBL cell lines
| Cell line | NMYC amplified (+/−) | ALK amplified (+/−) | ALK mutated (type of mutation) |
|---|---|---|---|
| AMC106C | + | − | F1174L |
| SK-N-FI | + | − | R1275Q |
| GI-ME-N | − | − | − |
| IMR-32 | + | − | − |
| KCNR | + | − | F1174L |
| LAN-5 | + | − | R1275Q |
| N206 | + | − | F1174L |
| SK-N-AS | − | − | − |
| NGP-C4 | + | − | − |
| NMB | + | + | − |
| SJNB-1 | − | − | − |
| SJNB-6 | + | − | − |
| SJNB-8 | + | − | − |
| SJNB-10 | + | − | − |
| SJNB-12 | − | − | − |
| SK-N-BE | + | + | − |
| SK-N-SH | − | − | F1174L |
| TR-14 | + | + | − |
| UHG-NP | + | + | − |
aAmplification was identified as having =/>5 gene copies; F1174L, p.Phe1174Leu; R1275Q, p.Arg1275Gln
Measurement of ALK and downstream signaling protein levels
To study the consequences of ALK point mutations and amplifications, we investigated ALK mRNA, ALK protein and activation of downstream signaling proteins. ALK point mutated cell lines have significantly higher ALK mRNA levels compared with amplified and WT cell lines (p < 0.01) (Fig. 1a, Supplementary Figure 1–2). Also, ALK 220 kDa (p < 0.01) and ALK 140 kDa (p < 0.01) protein levels measured by Western blot were significantly higher ALK in ALK mutated cell lines. The ALK amplified cell lines had similar ALK levels as wild type cell lines. ALK amplified cell lines were therefore co-analyzed with the wild type cell lines, unless otherwise stated. In contrast to total ALK 220 kDa protein levels, phosphorylation of the 220 kDa ALK protein at Y1604 was not significantly different between mutant and WT cell lines (Fig. 1d, Supplementary Figure 2C). Phosphorylation of the 140 kDa ALK product was very low as measured with the anti-Y1604 pALK antibody (Epitomics, Burlingame, U.S.A.) (Supplementary Figure 3). To understand the effect of increased ALK expression on the activation of downstream signaling molecules, we measured total protein levels and phosphorylation status of downstream signaling proteins of the MAPK and AKT pathway. ERK1 and ERK2 protein expression was significantly correlated with ALK 220 and 140 kDa expression in mutated and WT NBL cell lines (Fig. 2a-b, Supplementary Figure 2). The levels of total AKT and phosphorylated intermediates ERK1, ERK2 and AKT were not significantly different between four mutated and eight wild type NBL cell lines (Supplementary Figure 2 and 3).
Fig. 1.
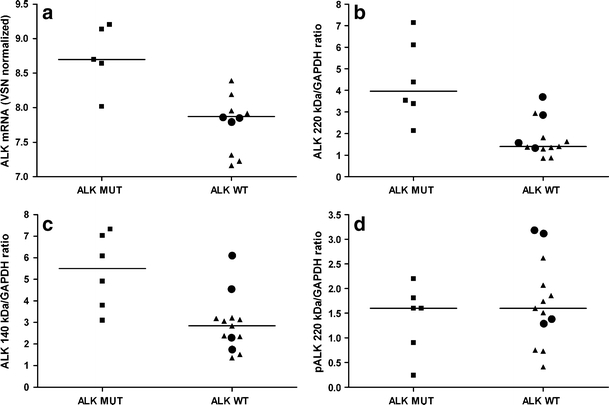
ALK levels are higher in ALK mutant than wild type NBL cell lines. Higher a ALK mRNA (p < 0.01) b ALK 220 kDa protein (p < 0.01) and c ALK 140 kDa protein (p < 0.01) levels in ALK mutant than WT NBL cell lines. d No significant difference of Y1604 pALK levels between mutant (squares) and wild type (dots) cell lines (p = 0.70). MUT, ALK point mutation; WT, ALK wild type gene, including copy number gains (amplified lines are indicated as dots). Western blots displayed in Supplementary Figure 2 and 3
Fig. 2.
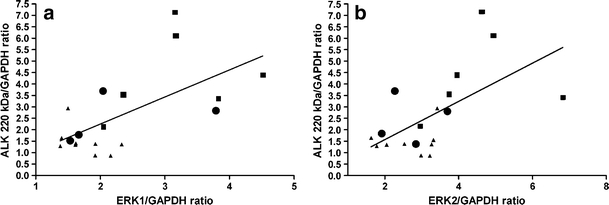
ERK1 and ERK2 correlate with ALK expression. a ERK1 protein expression correlates with ALK 220 kDa protein expression (Spearman’s rho r = 0.55, p = 0.02) and ALK 140 kDa protein expression (Spearman’s rho r = 0.56, p = 0.01, graph not shown). b ERK2 protein expression correlates with ALK 220 kDa protein expression (spearman’s rho r = 0.60, p < 0.01) and ALK 140 kDa protein expression (spearman’s rho r = 0.60, p < 0.01, graph not shown). ALK point mutated cell lines are indicated as black squares, ALK WT cell lines as black triangles (including amplified cell lines as black dots). Western blots displayed in Supplementary Figure 2
Cell viability assay after treatment with ALK inhibitor TAE684
As ALK point mutations were associated with higher ALK expression levels and increased levels of downstream proteins ERK1 and ERK2, we investigated whether ALK mutant cell lines would demonstrate a better response to the ALK inhibitor TAE684. ALK mutant cell lines (n = 4) displayed a significantly higher sensitivity towards the ALK inhibitor TAE684 than WT cell lines (n = 8) with 14.9 fold lower LC50 values (p < 0.01) (Fig. 3, Table 2). ALK amplified cell lines showed similar sensitivity to ALK inhibition as WT cell lines without an amplification (Supplementary Figure 4). The LC50 of TAE684 was highly correlated with ALK mRNA (r = −0.75, p = 0.01), ALK 220 (r = −0.81, p < 0.01) and 140 kDa (r = −0.84, p < 0.01) protein levels in all NBL cell lines (Fig. 4a-b) and remained significant after adjustment for MYCN status (Supplementary Table 1). This correlation tends to be present in both ALK mutated (n = 4) and wild type cell lines (n = 8) independently. A significant correlation was present between 140 kDa ALK protein levels and TAE684 response in only WT cell lines (r = 0.71, p = 0.05) (Fig. 4b). The correlation between pALK levels and responsiveness to TAE684 was not significant (r = 0.55, p = 0.08).
Fig. 3.
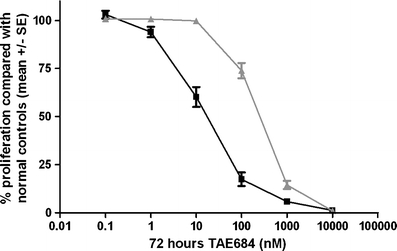
Responsiveness to ALK inhibitor TAE684 is correlated with ALK mutation status. Effect of ALK inhibitor treatment on proliferation of 12 NBL cell lines measured by MTS/PMS based viability assay. ALK mutant cell lines (black solid line, n = 4) show higher sensitivity to TAE684 than WT cell lines (grey solid line (n = 8)). Experiments were performed with triplicate measurements. The Y-axis displays the mean and standard error of all measurements in all mutated versus all wild type cell lines
Table 2.
Overview of ALK mutated and wild type NBL cell lines, displaying ALK levels and sensitivity to TAE684
| Cell line | ALK mutation status | ALK mRNAa | ALK (220 kDa)/GAPDH | ALK (140 kDa)/GAPDH | pALK (220 kda)/GAPDH | LC50 TAE684 |
|---|---|---|---|---|---|---|
| SK-N-SH | MUT | 8.7 | 4.4 | 7.0 | 0.9 | 9.4 |
| LAN-5 | MUT | 9.1 | 3.5 | 3.1 | 1.6 | 9.4 |
| KCNR | MUT | 9.2 | 6.1 | 6.1 | 1.8 | 27.5 |
| AMC106c | MUT | 8 | 2.1 | 3.8 | 2.2 | 66.6 |
| SK-N-BE | AMP | 7.8 | 1.8 | 1.8 | 1.4 | 348.9 |
| NMB | AMP | 7.9 | 1.4 | 2.4 | 3.2 | 591.2 |
| SJNB10 | WT | 7.6 | 1.6 | 3.1 | 1.6 | 239.9 |
| SJNB12 | WT | NAb | 1.3 | 3.2 | 0.8 | 268.4 |
| SJNB1 | WT | 8.2 | 1.4 | 2.9 | 0.7 | 282.7 |
| SK-N-AS | WT | 7.2 | 1.4 | 2.3 | 2.6 | 496.8 |
| SJNB6 | WT | NAb | 0.9 | 1.5 | 2.1 | 552.6 |
| SJNB8 | WT | 7.3 | 0.9 | 1.4 | 1.9 | 587.7 |
aALK mRNA from array, average two probesets 208211_s_at and 208212_s_at; bNA, not available.
Fig. 4.
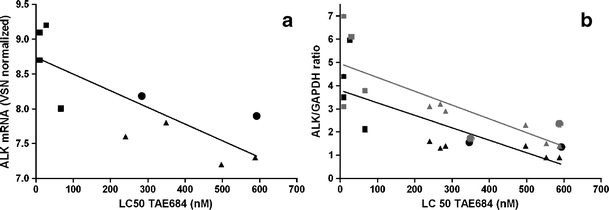
Responsiveness to ALK inhibitor TAE684 is correlated with ALK mRNA and ALK protein levels. a ALK mRNA levels relate to in-vitro sensitivity towards the ALK inhibitor TAE684 (Spearman’s rho r = −0.75, p = 0.01), the black line is a linear fit. ALK mRNA levels in mutated (black squares, Spearman’s rho r = −0.32, p = 0.68) and WT cell lines (black triangles are WT; dots are AMP, spearman’s rho r = −0.14, p = 0.79) independently tend to correlate with ALK inhibitor response. b ALK 220 kDa (black symbols, spearman’s rho r = −0.81, p < 0.01) and 140 kDa (grey symbols, spearman’s rho r = −0.84, p < 0.01) protein levels relate to the in vitro sensitivity towards the ALK inhibitor TAE684. A linear line was fitted (black for 220 kDa protein, grey for 140 kDa protein). ALK 220 kDa protein levels of mutants are displayed by black squares (spearman’s rho r = −0.32, p = 0.68) and WTs by black triangles (WT) and black dots (AMP) (spearman’s rho r = −0.43, p = 0.29). ALK 140 kDa protein levels mutants by grey squares (spearman’s rho r = −0.11, p = 0.90) and WTs by grey triangles (WT) and grey circles (AMP) (spearman’s rho r = −0.71, p = 0.05)
Effect of ALK inhibition on ALK, pALK and downstream signaling proteins
Four mutated and eight wild type NBL cell lines were treated for 72 h with ALK inhibitors, according to their corresponding LC50. ALK inhibitor treatment induced a significant decrease of phosphorylated AKT (p = 0.01, Fig. 5a-b). Interestingly, this decrease was only observed in ALK amplified and WT cell lines. ALK inhibitor treatment did not significant alter the expression of ALK, pALK or downstream target proteins. (Supplementary Figure 3A-B)
Fig. 5.
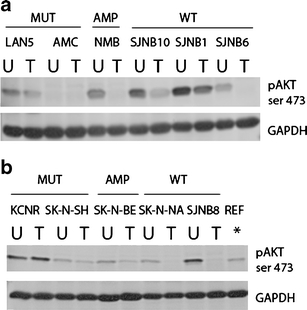
Treatment of NBL cell lines with TAE684 resulted in pAKT downregulation. Phosphorylation of AKT at ser473 was significantly downregulated in TAE684 treated (T) versus untreated (U) NBL cell lines (p = 0.01). SJNB-12 cell line was not included on western blot, due to a limited amount of protein that was available
Correlation of ALK expression with differentiation stage
The phenotypical differences between NBL cell lines is apparent in the NBL cell line SK-N-SH which contains mainly neuronal, but also some schwannian-type cells. We studied ALK inhibitor response in SK-N-SH and its schwannian derivatives SHEP-2 and SHEP-21N. Remarkably, these derivates carry the same mutation of the ALK gene (F1174L), however they do not have high ALK levels and did not show high ALK inhibitor sensitivity. This was in contrast with the biphenotypical cell line SK-N-SH that showed high ALK levels and ALK inhibitor response (Supplementary Table 2). This suggests that ALK mutations may have a specific function in neural-type cells (SK-N-SH) and not in schwannian-type cells (SHEP-2, SHEP-21N). Furthermore, these data confirm the specificity of the ALK inhibitor TAE684. We next examined if an early neural or schwannian phenotype correlated with ALK expression in WT cell lines. There was a strong positive correlation between ALK expression and expression of early neural differentiation marker PHOX2B in WT cell lines (r = 0.95, p < 0.01) (Fig. 6a). An inverse correlation tends to exist between schwannian marker S100A6 and ALK expression in WT cell lines (r = −0.48, p = 0.16) (Fig. 6b).
Fig. 6.
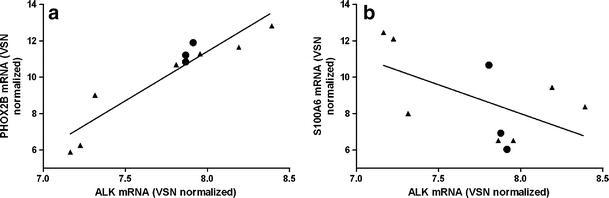
ALK and neuronal and schwannian markers in WT cell lines. a ALK mRNA expression correlates highly with PHOX2B mRNA expression in WT (triangles) and AMP (circles) NBL cell lines (spearman’s rho r = 0.95, p < 0.01). b ALK mRNA expression tends to show an inverse correlation with S100A6 mRNA expression in WT (triangles) and AMP (circles) NBL cell lines (spearman’s rho r = −0.48, p = 0.16)
Discussion
ALK gene mutations and high levels of ALK protein are characteristics of a subgroup of neuroblastoma. Here, we show that ALK mutated cell lines express ALK mRNA and ALK protein at significantly higher levels and also show higher responsiveness to ALK inhibition. ALK expression was further positively correlated with levels of downstream targets ERK1 and ERK2 and expression of neural differentiation marker PHOX2B. In addition, we show that response to ALK inhibitor TAE684 strongly correlates with ALK mutation status and ALK protein levels. These data may help to clarify the biological mechanism leading to high ALK expression and ALK inhibitor sensitivity in NBL patients.
A prognostic value for high ALK levels independent of mutation status was recently shown in NBL patients [18]. In accordance with others, our data show high ALK levels and ALK inhibitor response, especially in ALK point mutated NBL cell lines. These data may not be conflicting. Since high ALK levels are characteristic of fetal neural tissue it may be that high ALK expression in primary NBL is a characteristic of the early neural crest origin. Our data show that neural crest differentiation of the cell lines seems to be correlated with ALK levels and responsiveness to ALK inhibition. High ALK levels in WT NBL tumors and cell lines may therefore be explained by predominance of the neural-type versus schwannian-type of cells in poorly differentiated tumors.
Reduced levels of pAKT were observed upon treatment with the ALK inhibitor TAE684. This was most probably caused by reduced kinase activity of ALK. In contrast to others, who identified differential phosphorylation of ALK between ALK mutant and WT cell lines or between TAE684 treated and non-treated NBL cell lines, we did not observe differential phosphorylation of ALK at Y1604 [4, 5, 16, 18, 20, 22]. One possible explanation for this discrepancy could be that Y1604 is not regulated by ALK autophosphorylation but by another kinase. Another explanation could be that the anti-pALK y1604 antibody is directed at another epitope. Alternatively, the decrease of pALK was too transient to be detected in our assays. One important consideration is that TAE684 could also be targeting a different kinase than ALK, leading to the observed decrease of pAKT [24]. Although, in this study we observed an ALK inhibitor response that appeared ALK specific, as demonstrated by SK-N-SH and its schwannian derivatives. (Supplementary Table 1)
ALK gains, amplifications and mutations in NBL patients have previously been associated with inferior survival [1]. However, others did not identify an independent influence of ALK mutations on survival when correcting for MYCN status and stage of disease [2]. Similarly, high ALK levels irrespective of ALK mutation status have been associated with inferior prognosis by Passoni et al. (2009) in a univariate analysis [18, 20]. In our study, we were able to identify a similar correlation between high ALK levels and good response to ALK inhibition. This correlation did not seem to be influenced by MYCN status. In summary, both ALK mutation status and ALK levels seem to be important in ALK inhibitor response.
In our study, five of the six cell mutant cell lines showed amplification of the MYCN gene, which is in accordance with others [2, 20]. We further could not identify differences in ALK levels or ALK inhibitor response between R1275Q and F1174L ALK mutated cell lines, although numbers are low. MYCN amplification has been shown to be correlated with positive ALK mutation status and specifically with the F1174L mutation in NBL patients [2, 20]. De Brouwer et al. identified a positive association between the F1174L mutation and higher transforming capacity, higher frequency of MYCN amplification and advanced stage compared with R1275Q mutations and WT tumors [20]. In hereditary NBL, ALK mutations are very common and found in almost half of the families. The most common germline mutation is R1275Q, which is identified in approximately 45% of the familial cases. The F1174L has not yet been identified as germline mutation. It is the most common mutation in NBL cell lines and possibly carries a more aggressive phenotype. This led de Brouwer et al., to postulate that this might suggest embryonic lethality of the F1174L mutation in germ line mutated individuals. This could explain the absence of this mutation in hereditary neuroblastoma.
In conclusion, NBL cell lines and patients often express ALK at high levels, especially in ALK mutant cases. ALK mRNA and protein levels, ALK mutation status and differentiation status were strongly correlated with ALK inhibitor response. The most defining factor for the response to ALK inhibitors remains elusive. These data will be important in understanding ALK inhibitor response in ongoing patient trials with ALK inhibitors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
(PDF 458 kb)
Acknowledgments
This project was financially supported by Villa Joep (Dutch Neuroblastoma Foundation), the Dutch Cancer Society (KWF) and the ODAS foundation (Delft, The Netherlands).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Contributor Information
Floor A. M. Duijkers, Email: f.duijkers@erasmusmc.nl
Max M. van Noesel, Phone: +31-10-7032073, FAX: +31-10-7036801, Email: m.vannoesel@erasmusmc.nl
References
- 1.Caren H, Abel F, Kogner P, Martinsson T. High incidence of DNA mutations and gene amplifications of the ALK gene in advanced sporadic neuroblastoma tumours. Biochem J. 2008;416:153–159. doi: 10.1042/BJ20081834. [DOI] [PubMed] [Google Scholar]
- 2.Chen Y, Takita J, Choi YL, Kato M, Ohira M, Sanada M, Wang L, Soda M, Kikuchi A, Igarashi T, Nakagawara A, Hayashi Y, Mano H, Ogawa S. Oncogenic mutations of ALK kinase in neuroblastoma. Nature. 2008;455:971–974. doi: 10.1038/nature07399. [DOI] [PubMed] [Google Scholar]
- 3.George RE, Sanda T, Hanna M, Frohling S, Luther W, 2nd, Zhang J, Ahn Y, Zhou W, London WB, McGrady P, Xue L, Zozulya S, Gregor VE, Webb TR, Gray NS, Gilliland DG, Diller L, Greulich H, Morris SW, Meyerson M, Look AT. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature. 2008;455:975–978. doi: 10.1038/nature07397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Janoueix-Lerosey I, Lequin D, Brugieres L, Ribeiro A, de Pontual L, Combaret V, Raynal V, Puisieux A, Schleiermacher G, Pierron G, Valteau-Couanet D, Frebourg T, Michon J, Lyonnet S, Amiel J, Delattre O. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature. 2008;455:967–970. doi: 10.1038/nature07398. [DOI] [PubMed] [Google Scholar]
- 5.Mosse YP, Laudenslager M, Longo L, Cole KA, Wood A, Attiyeh EF, Laquaglia MJ, Sennett R, Lynch JE, Perri P, Laureys G, Speleman F, Kim C, Hou C, Hakonarson H, Torkamani A, Schork NJ, Brodeur GM, Tonini GP, Rappaport E, Devoto M, Maris JM. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature. 2008;455:930–935. doi: 10.1038/nature07261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iwahara T, Fujimoto J, Wen D, Cupples R, Bucay N, Arakawa T, Mori S, Ratzkin B, Yamamoto T. Molecular characterization of ALK, a receptor tyrosine kinase expressed specifically in the nervous system. Oncogene. 1997;14:439–449. doi: 10.1038/sj.onc.1200849. [DOI] [PubMed] [Google Scholar]
- 7.Pulford K, Lamant L, Morris SW, Butler LH, Wood KM, Stroud D, Delsol G, Mason DY. Detection of anaplastic lymphoma kinase (ALK) and nucleolar protein nucleophosmin (NPM)-ALK proteins in normal and neoplastic cells with the monoclonal antibody ALK1. Blood. 1997;89:1394–1404. [PubMed] [Google Scholar]
- 8.Dirks WG, Fahnrich S, Lis Y, Becker E, MacLeod RA, Drexler HG. Expression and functional analysis of the anaplastic lymphoma kinase (ALK) gene in tumor cell lines. Int J Cancer. 2002;100:49–56. doi: 10.1002/ijc.10435. [DOI] [PubMed] [Google Scholar]
- 9.Lamant L, Pulford K, Bischof D, Morris SW, Mason DY, Delsol G, Mariame B. Expression of the ALK tyrosine kinase gene in neuroblastoma. Am J Pathol. 2000;156:1711–1721. doi: 10.1016/S0002-9440(10)65042-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miyake I, Ohira M, Nakagawara A, Sakai R. Distinct role of ShcC docking protein in the differentiation of neuroblastoma. Oncogene. 2009;28:662–673. doi: 10.1038/onc.2008.413. [DOI] [PubMed] [Google Scholar]
- 11.Degoutin J, Brunet-de Carvalho N, Cifuentes-Diaz C, Vigny M. ALK (Anaplastic Lymphoma Kinase) expression in DRG neurons and its involvement in neuron-Schwann cells interaction. Eur J Neurosci. 2009;29:275–286. doi: 10.1111/j.1460-9568.2008.06593.x. [DOI] [PubMed] [Google Scholar]
- 12.Li R, Morris SW. Development of anaplastic lymphoma kinase (ALK) small-molecule inhibitors for cancer therapy. Med Res Rev. 2008;28:372–412. doi: 10.1002/med.20109. [DOI] [PubMed] [Google Scholar]
- 13.Chiarle R, Voena C, Ambrogio C, Piva R, Inghirami G. The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat Rev Cancer. 2008;8:11–23. doi: 10.1038/nrc2291. [DOI] [PubMed] [Google Scholar]
- 14.Palmer RH, Vernersson E, Grabbe C, Hallberg B. Anaplastic lymphoma kinase: signalling in development and disease. Biochem J. 2009;420:345–361. doi: 10.1042/BJ20090387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oberthuer A, Theissen J, Westermann F, Hero B, Fischer M. Molecular characterization and classification of neuroblastoma. Future Oncol. 2009;5:625–639. doi: 10.2217/fon.09.41. [DOI] [PubMed] [Google Scholar]
- 16.Osajima-Hakomori Y, Miyake I, Ohira M, Nakagawara A, Nakagawa A, Sakai R. Biological role of anaplastic lymphoma kinase in neuroblastoma. Am J Pathol. 2005;167:213–222. doi: 10.1016/S0002-9440(10)62966-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mosse YP, Wood A, Maris JM. Inhibition of ALK signaling for cancer therapy. Clin Cancer Res. 2009;15:5609–5614. doi: 10.1158/1078-0432.CCR-08-2762. [DOI] [PubMed] [Google Scholar]
- 18.Passoni L, Longo L, Collini P, Coluccia AM, Bozzi F, Podda M, Gregorio A, Gambini C, Garaventa A, Pistoia V, Del Grosso F, Tonini GP, Cheng M, Gambacorti-Passerini C, Anichini A, Fossati-Bellani F, Di Nicola M, Luksch R. Mutation-independent anaplastic lymphoma kinase overexpression in poor prognosis neuroblastoma patients. Cancer Res. 2009;69:7338–7346. doi: 10.1158/0008-5472.CAN-08-4419. [DOI] [PubMed] [Google Scholar]
- 19.Motegi A, Fujimoto J, Kotani M, Sakuraba H, Yamamoto T. ALK receptor tyrosine kinase promotes cell growth and neurite outgrowth. J Cell Sci. 2004;117:3319–3329. doi: 10.1242/jcs.01183. [DOI] [PubMed] [Google Scholar]
- 20.De Brouwer S, De Preter K, Kumps C, Zabrocki P, Porcu M, Westerhout EM, Lakeman A, Vandesompele J, Hoebeeck J, Van Maerken T, De Paepe A, Laureys G, Schulte JH, Schramm A, Van Den Broecke C, Vermeulen J, Van Roy N, Beiske K, Renard M, Noguera R, Delattre O, Janoueix-Lerosey I, Kogner P, Martinsson T, Nakagawara A, Ohira M, Caron H, Eggert A, Cools J, Versteeg R, Speleman F. Meta-analysis of neuroblastomas reveals a skewed ALK mutation spectrum in tumors with MYCN amplification. Clin Cancer Res. 2010;16:4353–4362. doi: 10.1158/1078-0432.CCR-09-2660. [DOI] [PubMed] [Google Scholar]
- 21.Garaventa A, Parodi S, De Bernardi B, Dau D, Manzitti C, Conte M, Casale F, Viscardi E, Bianchi M, D’Angelo P, Zanazzo GA, Luksch R, Favre C, Tamburini A, Haupt R. Outcome of children with neuroblastoma after progression or relapse. A retrospective study of the Italian neuroblastoma registry. Eur J Cancer. 2009;45:2835–2842. doi: 10.1016/j.ejca.2009.06.010. [DOI] [PubMed] [Google Scholar]
- 22.McDermott U, Iafrate AJ, Gray NS, Shioda T, Classon M, Maheswaran S, Zhou W, Choi HG, Smith SL, Dowell L, Ulkus LE, Kuhlmann G, Greninger P, Christensen JG, Haber DA, Settleman J. Genomic alterations of anaplastic lymphoma kinase may sensitize tumors to anaplastic lymphoma kinase inhibitors. Cancer Res. 2008;68:3389–3395. doi: 10.1158/0008-5472.CAN-07-6186. [DOI] [PubMed] [Google Scholar]
- 23.Janoueix-Lerosey I, Schleiermacher G, Delattre O. Molecular pathogenesis of peripheral neuroblastic tumors. Oncogene. 2010;29:1566–1579. doi: 10.1038/onc.2009.518. [DOI] [PubMed] [Google Scholar]
- 24.Webb TR, Slavish J, George RE, Look AT, Xue L, Jiang Q, Cui X, Rentrop WB, Morris SW. Anaplastic lymphoma kinase: role in cancer pathogenesis and small-molecule inhibitor development for therapy. Expert Rev Anticancer Ther. 2009;9:331–356. doi: 10.1586/14737140.9.3.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 458 kb)