

Abstract
Eosinophils contribute to the pathogenesis of bullous pemphigoid (BP) by secretion of proinflammatory cytokines and proteases. Trafficking of eosinophils into tissue in animal models and asthma depends on interleukin-5 and a family of chemokines named eotaxins, comprising CCL11, CCL24 and CCL26. Up-regulation of CCL11 has been described in BP, but the expression of the other two members of the eotaxin-family, CCL24 and CCL26, has not been investigated. In addition to these chemokines, expression of adhesion molecules associated with eosinophil migration to the skin should be analysed. We demonstrate that similar to CCL11, the concentration of CCL26 was up-regulated in serum and blister fluid of BP patients. In contrast, the concentration of CCL24 was not elevated in sera and blister fluid of the same BP patients. In lesional skin, CCL11 and CCL26 were detected in epidermis and dermis by immunohistochemistry. In contrast to CCL11, CCL26 was expressed strongly by endothelial cells. In line with these findings, eosinophils represented the dominating cell population in BP lesional skin outnumbering other leucocytes. The percentage of eosinophils expressing very late antigen (VLA): VLA-4 (CD49d) and CD11c correlated with their quantity in tissue. Macrophage antigen (MAC)-1 (CD11b/CD18) was expressed constitutively by tissue eosinophils. In conclusion, these data link the up-regulation of the eosinophil chemotactic factor CCL26 in BP to the lesional accumulation of activated eosinophils in the skin. Thereby they broaden the understanding of BP pathogenesis and might indicate new options for therapeutic intervention.
Keywords: bullous pemphigoid, CCL11, CCL26, chemokines, eosinophil
Introduction
Bullous pemphigoid (BP) is the most prevalent autoimmune blistering skin disease, presenting with pruritic erythema preceding urticarial infiltrates followed by tense blisters on erythematous skin, predominantly affecting elderly people [1]. The hallmark of BP is the deposition of immunoglobulin (Ig)G autoantibodies and/or complement component C3 at the basement membrane zone [2]. These autoantibodies target hemidesmosomal proteins, most notably a 180 kD transmembrane hemidesmosomal glycoprotein (BP180) whose non-collagenous stretch called NC16A harbours the major antigenic site [3]. Autoantibody titres to NC16A correlate with disease activity, while a correlation of disease activity to titres of autoantibodies to other BP antigens, such as BP230 or BP200, has not been demonstrated [4]. Autoantibody presence in the skin is a prerequisite, albeit not sufficient to induce a lesion, as autoantibody deposits can be demonstrated regularly in healthy skin of affected individuals.
In animal models, as well as in in-vitro models, blister formation upon autoantibody binding in BP in contrast to pemphigus vulgaris depends on the presence of granulocytes and their release of proteases [5]. In BP patients, lesional skin is characterized by the presence of a cellular, mainly granulocytic, infiltrate and complement deposition [5–7]. Eosinophils seem to play an essential role in the initiation and/or progression of human BP by secretion of eosinophil-derived proteases. These enzymes process basement membrane structural proteins [8] and activate neutrophilic elastase, a very potent cleaver of BP180 in human BP blister fluid [9,10] Their secretion and long persistence in the tissue may also explain the exquisite pruritus present in BP but not in pemphigus vulgaris [11].
The recruitment of eosinophils into tissues is a multi-step process. Tethering and rolling are followed by firm adhesion to endothelial cells and subsequent transmigration. Chemoattractants produced locally by endothelial or stromal cells can activate leucocytes and induce up-regulation of integrins. In BP and allergic conditions, interleukin (IL)-4 can induce production of CCL11 in fibroblasts [12]. This chemokine belongs to the eotaxin subfamily of CC-chemokines consisting of eotaxin/CCL11, eotaxin-2/CCL24 and eotaxin-3/CCL26. CCL11 binds to CCR3 expressed on eosinophils and has been shown to induce eosinophil chemotaxis in vitro and in vivo[13]. CCR3 is the main chemokine receptor expressed on all eosinophils in blood, whereas other receptors such as CCR1, CCR2, CCR5, CCR8, CXCR1, CXCR2, CXCR3 and CXCR4 were found only in a minor subpopulation of the cells [14–16]. Beside eotaxins, the ligands CCL5, CCL7, CCL8, CCL13 and CCL15 can induce CCR3-mediated migration of eosinophils [17,18]. Of those, CCL13 has been found to be up-regulated in BP [19], whereas the weaker CCR3 ligands CCL5, CCL7 and CCL8 are not elevated significantly in sera of BP patients [20]. Activation of eosinophils through CCR3 results in enhanced avidity of α4 and β2 integrins [21]. The β2 integrins (CD11a/CD18 (LFA-1), CD11b/CD18 [macrophage antigen (MAC)-1, CD11c/CD18] are expressed by all granulocytes and mediate binding to intercellular adhesion molecule (ICAM)-1. Unlike neutrophils, eosinophils express the ligand of vascular cell adhesion molecule (VCAM)-1, α4/β1 integrin [CD29/CD49d (very late antigen-4: VLA-4)], that is especially important for eosinophil arrest [22]. This selective expression of VLA-4 in eosinophils and the inducibility of the ligand VCAM-1 by IL-4 on endothelial cells might explain the predominance of eosinophils in diseases with high levels of IL-4-like allergic asthma or BP.
In order to investigate further the mechanism of eosinophil recruitment in BP, we analysed the serum of a large number of patients for expression of CCR3-specific chemotactic ligands, correlated the concentration of these ligands with blister fluid levels in selected patients and studied the expression of integrins on lesional eosinophils.
Materials and methods
Patients and samples
Blood serum samples were obtained for diagnostic purposes from caucasian individuals before systemic immunosuppressive treatment for autoimmune blistering diseases, including patients with BP (age 56–90 years, mean 77 years, 56% female), non-atopic healthy controls (age 16–87 years, mean 38 years, 58% female) and patients with pemphigus vulgaris (PV) (age 38–87 years, mean 62 years, 53% female). Included were only sera from BP patients with typical presentation with bullous disease and direct immunofluorescence of perilesional skin revealing linear deposits of IgG and/or C3 along the basement membrane zone, as well as indirect immunofluorescence (IIF) performed on monkey oesophagus and sodium chloride-split skin, revealing linear staining of the induced blister roof. Clinical severity was documented by the clinicians using a three-point clinical scoring system documenting the extension of patients lesions: mild = few lesions affecting maximally 10% of the body surface; moderate = fresh infiltrates and blisters covering up to 50% of the body; severe = heavy disease with infiltrates and blister formation on 50% of body surface or higher. In three patients with active blister formation, blister fluid and serum could be obtained. Diagnosis of PV was confirmed by staining intercellular IgG deposits in lesional epidermis and in indirect immunofluorescence on monkey oesophagus. Skin biopsies and blister fluid samples were obtained for diagnostic purposes. Patients did not take immunosuppressive drugs for other indications. Concomitant medication was taken in some patients for age-related diseases such as hypertension or diabetes.
Chemokine and cytokine measurement
CCL11, CCL24 and CCL26 levels in serum and blister fluid samples were determined using commercially available enzyme-linked immunosorbent assay (ELISA) kits (R&D Systems, Abingdon, UK), according to the manufacturer's instructions. The detection limits of the ELISAs were 5 pg/ml, 2·5 pg/ml and 2·3 pg/ml, respectively.
Immunofluorescence staining
Skin sections were stained with a panel of antibodies specific for cell markers and surface molecules (Table 1), as described previously [23]. Briefly, cryostat sections were stained with primary antibodies followed by secondary reagents labelled with fluorescence dye. Nuclei were stained by 4′,6-diamidine-2-phenylindole (DAPI). Analysis was performed using an Olympus IMT2 fluorescence microscope. Each antibody staining was controlled by staining with a respective unspecific antibody from the same species. For each eosinophil surface marker studied, five different subepidermal tissue areas of eight individual patients were photographed at 400-fold magnification. The cell density and percentage of double staining was calculated by manual counting of the cells in the photographed area (0·2 mm2). In order to minimize observer-related errors, all slides were examined by three independent observers. The results show the means and standard deviation of those data.
Table 1.
Antibodies used for immunofluorescence staining
| Antibody against | Clone | Isotype/form | Source | Specificity relevant to present study |
|---|---|---|---|---|
| Lineage-specific markers | ||||
| Major basic protein (MBP) | Polyclonal | Mouse IgG1 | Chemicon | Eosinophils |
| CD68 | EBM11 | Mouse IgG1 | Dako | Monocyte lineage |
| CD3 | UCHT1 | Mouse IgG1–biotin | Pharmingen | T cells |
| Neutrophil elastase | AHN-10 | Mouse IgG1 | Pharmingen | Neutrophils |
| Secondary antibodies for lineage-specific markers | ||||
| Mouse IgG, IgM, IgA | Polyclonal | Goat-Texas red | Biozol | |
| Mouse Ig | Goat-rhodamin | Biosource | ||
| Biotin | Streptavidin–FITC | Vector | ||
| Rabbit IgG | Polyclonal | Alexa Fluor 488 | Molecular Probes | |
| Mouse IgG1 | Polyclonal | Alexa Fluor 546 | Molecular Probes | |
| Antibodies against surface markers | ||||
| CD11c | EP1347Y | rabbit IgG | abcam | p150, integrin α |
| CD11c | 3·9 | Mouse IgG1-FITC | Biosource | p150, integrin α |
| CD49d | BU49 | Mouse IgG1-FITC | Biozol | VLA-4 |
| CD11b | LM2/1 | Mouse IgG1-FITC | Biosource | MAC-1α, CR3 |
| HLA-DR | EDU-1 | Mouse IgG2b-FITC | Biosource | Human MHC class II |
| Factor VIII | F8/86 | Mouse IgG1-FITC | Signet | Endothelial cells |
Ig: immunoglobulin; FITC: fluorescence activated cell sorter; HLA: human leucocyte antigen; MAC: macrophage antigen; MHC: major histocompatibility complex; VLA: very late antigen-4.
Immunohistochemistry
For immunohistochemistry, cryostat sections of BP lesonal skin were incubated with antibodies against CCL11 (mouse anti-human IgG1, MAB320; R&D Systems) or CCL26 (goat anti-human IgG, AF693; R&D Systems). Detection of the primary antibodies was performed with donkey anti-mouse IgG biotin (Southern Biotec, Birmingham, AL, USA) or horse anti-goat IgG biotin (Vector Laboratories, Burlingham, USA) followed by streptavidin alkaline phosphatase (Biolegend, San Diego, CA, USA) and the substrate from LSAB-Kit-AP (Dako, Glostrup, Denmark). Unspecific mouse IgG1 and goat IgG were used as isotype controls.
Statistics
Data are presented as bars [indicating averages ± standard deviation (s.d.)] or box-plots (indicating average, median, 25–75% percentiles and s.d. (Origin 7·0 software; OriginLab Co., Northampton, MA, USA]. The Kolmogorov–Smirnov-test was used to test whether the samples were distributed normally. Statistical analysis was performed using the Mann–Whitney U-test for analysis of significant differences and Spearman's rank correlation test for testing correlation using WINstat software. In all cases, P < 0·05 was considered to be significant.
Results
The eotaxins CCL11 and CCL26 but not CCL24 are up-regulated in serum and blister fluid of BP patients
In the search for chemotactic factors of the whole eotaxin family of chemokines, concentrations of CCL11, CCL24 and CCL26 were measured in serum and blister fluid of patients with BP compared to sera from patients with the autoimmune blistering disease pemphigus vulgaris (PV) and healthy controls. Values of CCL11 were higher in sera of BP patients than in healthy controls (mean values 133 pg/ml ± 108 versus 48 pg/ml ± 79, P = 1·5 × E-6) or patients with PV (53 pg/ml ± 29, P = 0·002) (Fig. 1a). The concentrations of CCL11 correlated with the extension of the disease indicated by the disease score. The highest levels of CCL11 were detected among patients with severe BP (Fig. 1b). CCL26 was up-regulated in 55% of the BP sera and mean values were 10-fold higher (34 pg/ml ± 69) compared to sera from patients with PV (3 pg/ml ± 6, P = 0·04, Fig. 1a). The difference from healthy controls (9 pg/ml ± 17) was not statistically significant, because only patients with severe BP displayed elevated concentrations of CCL26 in serum (Fig. 1b). The mean of this subpopulation (105 pg/ml) was elevated significantly compared to healthy controls (P = 4 × E-5).
Fig. 1.
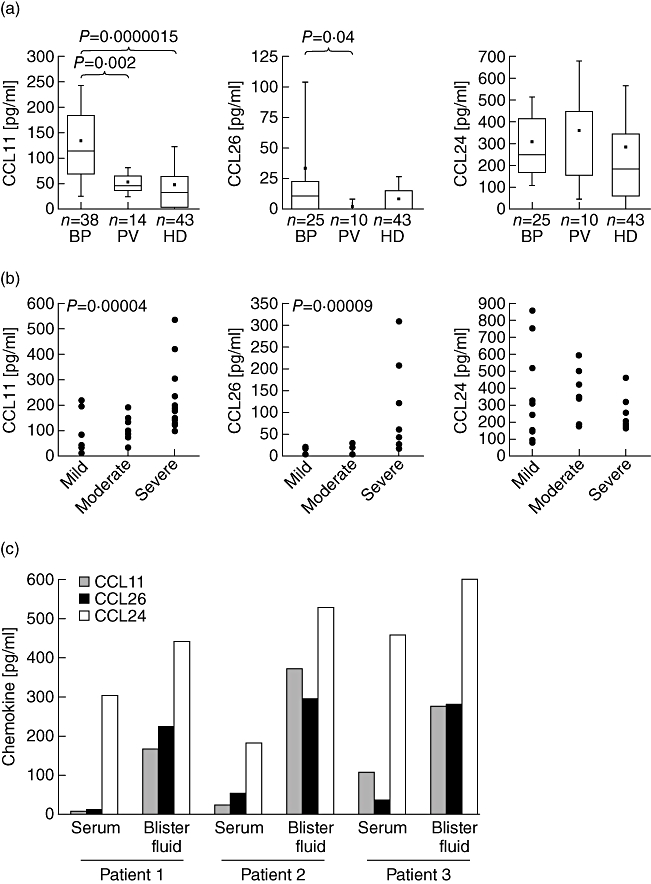
CCL11 and CCL26 are up-regulated in bullous pemphigoid (BP). (a) CCL11 and CCL26 were determined in serum of patients with BP, patients with PV and healthy donors (HD) by enzyme-linked immunosorbent assay (ELISA). P-values determined by Mann–Whitney U-test indicate significant differences. (b) Concentrations of serum chemokines are grouped to the disease score of the patient. Mild = lesions covering maximally 10% of the body surface; moderate = BP lesions affecting up to 50% of the body; severe = heavy disease with infiltrates and blister formation on 50% of the body or higher. Significant Spearman's rank correlation test is indicated by the P-value (c) concentrations of CCL11, CCL24 and CCL26 were determined by ELISA in blister fluid of three individual BP patients compared to the respective serum concentration (P-values of the mean of three patients between serum and blister concentration: 0·04 for CCL11; 0·04 for CCL26 and 0·12 for CCL24).
The levels of CCL24 were not significantly different in patients with BP (310 pg/ml ± 204) compared to patients with PV (362 pg/ml ± 317) or healthy controls (274 pg/ml ± 293; Fig. 1a). Concentrations of CCL24 did also not correlate with the disease score of BP (Fig. 1b).
Chemotaxis depends on a gradient of the chemotactic substance. To investigate the concentration-gradient from blood to tissue for the eotaxins we determined the concentration of the three eotaxins in blister fluid of three individuals with BP (Fig. 1c). In all blister fluid samples, the concentrations of CCL11 and CCL26 were increased, even in patients who presented with low serum concentrations of the respective chemokines. Blister fluid and serum of these two patients (patients 1 and 2, Fig. 1c) were collected during the first 48 h of blister formation. During this short disease period locally produced CCL11 might have not been distributed to the serum. We observed a ratio of blister fluid to serum for CCL11 of 2·5–33, with a mean of 45 pg/ml ± 55 in serum and 270 pg/ml ± 103 in blister fluid (P = 0·04). The concentration of CCL26 detected in sera was 37 pg/ml ± 18 compared to 269 pg/ml ± 37 in the matched blister fluid (P = 0·04). Thus, blister fluid concentration of CCL26 surpassed concentrations in matched sera at a ratio of 5·3–12·6. The blister fluid/serum ratios for CCL24 were only between 1·3 and 2·9 and the concentration was not increased significantly in blister fluid (525 pg/ml ± 80) compared to the sera (314 pg/ml ± 137, P = 0·12).
The up-regulation of the chemokines in blister fluid prompted us to investigate their expression in vivo. Immunohistochemistry staining of CCL26 in lesional BP skin was performed and demonstrated a strong expression of the chemokine in lesional BP epidermis and dermis (Fig. 2a). CCL11 was also detected by immunohistochemistry in lesional BP epidermal and dermal skin (Fig. 2a). In contrast to CCL11, CCL26 was co-expressed by endothelial cells in the dermis, as indicated by double immunofluorescence staining with the endothelial cell marker factor VIII (Fig. 2b).
Fig. 2.
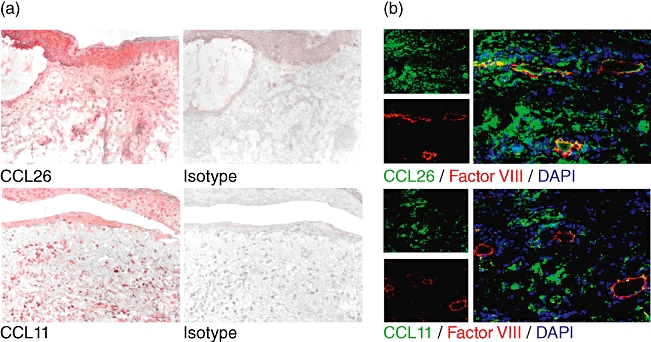
CCL26 and CCL11 are detected in lesional bullous pemphigoid (BP) skin. (a) Immunohistochemical staining revealed deposition of CCL26 and CCL11 in BP skin. Isotype staining was negative. Representative stainings of five individual patients with BP are shown. Original magnification ×200 (b) Double immunofluorescence staining of Factor VIII marking endothelial cells (red) and CCL26 or CCL11 (green) in BP. Yellow fluorescence indicates coexpression of CCL26 and Factor VIII by endothelial cells. Nuclei are stained with 4′,6-diamidine-2-phenylindole (DAPI). Representative stainings of three individual patients with BP are shown. Original magnification ×200.
Eosinophils predominate in the inflammatory infiltrate in BP lesions
To evaluate whether the expression of eotaxins in BP is associated with increased numbers of eosinophils, eosinophils were quantified and compared to other inflammatory cells in the lesional tissue (Fig. 3). BP lesional skin exhibited large numbers of eosinophils (51/mm2). Their amount was comparable to the total number of all cells of the CD68+ monocyte/myeloid origin (59/mm2) comprised of macrophages and dendritic cells. CD3+ T lymphocytes were less prominent, and eosinophils significantly outnumbered neutrophils (21/mm2).
Fig. 3.
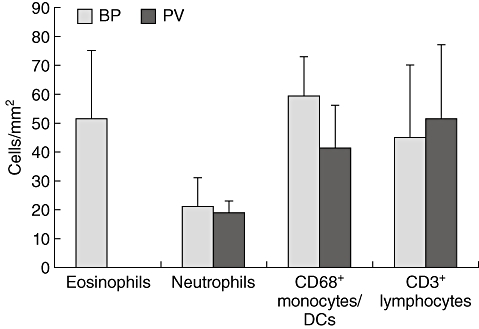
Eosinophils predominate bullous pemphigoid (BP) lesions. Cells were stained by immunofluorescence and counted per visual field. Numbers were calculated per mm2. The average densities and standard deviations of five different subepidermal tissue areas of nine individual patients with BP and four patients with pemphigus vulgaris (PV) for each cell type are shown.
In lesional skin of patients with PV, we did not detect eosinophils. The density of the other inflammatory cell populations was comparable to BP lesions (Fig. 3).
Lesional eosinophils show signs of activation
Next we aimed at analysing the activation status of lesional eosinophils and the expression of adhesion molecules responsible for their immigration into the skin. Eosinophils were stained by antibodies to major basic protein (MBP), a major eosinophil derived-protease. Double immunofluorescence staining with antibodies against human leucocyte antigen D-related (HLA-DR) demonstrated that 38 ± 12% of tissue eosinophils in lesional skin expressed HLA-DR (Fig. 4a,b). This indicated the activation status of the infiltrated eosinophils, as eosinophils in blood do not express HLA-DR.
Fig. 4.
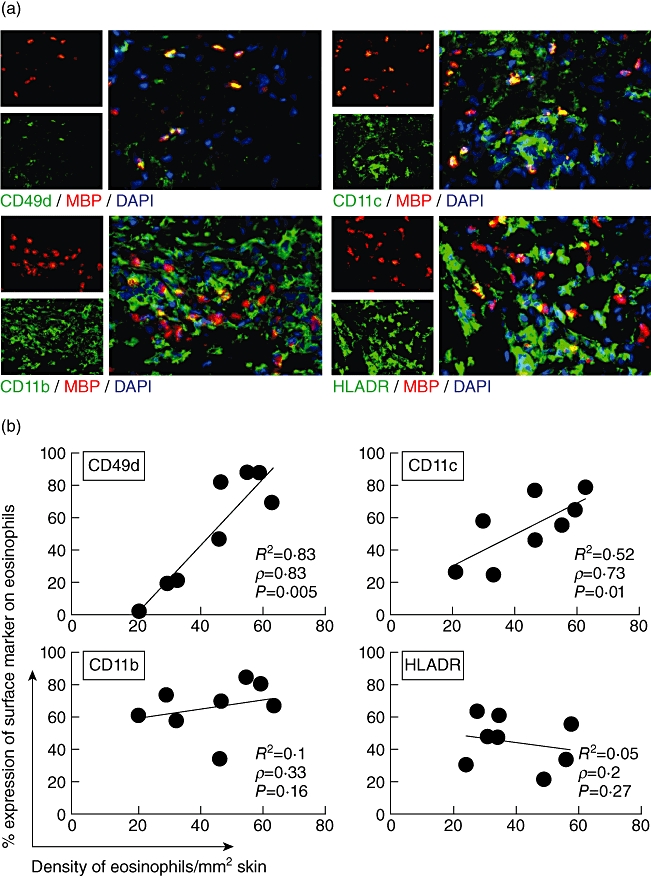
Eosinophils up-regulate activation markers in bullous pemphigoid (BP). Two-colour immunofluorescence staining for eosinophils and surface markers was performed in eight lesional biopsies from BP patients. (a) Using yellow fluorescence, double immunofluorescence stainings demonstrate the co-expression of surface molecules CD49d, CD11c, CD11b or human leucocyte antigen D-related (HLA-DR) on eosinophils major basic protein (MBP). Nuclei are stained with 4′,6-diamidine-2-phenylindole (DAPI) (blue). Original magnification ×400. (b) Eosinophils were counted in lesional BP skin and the percentage of double-positive eosinophils for each surface marker was determined (e.g. 27% of 21 eosinophils/mm2 express CD11c). Three sections for each marker were analysed by three independent investigators. The correlation of the density of eosinophils in tissue and the expression of adhesion molecules on the surface of eosinophils is given by Spearman's correlation coefficient ρ and the respective P-value, as well as the linear correlation coefficient R2. Each point represents the mean values of an individual patient.
Further double immunofluorescence stainings of cryostat sections of eosinophils and adhesion molecules (CD11c, CD11b, CD49d) demonstrated that the percentage of eosinophils expressing CD49d (VLA-4) and CD11c correlated with the density of eosinophils in the lesions (Fig. 4a,b) The detection limit of surface molecules by immunofluorescence staining in lesional skin depends on their expression number on the cell surface. A higher percentage of positive cells can therefore indicate higher expression levels on the respective cells. The correlation of expression levels of CD49d and CD11c may suggest that these surface molecules can be recognized as activation markers for eosinophils in lesional skin. CD11b was constitutively highly expressed on eosinophils (Fig. 4b).
Discussion
In this study we demonstrate that in addition to CCL11, the chemokine CCL26 is expressed in lesional BP skin. The concentration of CCL26 is elevated in blister fluid and serum of BP patients, establishing a gradient from tissue to vessel. Both eotaxins are known to induce CCR3-mediated eosinophil migration [24]. Their up-regulation can therefore explain the high density of activated eosinophils in cutaneous BP lesions, which are required for blister formation, as has been proved in animal and in-vitro models [5].
The proof of significantly up-regulated levels of CCL11 in a large group of BP patients compared to PV and healthy controls substantiates previous investigations on smaller numbers of patients [25,26]. In addition, up-regulation of CCL11 in association with dermal eosinophil accumulation was shown in pemphigoid gestationis, a variant of BP occurring in the peripartum period of pregnant women [23]. The serum values of CCL11 correlated with the disease severity [20].
CCL11 can be expressed by many cell types including eosinophils, keratinocytes, fibroblasts and macrophages [27]. Its expression is induced by IL-4 in human dermal fibroblasts [12] and by tumour necrosis factor (TNF)-α, IL-1α and interferon (IFN)-γ in human umbilical vein endothelial cells (HUVEC) [28]. Keeping in mind that IL-4 is up-regulated in lesional skin of BP patients, it is important that CCL26 but not CCL11 was inducible by this cytokine in HUVEC [24]. In line with these in-vitro data, immunohistochemical stainings in our study displayed the predominant expression of CCL26 by endothelial cells. It can therefore be proposed that eosinophils are activated by CCL26 expressed on endothelial cells in BP, and then transmigrate to the centre of the inflamed tissue through the action of CCL26 and CCL11. This sequential activation has been suggested previously to explain eosinophil migration into skin lesions in atopic dermatitis. Like BP, atopic dermatitis lesions are characterized by IL-4 expression during development of the disease [24]. CCL26 is also induced by IL-4 and IL-13 in keratinocytes [29,30], which supports our immunohistochemical staining of CCL26 in the epidermis.
The concentrations of CCL11, CCL24 and CCL26 in blister fluid reflect the intercellular concentration in tissue. Their several-fold higher levels in blister fluid compared to the serum of BP patients constitute a concentration gradient that enables the eosinophils to migrate further from the vessels to the basement membrane, where blister formation is induced. The up-regulation of CCL24 only in blister fluid but not in the patients' sera accounts for a limited impact of CCL24 in eosinophil migration in BP. However, one possible function of up-regulated CCL24 in tissue can be its influence on the eosinophil adhesion molecule usage from VCAM-1 to ICAM-1 [31]. CCL24 was the only one of the three eotaxins not up-regulated in serum, but its total concentration in healthy controls as well as BP patients exceeded that of CCL11 and CCL26. This pattern has been described previously in atopic dermatitis, Churg–Strauss syndrome and leprosy [32–34]. Effects exerted by lower concentrations of CCL11 and CCL26 in the presence of higher concentrations of CCL24, which binds to the same receptor CCR3, may be a function of the receptor's affinity for the three chemokines. In Chinese hamster ovary (CHO) cells transfected with CCR3, CCL11 and CCL26 bound with higher affinity to CCR3 compared to CCL24 [35]. The base for this difference in affinity may be the three-dimensional structure of CCR3 after post-translational modification by sulphation, as Zhu et al. [36] described recently that double tyrosine sulphation of an N-terminal CCR3-derived peptide resulted in a 10-fold higher binding affinity of CCL11 and CCL26 to CCR3 compared to CCL24 [36]. Tyrosine sulphation of cell surface proteins occurs commonly in the trans-Golgi network, and has been detected in other chemokine receptors. Moreover, tyrosine residues have been modelled to be present in the binding site pocket of CCR3 [37,38]. The resulting higher affinity of CCR3 for CCL11 and CCL26 could be a reason for the effectiveness of CCL11 and CCL26 in the recruitment of eosinophils, despite the higher total concentration of CCL24.
Transmigration of eosinophils through the endothelium requires the expression of adhesion molecules and results in activation and up-regulation of specific eosinophil functions [39]. Consequently, eosinophils in BP expressed HLA-DR.
Selective expression of the adhesion molecule VLA-4 (CD49d/CD29) on eosinophils in contrast to neutrophils is one suggested mechanism by which predominant infiltration of eosinophils in asthma, atopic dermatitis and also BP is achieved [40]. VLA-4 interacts with VCAM-1 expressed by endothelial cells in skin [41] and lung [42], and mediates rolling, adhesion and transmigration of eosinophils.
In contrast to the suggested importance of the VLA-4–VCAM-1 interaction for emigration of eosinophils into tissue, therapeutic blockade of the α4-integrin chain by specific antibodies failed to prevent airway eosinophilia in humans [43]. In BP patients with low lesional eosinophil numbers, the percentage of CD49d was lower compared to highly infiltrated lesions. This low expression of CD49d might indicate that this integrin is not essential for transmigration, and could partially explain the lack of effectiveness of antagonizing drugs in vivo.
CD11b/CD18 is expressed on purified blood eosinophils [40]. Its interaction with ICAM-1 is influenced greatly by activation. CD11b/CD18 can also mediate a constitutive low-level adhesion of VCAM-1 and supports adhesion and transmigration together with α4β1[40]. Interaction of CD11b with its ligands on endothelial cells can also induce activation of eosinophils, as shown by CD11b-dependent CD69 up-regulation on eosinophils [44]. In its function as complement receptor 3, CD11b can support eosinophil degranulation [45]. The levels of CD11b expression on tissue eosinophils in BP were constantly high and did not correlate with the number of eosinophils. This might indicate a requirement of CD11b for transmigration [46] and function of eosinophils in BP [47].
All other integrins expressed on eosinophils, including CD11c/CD18, did not seem to play significant roles in eosinophil adhesion because antibodies to this ligand did not block adhesion to ICAM-1, albumin or fibrinogen in vitro[40]. In BP lesions we observed a weak correlation between the number of eosinophils infiltrated and CD11c. This could point to CD11c as a possible activation marker on these cells.
The findings of this study can be integrated into the current pathogenetic model of BP [48]: hemidesmosomal antigens are presented aberrantly by dendritic cells and induce a specific T and B cell response. Circulating antibodies bind to BP180 predominately in the skin of the great flexures, where most BP180 is expressed and the disease often starts [49]. A high density of these antibodies might induce complement binding and IL-5, IL-4, TNF-α and eotaxin production in mast cells, followed by additional chemokine secretion by fibroblasts and luminal presentation of chemotactic factors by endothelial cells. IL-5 can mediate release of eosinophils [50] from bone marrow and CCL26 supports adhesion of eosinophils to endothelial cells. Upon activation, eosinophils transmigrate mediated by VLA-4 (CD49d) and MAC-1 (CD11b) and follow a chemotactic gradient of CCL11, CCL26 and CCL13. Beside CCR3-induced chemotaxis, the chemokine receptor CCR1, highly expressed on eosinophils in about 20% of atopic and non-atopic donors [16], might contribute to eosinophil chemotaxis in some BP patients, in which the ligand CCL3 is up-regulated [20]. Other non-specific chemoattractants might also contribute to eosinophil migration: prostaglandin D2 by acting on the receptor CRTH2 on eosinophils, complement components C3a and C5a or platelet-activating factor through their respective receptors [51,52]. After transmigration eosinophils up-regulate HLA-DR and stimulate Th2 cells to further secrete IL-4 and IL-5 [53]. Concomitant immigration of further eosinophils leads to secretion of large amounts of eosinophil-derived proteases such as matrix metalloproteinases, eosinophil cationic protein and MBP [9,48]. These enzymes promote blister formation and induction of concomitant inflammation [8]. Because of their important function, eosinophil immigration is essential for the development of BP. Regulation of eosinophil influx might be achieved by sequestration of CCL11 by CXCR3-expressing T cells located in BP-skin [54,55] and the antagonistic function of CCL18 and CXCL10 on CCR3 [54,56], both of which have been shown to be up-regulated in BP lesional skin [20,57]. In addition, CCL26 could exert antagonistic functions on eosinophils expressing CCR1 [58].
Our data on the expression of CCL26, CCL11 and CCL24 in BP lesions and the up-regulation of adhesion molecules on eosinophils contribute to the understanding of the recruitment process for eosinophils in BP, and might open new possibilities for future therapeutic intervention in this autoimmune blistering skin disease. They further enhance the current knowledge of the fine interplay of chemokines and integrins in facilitating transmigration, tissue invasion and in-situ activation, linking biological knowledge and clinical observation to establish pathogenetic significance of immunological findings.
Acknowledgments
We thank Andreas Herrmann for collecting sera of healthy controls and highly acknowledge the technical support from Katharina Blümlein, Lysann Mehlig, Nick Zimmermann and Jana Phillip. This study was funded by a MeDDrive and a habilitation grant of the Medical Faculty of the Technical University of Dresden to C.P. and C.G., respectively.
Disclosure
None.
References
- 1.Korman NJ. Bullous pemphigoid. The latest in diagnosis, prognosis, and therapy. Arch Dermatol. 1998;134:1137–41. doi: 10.1001/archderm.134.9.1137. [DOI] [PubMed] [Google Scholar]
- 2.Jordon RE, Beutner EH, Witebsky E, Blumental G, Hale WL, Lever WF. Basement zone antibodies in bullous pemphigoid. JAMA. 1967;200:751–6. [PubMed] [Google Scholar]
- 3.Labib RS, Anhalt GJ, Patel HP, Mutasim DF, Diaz LA. Molecular heterogeneity of the bullous pemphigoid antigens as detected by immunoblotting. J Immunol. 1986;136:1231–5. [PubMed] [Google Scholar]
- 4.Kobayashi M, Amagai M, Kuroda-Kinoshita K, et al. BP180 ELISA using bacterial recombinant NC16a protein as a diagnostic and monitoring tool for bullous pemphigoid. J Dermatol Sci. 2002;30:224–32. doi: 10.1016/s0923-1811(02)00109-3. [DOI] [PubMed] [Google Scholar]
- 5.Liu Z. Immunopathology of bullous pemphigoid, an autoimmune and inflammatory skin blistering disease. Keio J Med. 2003;52:128–33. doi: 10.2302/kjm.52.128. [DOI] [PubMed] [Google Scholar]
- 6.Liu Z, Diaz LA. Bullous pemphigoid: end of the century overview. J Dermatol. 2001;28:647–50. doi: 10.1111/j.1346-8138.2001.tb00055.x. [DOI] [PubMed] [Google Scholar]
- 7.Liu Z, Giudice GJ, Zhou X, et al. A major role for neutrophils in experimental bullous pemphigoid. J Clin Invest. 1997;100:1256–63. doi: 10.1172/JCI119639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Borrego L, Maynard B, Peterson EA, et al. Deposition of eosinophil granule proteins precedes blister formation in bullous pemphigoid. Comparison with neutrophil and mast cell granule proteins. Am J Pathol. 1996;148:897–909. [PMC free article] [PubMed] [Google Scholar]
- 9.Stahle-Backdahl M, Inoue M, Guidice GJ, Parks WC. 92-kD gelatinase is produced by eosinophils at the site of blister formation in bullous pemphigoid and cleaves the extracellular domain of recombinant 180-kD bullous pemphigoid autoantigen. J Clin Invest. 1994;93:2022–30. doi: 10.1172/JCI117196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Verraes S, Hornebeck W, Polette M, Borradori L, Bernard P. Respective contribution of neutrophil elastase and matrix metalloproteinase 9 in the degradation of BP180 (type XVII collagen) in human bullous pemphigoid. J Invest Dermatol. 2001;117:1091–6. doi: 10.1046/j.0022-202x.2001.01521.x. [DOI] [PubMed] [Google Scholar]
- 11.Davis MD, Plager DA, George TJ, Weiss EA, Gleich GJ, Leiferman KM. Interactions of eosinophil granule proteins with skin: limits of detection, persistence, and vasopermeabilization. J Allergy Clin Immunol. 2003;112:988–94. doi: 10.1016/j.jaci.2003.08.028. [DOI] [PubMed] [Google Scholar]
- 12.Mochizuki M, Schroder J, Christophers E, Yamamoto S. IL-4 induces eotaxin in human dermal fibroblasts. Int Arch Allergy Immunol. 1999;120(Suppl 1):19–23. doi: 10.1159/000053587. [DOI] [PubMed] [Google Scholar]
- 13.Wardlaw AJ. Molecular basis for selective eosinophil trafficking in asthma: a multistep paradigm. J Allergy Clin Immunol. 1999;104:917–26. doi: 10.1016/s0091-6749(99)70069-2. [DOI] [PubMed] [Google Scholar]
- 14.Liu LY, Jarjour NN, Busse WW, Kelly EA. Chemokine receptor expression on human eosinophils from peripheral blood and bronchoalveolar lavage fluid after segmental antigen challenge. J Allergy Clin Immunol. 2003;112:556–62. doi: 10.1016/s0091-6749(03)01798-6. [DOI] [PubMed] [Google Scholar]
- 15.Silveira-Lemos D, Teixeira-Carvalho A, Martins-Filho OA, et al. Seric chemokines and chemokine receptors in eosinophils during acute human Schistosomiasis mansoni. Mem Inst Oswaldo Cruz. 2010;105:380–6. doi: 10.1590/s0074-02762010000400006. [DOI] [PubMed] [Google Scholar]
- 16.Phillips RM, Stubbs VE, Henson MR, Williams TJ, Pease JE, Sabroe I. Variations in eosinophil chemokine responses: an investigation of CCR1 and CCR3 function, expression in atopy, and identification of a functional CCR1 promoter. J Immunol. 2003;170:6190–201. doi: 10.4049/jimmunol.170.12.6190. [DOI] [PubMed] [Google Scholar]
- 17.Heath H, Qin S, Rao P, et al. Chemokine receptor usage by human eosinophils. The importance of CCR3 demonstrated using an antagonistic monoclonal antibody. J Clin Invest. 1997;99:178–84. doi: 10.1172/JCI119145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coulin F, Power CA, Alouani S, et al. Characterisation of macrophage inflammatory protein-5/human CC cytokine-2, a member of the macrophage-inflammatory-protein family of chemokines. Eur J Biochem. 1997;248:507–15. doi: 10.1111/j.1432-1033.1997.00507.x. [DOI] [PubMed] [Google Scholar]
- 19.Gounni AS, Wellemans V, Agouli M, et al. Increased expression of Th2-associated chemokines in bullous pemphigoid disease. Role of eosinophils in the production and release of these chemokines. Clin Immunol. 2006;120:220–31. doi: 10.1016/j.clim.2006.03.014. [DOI] [PubMed] [Google Scholar]
- 20.Nakashima H, Fujimoto M, Asashima N, et al. Serum chemokine profile in patients with bullous pemphigoid. Br J Dermatol. 2007;156:454–9. doi: 10.1111/j.1365-2133.2006.07601.x. [DOI] [PubMed] [Google Scholar]
- 21.Kitayama J, Mackay CR, Ponath PD, Springer TA. The C-C chemokine receptor CCR3 participates in stimulation of eosinophil arrest on inflammatory endothelium in shear flow. J Clin Invest. 1998;101:2017–24. doi: 10.1172/JCI2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weber C, Springer TA. Interaction of very late antigen-4 with VCAM-1 supports transendothelial chemotaxis of monocytes by facilitating lateral migration. J Immunol. 1998;161:6825–34. [PubMed] [Google Scholar]
- 23.Gunther C, Wozel G, Dressler J, Meurer M, Pfeiffer C. Tissue eosinophilia in pemphigoid gestationis: association with eotaxin and upregulated activation markers on transmigrated eosinophils. Am J Reprod Immunol. 2004;51:32–9. doi: 10.1046/j.8755-8920.2003.00118.x. [DOI] [PubMed] [Google Scholar]
- 24.Shinkai A, Yoshisue H, Koike M, et al. A novel human CC chemokine, eotaxin-3, which is expressed in IL-4-stimulated vascular endothelial cells, exhibits potent activity toward eosinophils. J Immunol. 1999;163:1602–10. [PubMed] [Google Scholar]
- 25.Frezzolini A, Teofoli P, Cianchini G, et al. Increased expression of eotaxin and its specific receptor CCR3 in bullous pemphigoid. Eur J Dermatol. 2002;12:27–31. [PubMed] [Google Scholar]
- 26.Shrikhande M, Hunziker T, Braathen LR, Pichler WJ, Dahinden CA, Yawalkar N. Increased coexpression of eotaxin and interleukin 5 in bullous pemphigoid. Acta Derm Venereol. 2000;80:277–80. doi: 10.1080/000155500750012162. [DOI] [PubMed] [Google Scholar]
- 27.Amerio P, Frezzolini A, Feliciani C, et al. Eotaxins and CCR3 receptor in inflammatory and allergic skin diseases: therapeutical implications. Curr Drug Targets Inflamm Allergy. 2003;2:81–94. doi: 10.2174/1568010033344480. [DOI] [PubMed] [Google Scholar]
- 28.Garcia-Zepeda EA, Rothenberg ME, Ownbey RT, Celestin J, Leder P, Luster AD. Human eotaxin is a specific chemoattractant for eosinophil cells and provides a new mechanism to explain tissue eosinophilia. Nat Med. 1996;2:449–56. doi: 10.1038/nm0496-449. [DOI] [PubMed] [Google Scholar]
- 29.Nishi N, Yamamoto S, Ou W, Muro E, Inada Y, Hamasaki Y. Enhanced CCL26 production by IL-4 through IFN-gamma-induced upregulation of type 1 IL-4 receptor in keratinocytes. Biochem Biophys Res Commun. 2008;376:234–40. doi: 10.1016/j.bbrc.2008.08.136. [DOI] [PubMed] [Google Scholar]
- 30.Kagami S, Saeki H, Komine M, et al. Interleukin-4 and interleukin-13 enhance CCL26 production in a human keratinocyte cell line, HaCaT cells. Clin Exp Immunol. 2005;141:459–66. doi: 10.1111/j.1365-2249.2005.02875.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tachimoto H, Burdick MM, Hudson SA, Kikuchi M, Konstantopoulos K, Bochner BS. CCR3-active chemokines promote rapid detachment of eosinophils from VCAM-1 in vitro. J Immunol. 2000;165:2748–54. doi: 10.4049/jimmunol.165.5.2748. [DOI] [PubMed] [Google Scholar]
- 32.Kagami S, Kakinuma T, Saeki H, et al. Significant elevation of serum levels of eotaxin-3/CCL26, but not of eotaxin-2/CCL24, in patients with atopic dermatitis: serum eotaxin-3/CCL26 levels reflect the disease activity of atopic dermatitis. Clin Exp Immunol. 2003;134:309–13. doi: 10.1046/j.1365-2249.2003.02273.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Polzer K, Karonitsch T, Neumann T, et al. Eotaxin-3 is involved in Churg–Strauss syndrome – a serum marker closely correlating with disease activity. Rheumatology (Oxf) 2008;47:804–8. doi: 10.1093/rheumatology/ken033. [DOI] [PubMed] [Google Scholar]
- 34.Mendonca VA, Malaquias LC, Brito-Melo GE, et al. Differentiation of patients with leprosy from non-infected individuals by the chemokine eotaxin/CCL11. Am J Trop Med Hyg. 2007;77:547–50. [PubMed] [Google Scholar]
- 35.Parody TR, Stone MJ. High level expression, activation, and antagonism of CC chemokine receptors CCR2 and CCR3 in Chinese hamster ovary cells. Cytokine. 2004;27:38–46. doi: 10.1016/j.cyto.2004.03.013. [DOI] [PubMed] [Google Scholar]
- 36.Zhu JZ, Millard CJ, Ludeman JP, et al. Tyrosine sulfation influences the chemokine binding selectivity of peptides derived from chemokine receptor CCR3. Biochemistry. 2011;50:1524–34. doi: 10.1021/bi101240v. [DOI] [PubMed] [Google Scholar]
- 37.Wise EL, Duchesnes C, da Fonseca PC, Allen RA, Williams TJ, Pease JE. Small molecule receptor agonists and antagonists of CCR3 provide insight into mechanisms of chemokine receptor activation. J Biol Chem. 2007;282:27935–43. doi: 10.1074/jbc.M703255200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jain V, Saravanan P, Arvind A, Mohan CG. First pharmacophore model of CCR3 receptor antagonists and its homology model-assisted, stepwise virtual screening. Chem Biol Drug Des. 2011;77:373–87. doi: 10.1111/j.1747-0285.2011.01088.x. [DOI] [PubMed] [Google Scholar]
- 39.Chihara J, Yamamoto T, Kurachi D, Kakazu T, Higashimoto I, Nakajima S. Possible release of eosinophil granule proteins in response to signaling from intercellular adhesion molecule-1 and its ligands. Int Arch Allergy Immunol. 1995;108(Suppl. 1):52–4. doi: 10.1159/000237204. [DOI] [PubMed] [Google Scholar]
- 40.Barthel SR, Johansson MW, McNamee DM, Mosher DF. Roles of integrin activation in eosinophil function and the eosinophilic inflammation of asthma. J Leukoc Biol. 2008;83:1–12. doi: 10.1189/jlb.0607344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dahlman-Ghozlan K, Ortonne JP, Heilborn JD, Stephansson E. Altered tissue expression pattern of cell adhesion molecules, ICAM-1, E-selectin and VCAM-1, in bullous pemphigoid during methotrexate therapy. Exp Dermatol. 2004;13:65–9. doi: 10.1111/j.0906-6705.2004.00113.x. [DOI] [PubMed] [Google Scholar]
- 42.Popper HH, Pailer S, Wurzinger G, Feldner H, Hesse C, Eber E. Expression of adhesion molecules in allergic lung diseases. Virchows Arch. 2002;440:172–80. doi: 10.1007/s004280100507. [DOI] [PubMed] [Google Scholar]
- 43.Diamant Z, Kuperus J, Baan R, et al. Effect of a very late antigen-4 receptor antagonist on allergen-induced airway responses and inflammation in asthma. Clin Exp Allergy. 2005;35:1080–7. doi: 10.1111/j.1365-2222.2005.02296.x. [DOI] [PubMed] [Google Scholar]
- 44.Dallaire MJ, Ferland C, Page N, Lavigne S, Davoine F, Laviolette M. Endothelial cells modulate eosinophil surface markers and mediator release. Eur Respir J. 2003;21:918–24. doi: 10.1183/09031936.03.00102002. [DOI] [PubMed] [Google Scholar]
- 45.Egesten A, Blom M, Calafat J, Janssen H, Knol EF. Eosinophil granulocyte interaction with serum-opsonized particles: binding and degranulation are enhanced by tumor necrosis factor alpha. Int Arch Allergy Immunol. 1998;115:121–8. doi: 10.1159/000023891. [DOI] [PubMed] [Google Scholar]
- 46.Fernvik E, Lundahl J, Hallden G. The impact of eotaxin- and IL-5-induced adhesion and transmigration on eosinophil activity markers. Inflammation. 2000;24:73–87. doi: 10.1023/a:1006940109869. [DOI] [PubMed] [Google Scholar]
- 47.Lundahl J, Moshfegh A, Gronneberg R, Hallden G. Eotaxin increases the expression of CD11b/CD18 and adhesion properties in IL5, but not fMLP-prestimulated human peripheral blood eosinophils. Inflammation. 1998;22:123–35. doi: 10.1023/a:1022379821130. [DOI] [PubMed] [Google Scholar]
- 48.Kasperkiewicz M, Zillikens D. The pathophysiology of bullous pemphigoid. Clin Rev Allergy Immunol. 2007;33:67–77. doi: 10.1007/s12016-007-0030-y. [DOI] [PubMed] [Google Scholar]
- 49.Sison-Fonacier L, Bystryn JC. Regional variations in antigenic properties of skin. A possible cause for disease-specific distribution of skin lesions. J Exp Med. 1986;164:2125–30. doi: 10.1084/jem.164.6.2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.D'Auria L, Pimpinelli F, Ferraro C, et al. Relationship between theoretical molecular weight and blister fluid/serum ratio of cytokines and five other molecules evaluated in patients with bullous pemphigoid. J Biol Regul Homeost Agents. 1998;12:76–80. [PubMed] [Google Scholar]
- 51.Daffern PJ, Pfeifer PH, Ember JA, Hugli TE. C3a is a chemotaxin for human eosinophils but not for neutrophils. I. C3a stimulation of neutrophils is secondary to eosinophil activation. J Exp Med. 1995;181:2119–27. doi: 10.1084/jem.181.6.2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Satoh T, Shimura C, Miyagishi C, Yokozeki H. Indomethacin-induced reduction in CRTH2 in eosinophilic pustular folliculitis (Ofuji's disease): a proposed mechanism of action. Acta Derm Venereol. 2010;90:18–22. doi: 10.2340/00015555-0759. [DOI] [PubMed] [Google Scholar]
- 53.Rico MJ, Benning C, Weingart ES, Streilein RD, Hall RP., III Characterization of skin cytokines in bullous pemphigoid and pemphigus vulgaris. Br J Dermatol. 1999;140:1079–86. doi: 10.1046/j.1365-2133.1999.02907.x. [DOI] [PubMed] [Google Scholar]
- 54.Xanthou G, Duchesnes CE, Williams TJ, Pease JE. CCR3 functional responses are regulated by both CXCR3 and its ligands CXCL9, CXCL10 and CXCL11. Eur J Immunol. 2003;33:2241–50. doi: 10.1002/eji.200323787. [DOI] [PubMed] [Google Scholar]
- 55.Kakinuma T, Wakugawa M, Nakamura K, Hino H, Matsushima K, Tamaki K. High level of thymus and activation-regulated chemokine in blister fluid and sera of patients with bullous pemphigoid. Br J Dermatol. 2003;148:203–10. doi: 10.1046/j.1365-2133.2003.05066.x. [DOI] [PubMed] [Google Scholar]
- 56.Nibbs RJ, Salcedo TW, Campbell JD, et al. C-C chemokine receptor 3 antagonism by the beta-chemokine macrophage inflammatory protein 4, a property strongly enhanced by an amino-terminal alanine–methionine swap. J Immunol. 2000;164:1488–97. doi: 10.4049/jimmunol.164.3.1488. [DOI] [PubMed] [Google Scholar]
- 57.Gunther C, Carballido-Perrig N, Kopp T, Carballido JM, Pfeiffer C. CCL18 is expressed in patients with bullous pemphigoid and parallels disease course. Br J Dermatol. 2009;160:747–55. doi: 10.1111/j.1365-2133.2008.08979.x. [DOI] [PubMed] [Google Scholar]
- 58.Petkovic V, Moghini C, Paoletti S, Uguccioni M, Gerber B. Eotaxin-3/CCL26 is a natural antagonist for CC chemokine receptors 1 and 5. A human chemokine with a regulatory role. J Biol Chem. 2004;279:23357–63. doi: 10.1074/jbc.M309283200. [DOI] [PubMed] [Google Scholar]