

Abstract
Mechanisms associated with the progression of simple steatosis to non-alcoholic fatty liver disease (NAFLD) remain undefined. Regulatory T cells (Tregs) play a critical role in regulating inflammatory processes in non-alcoholic steatohepatitis (NASH) and because T helper type 17 (Th17) functionally oppose Treg-mediated responses, this study focused on characterizing the role of Th17 cells using a NAFLD mouse model. C57BL/6 mice were fed either a normal diet (ND) or high fat (HF) diet for 8 weeks. Mice in the HF group had a significantly higher frequency of liver Th17 cells compared to ND-fed mice. Neutralization of interleukin (IL)-17 in HF mice ameliorated lipopolysaccharide (LPS)-induced liver injury reflected by decreased serum alanine aminotransferase (ALT) levels and reduced inflammatory cell infiltrates in the liver. In vitro, HepG2 cells cultured in the presence of free fatty acids (FFA; oleic acid and palmitic acid) for 24 h and IL-17 developed steatosis via insulin-signalling pathway interference. IL-17 and FFAs synergized to induce IL-6 production by HepG2 cells and murine primary hepatocytes which, in combination with transforming growth factor (TGF-β), expanded Th17 cells. It is likely that a similar process occurs in NASH patients, as there were significant levels of IL-17+ cell infiltrates in NASH patient livers. The hepatic expression of Th17 cell-related genes [retinoid-related orphan receptor gamma (ROR)γt, IL-17, IL-21 and IL-23] was also increased significantly in NASH patients compared to healthy controls. Th17 cells and IL-17 were associated with hepatic steatosis and proinflammatory response in NAFLD and facilitated the transition from simple steatosis to steatohepatitis. Strategies designed to alter the balance between Th17 cells and Tregs should be explored as a means of preventing progression to NASH and advanced liver diseases in NAFLD patients.
Keywords: inflammation, insulin resistance, non-alcoholic steatohepatitis, signalling pathways, Th17 cells
Introduction
Up to one-third of patients presenting with non-alcoholic fatty liver disease (NAFLD) present with non-alcoholic steatohepatitis (NASH), characterized by lobular inflammation and hepatocyte ballooning. Those patients have a risk of progressive fibrosis, cirrhosis and hepatic cellular carcinoma [1–3]. Understanding the mechanisms that lead to progression from simple steatosis to NASH (and potentially more advanced disease) is critical to the design of rational treatment strategies directed towards patients who develop progressive disease [4]. Lipopolysaccharide (LPS), oxidative stress, cytokine production and other proinflammatory mediators may each play roles in delivering a ‘second hit’ during the transition from simple steatosis to NASH [1]. Because hepatic inflammation is one of the most distinguished features of NASH, hepatic immune responses may also play critical roles in the pathogenesis of NASH and other progressive diseases. Fatty liver may be more susceptible to injury because certain subsets of hepatic lymphocytes have changed, unbalancing the production of anti-inflammatory T helper type 2 (Th2) cytokines to proinflammatory Th1 cytokines [5].
Recently, the Th1/Th2 paradigm has been expanded following the discovery of a distinct T helper cell subset that produces interleukin (IL)-17 (Th17 cells) [6–8]. Th17 cells produce IL-17 (also referred to as IL-17A), IL-17F, IL-21 and IL-22, and mediate potent inflammatory immune responses [9]. Th17 cells play important roles not only in mediating pathogen clearance but also contribute to tissue inflammatory responses and autoimmune disease progression [10]. Polarization of Th17 cell responses requires the production of cytokines associated with the innate immune system, similar to the development of adaptive Th1 and Th2 responses. However, the requirement of transforming growth factor (TGF)-β for the differentiation of Th17 cells is one of the fundamental differences between the induction of this effector T cell subset [9]. Interestingly, TGF-β induces forkhead box P3 (FoxP3) expression and the development of regulatory T cells (Tregs), inhibits inflammation and maintains self-tolerance. However, a combination of the immunoregulatory cytokine TGF-β and proinflammatory cytokine IL-6 inhibits Treg generation, elicits Th17 cell production and leads proinflammatory response [9]. The reciprocal relationship between Treg cells and Th17 cells represents a delicate balance between tolerance and elicitation of immune responses [11].
Accumulating evidence has shown that IL-17 and Th17 cells may play a critical role in various kinds of liver diseases, including chronic viral hepatitis, autoimmune liver diseases, alcoholic liver disease and hepatocellular carcinoma [10,12,13]. Previously, we demonstrated that increased oxidative stress in fatty livers induced apoptosis of CD4+CD25+ Treg cells, reduced the number of hepatic Treg cells and led to impaired suppression of inflammatory responses [14]. Therefore, it was of interest to determine whether Th17 cells played a key role in NAFLD-associated inflammatory processes. In the current study, we hypothesized that Th17 cells and their production of IL-17 may influence inflammatory responses that determine the progression of simple hepatic steatosis to NASH. In addition, the interaction between hepatocytes and Th17 cells were investigated in order to identify potential mechanisms associated with the vicious cycle associated with hepatic steatosis and liver inflammation. The data presented in this report not only shed light on the pathogenic mechanisms resulting in NASH, but also identified potential therapeutic targets that can be targeted for the prevention of steatosis and hepatic inflammation.
Materials and methods
Animal experiments
Adult male C57BL/6 mice (6–8 weeks of age) were purchased from Shanghai SLAC Laboratory Animal Co., Ltd (Shanghai, China) and housed under pathogen-free conditions in the animal facility of the Shanghai Jiao Tong University School of Medicine. Mice were fed either a high fat (HF) diet (SLAC Laboratory Animal Co., Shanghai, China), providing 59% of calories from fat, 25% from carbohydrates and 16% from protein for 8 weeks, or an isocaloric normal diet (ND) (control mice) containing less fat (12% fat, 59% total carbohydrate and 29% protein). All mice were maintained in a temperature- and light-controlled facility with ad libitum access to food and water. For the LPS experiments, mice were injected intraperitoneally with a single dose of Escherichia coli LPS (50 µg/mouse; Sigma, St Louis, MO, USA) and killed 6 h later to obtain serum and liver samples. Neutralizing IL-17 antibody (100 µg/mice; Pharmingen, San Diego, CA, USA) was administrated intravenously through the tail vein in a group of mice prior to LPS injection. Control animals were injected with vehicle alone. All animal experiments were approved by the Institutional Animal Care and Use Committee of Shanghai Jiao Tong University School of Medicine and were conducted in accordance with the National Research Council Guide for Care and Use of Laboratory Animals. A lobular inflammatory grade was determined for each mouse according to reference with some modification. The number of inflammatory foci per ×10 field was counted in sections from every mouse. The inflammatory grades were defined as 0–3: 0, no inflammatory foci per ×10 field; 1: 1–2 inflammatory foci per ×10 field; 2: 3–4 inflammatory foci per ×10 field; and 3: >4 inflammatory foci per ×10 field [15].
Hepatic mononuclear cell (HMNC) isolation and cell surface and intracellular cytokine labelling
HMNCs were isolated and labelled as described previously, with minor modifications [16]. For intracellular cytokine labelling, HMNCs were incubated with phorbol 1,2-myristate 1,3-acetate (PMA, 50 ng/ml; Sigma), ionomycin (500 ng/ml) and GolgiPlug (1 µg/ml; Pharmingen, San Diego, CA, USA). Cells were then labelled with anti-mouse CD3 and CD4 antibodies (Pharmingen) prior to permeabilization with Cytoperm/Cytofix (Pharmingen), according to the manufacturer's instructions. After permeabilization, cells were incubated with labelled antibodies specific for either mouse IL-17 or interferon (IFN)-γ (Pharmingen). Cells were then centrifuged and pellets washed to remove unbound antibodies. After surface and intracellular labelling, HMNCs were evaluated by flow cytometry (Calibrate; Becton Dickinson, Palo Alto, CA, USA) and data were analysed using CellQuest software (Becton Dickinson).
Serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels
Serum ALT and AST levels were measured using a multi-channel autoanalyser at the Chinese Academy of Sciences Department of Biological Sciences Clinical Chemistry Laboratory.
Murine primary hepatocyte isolation, cell line and culture
Primary hepatocytes were isolated from 6-week-old male C57BL/6 mice by in-situ collagenase liver perfusion [17]. Briefly, livers were perfused through the portal vein with calcium and magnesium-free Hanks's balanced salt solution (HBSS; Invitrogen, Carlsbad, CA, USA) followed by Dulbecco's modified Eagle's medium (DMEM; Invitrogen) containing collagenase (collagenase types I and IV; Sigma). Livers were harvested from mice, minced in 37°C DMEM and filtered through a 100 µm nylon cell strainer. Hepatocytes were separated from non-parenchymal cells by centrifugation at 50 g for 5 min. The human hepatoma HepG2 cell line was purchased from the Shanghai Institute of Cell Biology (Chinese Academy of Sciences). Primary hepatocytes were cultured in type I collagen-coated plates (Canspec S&I Co. Ltd, Shanghai, China). Both primary hepatocytes and HepG2 were cultured in DMEM supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin and 100 mg/ml streptomycin (Invitrogen) in a humidified atmosphere (5% CO2 at 37°C). Cells were either left untreated or stimulated with 0·1–100 ng/ml recombinant mouse or human IL-17 (R&D Systems, Minneapolis, MN, USA), with or without OP21 (0·5 mm; mixture of oleic acid and palmitic acid at a ratio of 2:1; Sigma) for 24 h. In other experiments, insulin (100 nm) was also added to the culture media 30 min prior to harvest. IL-6 levels were measured in cell-free culture supernatants using an IL-6-specific enzyme-linked immunosorbent assay (ELISA) kit (R&D Systems) following the manufacturer's instructions.
Oil-Red-O staining and triglyceride assay
Cells were stained with Oil-Red-O to determine the intracellular fat accumulation and counterstained with haematoxylin. Triglyceride (TG) content was determined using an enzymatic kit (Jiancheng-Bio, Nanjing, China).
Western blot analysis
Cultured HepG2 cells or murine liver tissues were lysed in cell lysis buffer (Cell Signaling Technology, Beverly, MA, USA) containing 1 mm phenylmethylsulphonyl fluoride (PMSF). Cells or tissues were briefly homogenized, incubated on ice for 5 min and then centrifuged at 14 000 g at 4°C. Total protein was separated on 10% Ready Gels (Bio-Rad, Hercules, CA, USA) and electrotransferred onto nitrocellulose membranes (0·45 µm; Bio-Rad). Membranes were blocked with 5% (w/v) dried non-fat milk in Tris-buffered saline 0·05% Tween 20 (TBS–T) for 1 h at room temperature, washed three times for 5 min with TBS-T and then incubated with either rabbit polyclonal antibodies to phosphorylated or total mitogen-activated protein kinase (MAPK), protein kinase B (AKT) and IκB-α (Cell Signaling Technology) at 4°C overnight. After repeated washing, membranes were incubated with horseradish peroxidase-conjugated goat anti-rabbit immunoglobulin (Ig)G (1:3000; Amersham Bioscience, Pittsburg, PA, USA) at room temperature for 1 h. Membranes were then washed as above and visualized using the enhanced chemiluminescence reaction (Pierce, Rockford, IL, USA).
Electrophoretic mobility shift assay (EMSA)
Liver nuclear extracts were prepared using the CelLytic nuclear extraction kit (Sigma). Double-stranded oligonucleotides containing the consensus sequence for the nuclear factor (NF)-κB/DNA binding site (5′-TAGTTGAGGGGACTTTCCCAGG-3′) (Santa Cruz Biotechnology, Santa Cruz, CA, USA) were radiolabelled with (γ-32P)-adenosine triphosphate (ATP) (Amersham Biosciences, Little Chalfont, Bucks, UK). After purification over a Sephadex G-25 column (Amersham Biosciences), 32P-labelled oligonucleotides [20 000 counts per minute (cpm)] were incubated with nuclear extracts at room temperature for 20 min in binding buffer containing 12 mm HEPES (pH 7·9), 4 mm Tris HCl (pH 7·9), 60 mm KCl, 1 mm ethylenediamine tetraacetic acid (EDTA), 1 mm dithiothreitol (DTT), 1 mm PMSF, 12% glycerol, 5 µg of bovine serum albumin (BSA) and 2 µg poly(dI/dC) poly(dI/dC) (Amersham Biosciences). DNA–protein complexes were separated using 6% non-denaturing polyacrylamide gel electrophoresis. Signals were visualized by exposing the dried gel to X-ray film.
Human liver specimens and immunohistochemistry staining
Human liver biopsy specimens or resected liver tissues were obtained from four healthy controls (the liver tissues besides haemangiomas) and 14 patients with NASH. Each specimen contained at least six portal tracts. All samples were fixed in 10% neutral buffered formalin embedded in paraffin and 5-µm-thick sections were obtained. After deparaffinization, antigen retrieval was carried out by boiling sections in citrate buffer (pH 6·1) in a microwave and formalin-fixed paraffin-embedded sections blocked with 5% BSA for 10 min. Specimens were then incubated with a primary anti-human IL-17 antibody (1:50 dilution; R&D Systems) for 12 h at 4°C in a wet chamber. After rinsing with PBS, sections were incubated with an anti-goat secondary antibody (Zhongshan Golden Bridge Biotech, Beijing, China) for 20 min at room temperature. PBS was then used to rinse sections that were stained with 3,3′-diaminobenzidine (DAB; Maixin-Bio, Guangzhou, China). Sections were then stained with haematoxylin and visualized using light microscopy.
RNA extraction and real-time–polymerase chain reaction (RT–PCR)
Liver biopsy samples taken from eight NASH patients and five healthy controls were processed for RNA extraction using Trizol reagent (Invitrogen) and complementary DNA synthesized according to the manufacturer's instructions (Fermentas, Burlington, Canada). PCR was performed using a SYBR Green-based PCR approach using SYBR Premix Ex Taq™ (Takara, Shiga, Japan) using a Roche LightCycler 480 (Roche, Indianapolis, IN, USA). The primer sequences used to amplify the respective IL-17, IL-21, IL-23, retinoid-related orphan receptor (ROR)γt and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) sequences are described in Table 1.
Table 1.
Primer sequences of T helper type 17 (Th17)-related human cytokines
| Gene* | Forward primer | Reverse primer |
|---|---|---|
| IL-17 | TGTCCACCATGTGGCCTAAGAG | GTCCGAAATGAGGCTGTCTTTGA |
| IL-23 | CCAAGGACTCAGGGACAACA | ATCAGGGAGCAGAGAAGGCT |
| IL-21 | GCCACATGATTAGAATGCGTCAAC | TGGAGCTGGCAGAAATTCAGG |
| ROR-γt | CATCTCCAGCCTCAGCTTTGA | CATCTCCAGCCTCAGCTTTGA |
| GAPDH | GAAGGTGAAGGTCGGAGTC | GAAGATGGTGATGGGATTTC |
For each sample, mRNA expression levels were normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) using the ΔΔCt algorithm. IL: interleukin; ROR: retinoid-related orphan receptor.
Statistical analysis
Data are expressed as the mean + standard deviation (s.d.). The group means were compared using analysis of variance (anova). All statistical analysis was performed using the spss statistical software version 13·0 (SPSS Inc., Chicago, IL, USA). P-values <0·05 were considered statistically significant.
Results
HF increased the frequency of liver Th17 cells in mice
Our previous studies demonstrated that mice fed HF diets became obese and developed hepatic steatosis [18]. Furthermore, HF diets also decreased the number of Treg cells comprising the HMNC compartment of mice [14]. In this study we evaluated the changes of hepatic Th17 cells (which have an opposing function to Treg cells) by their surface markers (CD3 and CD4) and intracellular cytokines (IFN-γ and IL-17) using flow cytometry. Th17 cells were defined as IL-17+ IFN-γ- T cells. HF mice developed significant hepatic steatosis characterized by the presence of macrovesicular and microvesicular lipid drops and hepatocyte ballooning. HF mice had significantly higher frequencies of hepatic CD3+ or CD4+ Th17 than ND-fed mice (Fig. 1a,b). The number of hepatic Th17 cells present in HF mice at week 8 were twice those present in ND mice. In contrast, ND and HF mice had a similar number of splenic Th17 cells. In addition, the liver had significantly higher percentages of Th17 cells than spleens in either ND or HF mice (Fig. 1b). These results indicate that HF induced hepatic Th17 cell expansion in an organic-specific manner.
Fig. 1.
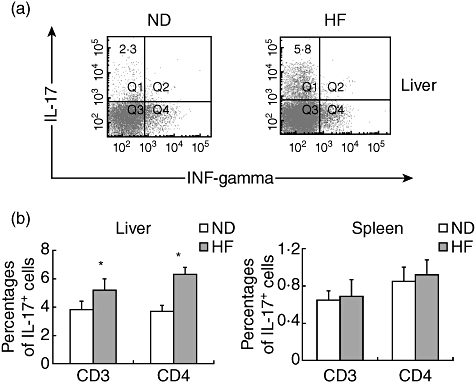
Increase in T helper type 17 (Th17) cells in the livers of mice fed high fat (HF) diets. C57BL/6 mice were fed normal diet (ND) or HF diets for 8 weeks. Hepatic mononuclear cells (HMNCs) were isolated and incubated with labelled antibodies specific to CD3 or CD4 as well as antibodies specific for intracellular interleukin (IL)-17 or interferon (IFN)-γ. Total liver mononuclear cell numbers were similar for mice fed either diet. (a) Representative dot-plots of gated CD4+ cells. (b) Mean [standard deviation (s.d.)] values of intracellular IL-17+ IFN-γ- staining HMNC (gated CD3+ or CD4+ cells) from murine livers or spleens (n = 6 in each group, *P < 0·05).
Neutralization of IL-17 attenuated LPS-induced liver injury in HF mice
To confirm the importance of IL-17 in mediating liver injury, anti-IL-17 antibodies were used to block LPS-induced liver injury in mice. Mice were injected intravenously with neutralizing anti-IL-17 antibodies (0·1 mg/mouse) or isotype control IgG 2 h prior to LPS injection. LPS treatment induced hepatic inflammatory responses in both ND and HF mice (Fig. 2a). HF mice treated with LPS had a trend towards more severe hepatic inflammation when compared with ND mice treated with LPS, although the difference did not reach statistical significance (Fig. 2b). Similarly, LPS treatment increased serum ALT levels in both ND and HF mice. HF mice treated with LPS had significantly higher ALT compared with ND mice treated with LPS (Fig. 2c), indicating that they are more susceptible to LPS-induced injury. In both ND and HF mice, anti-IL-17 pretreatment significantly decreased inflammatory cell infiltrates into the portal tracts compared to mice treated with the isotype controls (Fig. 2a,b). Anti-IL-17 antibody treatment also significantly decreased serum ALT levels in both LPS-treated ND and HF mice (Fig. 2c).
Fig. 2.
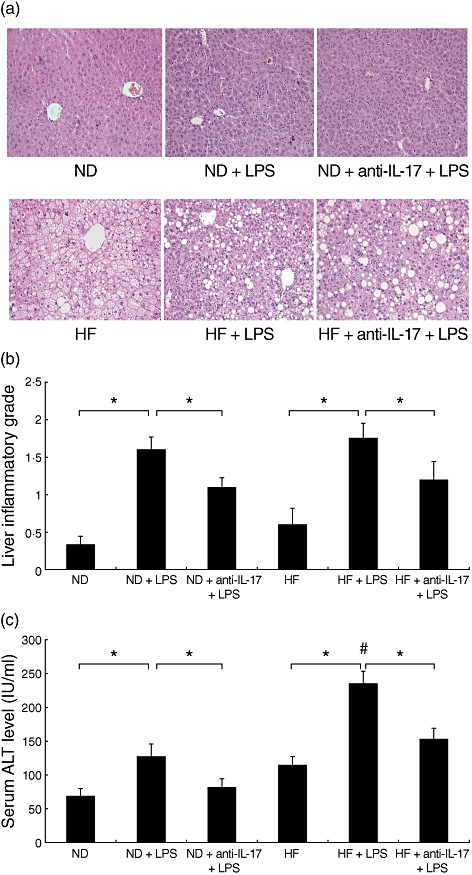
Neutralization of interleukin (IL)-17 attenuates lipopolysaccharide (LPS)-induced liver injury in high fat (HF) diet mice. C57BL/6 mice were fed a normal diet (ND) or HF diet for 8 weeks. Mice were injected intraperitoneally with a single dose of LPS (50 µg/mouse). Neutralizing IL-17 antibody (100 µg/mice) was administrated intravenously in some mice from each group prior to LPS injection. Serum and liver tissue samples were obtained 6 h after LPS injection. (a) Representative haematoxylin and eosin-stained liver sections (magnification ×200) from ND and HF mice. (b) Mean [standard deviation (s.d.)] liver inflammatory grade of triplicate experiments (n = 6/experiment). (c) Mean (s.d.) serum alanine aminotransferase (ALT) levels of triplicate experiments (n = 6/experiment). *P ≤ 0·05, #P ≤ 0·05 versus ND mice treated with LPS.
In order to elucidate potential signalling pathways mediating IL-17-induced hepatic inflammation, we assessed the phosphorylation status of intracellular components associated with three distinct MAPK signalling pathways [i.e. c-Jun N-terminal kinase (JNK), extracellular kinase (ERK) and p38 MAPK] by Western blot analysis. LPS increased JNK phospholyration in livers of both ND and HF mice; however, HF mice had significantly higher JNK activation levels than ND mice. In contrast, IL-17 neutralization attenuated LPS-mediated hepatic activation of JNK in both ND and HF mice (Fig. 3a). However, LPS and anti-IL-17 treatment had no discernable impact on liver ERK and p38 MAPK activation (data not shown). LPS also increased inhibitor of nuclear factor kappa-B (IKBα) activation in both HF and ND mouse livers. In contrast, IL-17 neutralization decreased LPS-mediated hepatic activation of IKBα in ND and HF mice (Fig. 3a) and the DNA binding activity of NF-κB paralleled IKBα activation (Fig. 3b). These data suggest that IL-17 neutralization attenuated LPS-induced hepatic inflammation via the JNK–NF-κB signalling pathway.
Fig. 3.
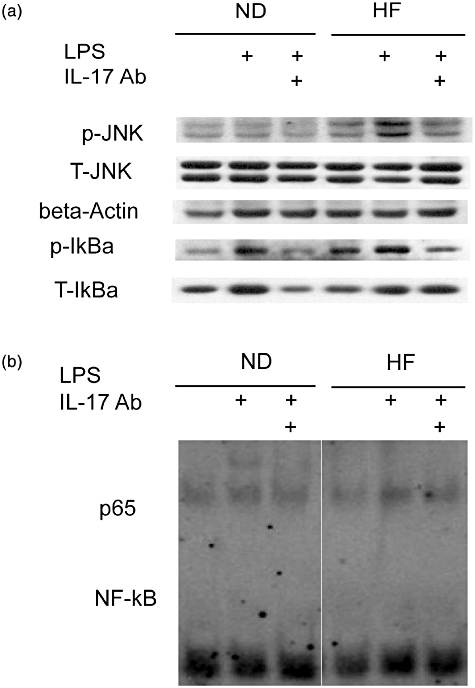
Neutralization of IL-17 attenuates lipopolysaccharide (LPS)-induced mitogen-activated protein kinase (MAPK) and nuclear factor (NF)-κB signalling pathways in normal diet (ND) and high fat (HF) mice. Animal experiments were performed as described in Fig. 2. Western blots were performed on whole liver protein extracts with antibodies specific to phosphorylated c-Jun N-terminal kinase (JNK) and on nuclear extracts to detect activation of the NF-κB signalling pathway [inhibitor of nuclear factor kappa-B (IKBα)]. Membranes were then stripped and reprobed with antibodies specific to JNK and IKBα. β-actin expression was used a loading control. (a) Representative autoradiographs of JNK and IKBα. (b) Representative autoradiographs of NF-κB binding activity from liver nuclear extracts.
IL-17 exacerbates HepG2 steatosis by blunting insulin signalling pathways in vitro
An in vitro cellular steatosis model was developed by incubating HepG2 cells in the presence of palmitic acid and oleic acid at a ratio of 2:1 for 24 h [19]. Three images were photographed per dish and representative images are shown (Fig. 4a). HepG2 cells developed significant steatosis determined by Oil-Red-O staining, and IL-17 (100 ng/ml) exacerbated intracellular lipid accumulation induced by free fatty acid (FFA) (Fig. 4a). We quantified the intracellular HepG2 TG content of cells cultured in the presence or absence of FFA using an enzymatic kit. IL-17 (100 ng/ml) treatment increased TG accumulation significantly in hepatocytes compared to hepatocytes incubated with FFA alone (Fig. 4b). In order to observe the efficacy of more physiological concentration of IL-17 on the TE contents of HepG2 cells, the effects of lower concentrations of IL-17 (0·1–10 ng/ml) on TE levels in HepG2 cells treated with FFA were tested. IL-17 in a dosage of 10 ng/ml increased significantly intracellular TG contents in HepG2 cells treated with FFA. Although lower dosages of IL-17 (0·1 and 1 ng/ml) treatment had a trend towards increased TG content in HepG2 cells, there were no significant differences when compared with the FFA-treated group (Fig. 4c).
Fig. 4.
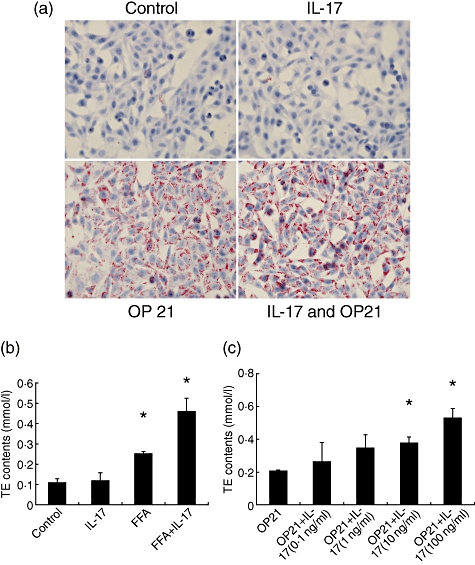
Interleukin (IL)-17 exacerbated hepatocyte steatosis in HepG2 cells in vitro. The free fatty acids (FFA) mixture (oleate acid and palmitate acid at 1:2 ratio, OP21) was added to HepG2 cells to induce fat overloading for 24 h. IL-17 (100 ng/ml) was added to the culture medium with or without FFA for 24 h. (a) Representative Oil-Red staining of HepG2 cells treated with FFA with or without IL-17. (b) Intracellular triglyceride (TG) content was determined using an enzymatic kit. Mean [standard deviation (s.d.)] intracellular TG content of triplicate experiments (n = 4/experiment). *P < 0·05 versus controls. (c) The OP21 was added to HepG2 cells with different dosages of IL-17 (0·1–100 ng/ml) to induce fat overloading for 24 h. Intracellular TG content was determined using an enzymatic kit. *P < 0·05 versus OP21 treated without IL-17 group.
The effect of IL-17 on the insulin-signalling pathway was determined using HepG2 cells cultured in the presence or absence of FFA. IL-17 (100 ng/ml) or FFA treatment alone did not significantly alter insulin-induced activation of IRS-1 or AKT; however, IL-17 in combination with FFA decreased significantly insulin-induced phosphorylation of IRS-1 and AKT in HepG2 cells (Fig. 5). In contrast, IL-17 and FFA treatment (either individually or together) had no significant impact on SREBP-1c or PPAR-α expression with or without insulin.
Fig. 5.
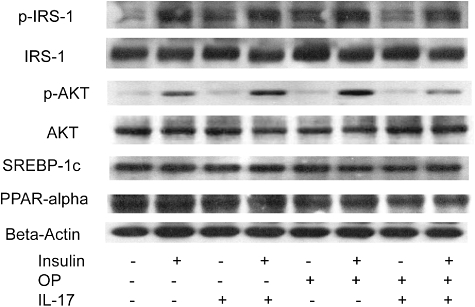
Interleukin (IL)-17 interfered with the insulin-signalling pathway in vitro. Representative autoradiographs of insulin pathway [insulin substrate receptor 1 (IRS-1) and protein kinase B (AKT) and fatty acid metabolism [sterol receptor element-binding protein-1c and peroxisome proliferator-activated receptor-α (SREBP-1c and PPAR-α)].
IL-17 and FFA induced hepatocyte production of IL-6
IL-6, in conjunction with TGF-β, promotes Th17 cell generation and differentiation [20]. Conversely, IL-6 also plays a critical role in mediating liver inflammation associated with NASH [21]. As hepatocytes are primary sources of IL-6 during the liver inflammatory process [22], we hypothesized that a positive-feedback loop between Th17 cells and hepatocytes is mediated through IL-6 and IL-17 signals. To test this hypothesis, IL-17 was added to HepG2 cell cultures. There were no IL-17-mediated effects on HepG2 cell growth at doses up to 100 ng/ml. Treatment with either IL-17 or FFA induced HepG2 cells to produce IL-6. More importantly, IL-17 and FFA acted synergistically in mediating HepG2 production of IL-6 (Fig. 6a). To confirm these results in normal hepatocytes, primary mouse hepatocytes were isolated and treated with IL-17 or FFA. Similar to HepG2 cells, IL-17 induced IL-6 production by primary mouse hepatocytes with synergetic effects when combined with FFA (Fig. 6b).
Fig. 6.
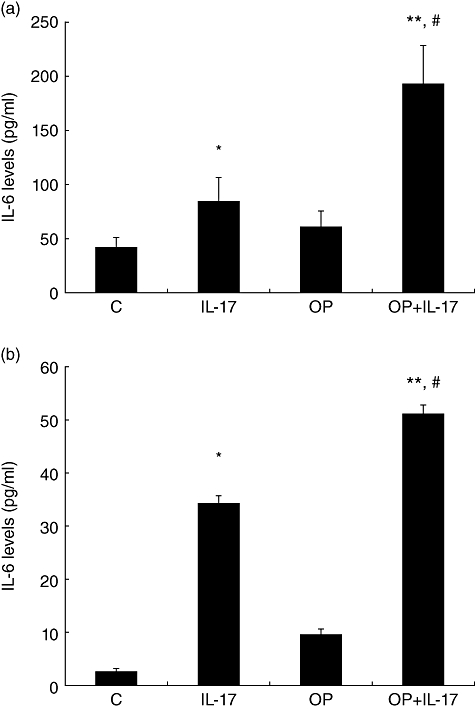
Interleukin (IL)-6 production by HepG2 cells and murine primary hepatocytes. IL-6 levels in HepG2 supernatants were measured by enzyme-linked immunosorbent assay (ELISA). (a) Mean [standard deviation (s.d.)] IL-6 levels in the supernatants of HepG2 cells treated with IL-17 or/and free fatty acid (FFA) (n = 4/experiment). (b) Mean [standard deviation (s.d.)] IL-6 levels in the supernatants of murine primary hepatocytes treated with IL-17 or/and FFA (n = 4/experiment), *P < 0·05 versus control; **P < 0·01 versus control; #P < 0·05 versus IL-17 group.
Increased IL-17+ cells and Th17 cell-related gene expression in the lives of NASH patients
Th17 cell distribution in human livers from NASH patients was examined by staining IL-17 using immunohistochemistry. IL-17+ cells could not be detected in healthy control livers. Similar to chronic hepatitis B and autoimmune hepatitis patients, IL-17+ cells were distributed among lymphoid infiltrates in the portal tracts of tissues examined from NASH patients (Fig. 7a). Furthermore, IL-17+ cell infiltrates were in proximity to ballooning or macrovesical hepatocytes in the lobular area of NASH patients. There were significantly more IL-17+ cell infiltrates in the livers of NASH when compared with normal controls (Fig. 7b). When additional Th17-related cytokines were examined in liver tissue using RT–PCR, gene expression of RORγt, IL-17, L-21 and IL-23 were increased significantly in patients with NASH compared to expression levels observed in healthy controls (Fig. 7c).
Fig. 7.
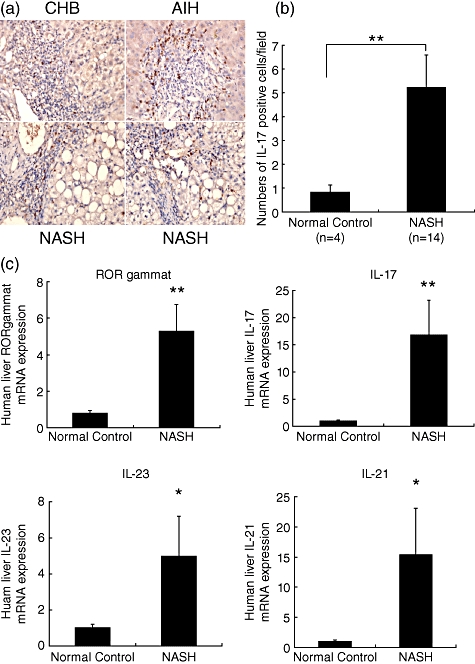
Interleukin (IL)-17 and other T helper type 17 (Th17)-related cytokine profile analysis of human liver tissues. (a) Representative liver staining for the presence of Th17 cells (IL-17+, brown-stained cells, magnification (×400). (b) Mean [standard deviation (s.d.)] numbers of IL-17 positive cells/field. Liver tissues were obtained from healthy controls (HC, n = 4) or from non-alcoholic steatohepatitis (NASH, n = 14) patients. Th17 cells in the liver were evaluated by immunohistochemical staining of IL-17. (c) Mean (s.d.) expression levels of the Th17-related cytokines retinoid-related orphan receptor (ROR)γt, IL-17, IL-21 and IL-23. Th17-related cytokines expressions were determined by quantitative reverse transcription–polymerase chain reaction (qRT–PCR) and normalized against glyceraldehyde 3-phosphate dehydrogenase (GAPDH) expression. Liver tissues were obtained from healthy controls (HC, n = 5) or NASH (n = 8) patients. *P < 0·05; **P < 0·01 compared with normal controls.
Discussion
In this study, we have demonstrated that IL-17 blunted the insulin-signalling pathway and exacerbated HepG2 steatosis when cultured in the presence of FFA. IL-17 and FFA induced IL-6 production, a cytokine critical to Th17 cell differentiation and hepatic insulin resistance, in HepG2 cells and primary murine hepatocytes. In contrast, neutralization of IL-17 in mice fed an HF diet presented with attenuated LPS-induced liver injury (i.e. mice presented with reduced liver damage and decreased serum ALT levels). Characterization of NASH in humans revealed massive IL-17+ cell infiltrates in the liver. To our knowledge, this study demonstrates for the first time that IL-17 plays a critical role both in hepatocyte steatosis and inflammatory processes associated with NASH.
Obesity is an inflammatory disorder characterized by heightened innate immune activity [23]. Innate immune activation is central to the development of obesity-related insulin resistance and obesity-related tissue damage such as NAFLD [23,24]. In addition, innate immune cytokines are critical to the development of Th17 cells [9]. In 2006, three groups demonstrated independently that the combination of TGF-β and IL-6 induced the differentiation of naive T cells into Th17 cells in mice [6–8]. In humans, combinations of TGF-β and IL-21, TGF-β, IL-6 and IL-23 or IL-6 and IL-21 were demonstrated to induce Th17 cell development [25–27]. These experiments demonstrated the central role of IL-6 in Th17 cell differentiation. IL-6 can be produced by various cells of the innate immune system, including monocytes, and IL-6 possess pleiotropic effects [9]. Accumulating data suggest that hepatic IL-6 may play an important role in NASH development, as well as in systemic insulin resistance and diabetics. In humans, liver IL-6 expression is correlated positively with the severity of inflammation and the degree of fibrosis observed in NASH patients [28]. Saturated FFA have been demonstrated to induce IL-6 production by hepatocytes in vitro[28] and IL-6 knock-out mice presented with attenuated hepatic inflammation in a methionine- and choline-deficient diet-induced NASH model [21]. In addition, IL-6-deficient mice were impaired in their ability to generate Th17 cells, and these animals possessed increased numbers of peripheral Tregs[29,30]. We demonstrated that IL-17 induced hepatocytes to secrete IL-6 which, in combination with TGF-β, is essential for Th17 cell differentiation and expansion [29,31,32]. Because IL-6 was demonstrated to induce IL-21 expression in naive CD4+ T cells and amplified RORγt-dependent differentiation of Th-17 cells in concert with TGF-β[20], this suggested that Th17 cell expansion and hepatocyte-generated IL-6 resulted in a vicious cycle that resulted in hepatic inflammation amplification. This interaction may be especially important in the fatty liver milieu, as our study showed that FFA synergized with IL-17 in increasing hepatocellular IL-6 production. This positive-feedback loop between IL-17/FFA and IL-6 may partially explain the increase in hepatic Th17 cells in HF diet-fed mice.
Several studies have demonstrated that peripheral and intrahepatic Th17 cells were increased in liver diseases such as primary biliary cirrhosis [33,34], chronic hepatitis B [35], alcoholic liver disease [36] and hepatocellular carcinoma [37]. However, the mechanisms resulting in Th17 cell expansion and their effector functions in the liver remain incompletely understood. Most research in this area has focused upon examining the interactions between Th17 and mesenchymal cells, including hepatic monocytes, dendritic cells (DCs) and hepatic stellate cells (HSCs). The increased numbers of circulating and intrahepatic Th17 cells correlated positively with plasma viral loads, serum ALT levels and the histological activity index of chronic hepatitis B patients. The increased numbers of Th17 cells may have also led to further activation of DCs and monocytes, resulting in the release of proinflammatory cytokines that, in turn, amplified hepatic inflammation and injury [35]. In the present study, we attempted to illustrate the interaction between Th17 cells and the hepatocytes. As hepatocytes are a major target of lipotoxicity in NAFLD, our finding may have implications for specific mechanisms in the NAFLD condition.
Human alcoholic liver disease is also characterized by the activation of the IL-17 pathway, both peripherally and in the liver. Moreover, IL-17 may contribute to liver neutrophil recruitment by inducing human HSCs to produce IL-8 and growth-related oncogen α[36]. In addition, IL-17-producing cells were enriched predominantly in the peritumoral stroma of hepatocellular carcinoma tissues, and their levels correlated with monocyte/macrophage densities in the same area. Moreover, tumour-activated monocytes secreted various proinflammatory cytokines that triggered Th17 cell proliferation. Inhibition of monocyte/macrophage-mediated liver inflammation markedly reduced the levels of tumour-infiltrating Th17 cells and tumour growth in vivo[37]. In our study, we found that hepatic parenchymal cells, i.e. hepatocytes, which account for 60% of the total cell population and 80% of the liver volume, interacted with Th17 cells. High fat diet-induced fatty liver in a murine model presents simple steatosis. In order to mimic steatohepatitis, LPS is administrated peritoneally to induce hepatic inflammation [18]. Therefore, the NASH model we used may be different from human NASH, which is a chronic disease without any clinical manifestation of acute liver inflammatory exacerbation. Neutralization of IL-17 before LPS administration gave us an opportunity to observe the potential role of IL-17 in the pathogenesis of NASH. However, it is of interest to study the effects of chronic IL-17 blocking using the IL-17 knock-out method on hepatocyte steatosis, inflammation and fibrosis in the future.
A reciprocal relationship exists between Treg and Th17 cells, i.e. TGF-β, induces FoxP3 expression in naive T cells, IL-6 suppresses TGF-β-driven induction of FoxP3 and TGF-β and IL-6 together induce RORγt and Th17 cell development [29]. Therefore, IL-6 produced during an acute-phase response could play a pivotal role in directing the generation of either Treg or Th17 cells [9]. At the molecular level, the antagonistic interaction of the transcription factors FoxP3 and RORγt contributes to the balance between Th17 and Treg cells [9,38,39]. Our previous study showed that the number of hepatic Treg cells was decreased significantly by HF diets in mice, resulting in diminished inflammatory responses when mice were challenged with exogenous LPS. Oxidative stress-induced apoptosis contributed to reductions in hepatic Treg cell numbers which expressed relatively lower Bcl-2 (anti-apoptosis gene) levels compared to CD4+CD25- T cells [14]. Therefore, the potential roles of imbalances between Treg and Th17 cells in NASH are worthy of further study. It is also interesting to ask whether adjusting the balance between Treg and Th17 cells would improve inflammation in NASH.
The data presented in this report demonstrate that Th17 cells play a critical role in NAFLD hepatic inflammation. Acting in synergy with FFA, IL-17 induce the production of hepatocyte IL-6 levels. Th17 cells and their production of IL-17 may favour hepatic steatosis and a proinflammatory response in NAFLD, facilitating the transition from simple steatosis to steatohepatitis. These results also suggest that strategies designed to alter the Th17/Treg balance should be explored as a means of preventing progression to NASH and/or advanced liver diseases in patients with NAFLD.
Acknowledgments
This work was supported by the Awards from the National Natural Science Foundation of China (no. 30770963, no. 30972751, X.M.), Shanghai Pujiang Program and Program for Innovative Research Team of Shanghai Municipal Education Commission (X.M.) and a research grant from NIH/NIDDK R01DK075990 (Z.L.).
Disclosure
The authors have no conflicts of interest.
References
- 1.Farrell GC, Larter CZ. Nonalcoholic fatty liver disease: from steatosis to cirrhosis. Hepatology. 2006;43(Suppl. 1):S99–S112. doi: 10.1002/hep.20973. [DOI] [PubMed] [Google Scholar]
- 2.Kleiner DE, Brunt EM, Van Natta M, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–21. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 3.Brunt EM. Nonalcoholic steatohepatitis. Semin Liver Dis. 2004;24:3–20. doi: 10.1055/s-2004-823098. [DOI] [PubMed] [Google Scholar]
- 4.Cortez-Pinto H, de Moura MC, Day CP. Non-alcoholic steatohepatitis: from cell biology to clinical practice. J Hepatol. 2006;44:197–208. doi: 10.1016/j.jhep.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 5.Kremer M, Hines IN, Milton RJ, Wheeler MD. Favored T helper 1 response in a mouse model of hepatosteatosis is associated with enhanced T cell-mediated hepatitis. Hepatology. 2006;44:216–27. doi: 10.1002/hep.21221. [DOI] [PubMed] [Google Scholar]
- 6.Langrish CL, Chen Y, Blumenschein WM, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–40. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Park H, Li Z, Yang XO, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–41. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harrington LE, Hatton RD, Mangan PR, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–32. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 9.Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 10.Zhao L, Qiu D, Ma X. Th17 cells: the emerging reciprocal partner of regulatory T cells in the liver. J Dig Dis. 2010;11:126–33. doi: 10.1111/j.1751-2980.2010.00428.x. [DOI] [PubMed] [Google Scholar]
- 11.Weaver CT, Hatton RD. Interplay between the TH17 and Treg cell lineages: a (co-)evolutionary perspective. Nat Rev Immunol. 2009;9:883–9. doi: 10.1038/nri2660. [DOI] [PubMed] [Google Scholar]
- 12.Ye C, Li WY, Zheng MH, Chen YP. T-helper 17 cell: a distinctive cell in liver diseases. Hepatol Res. 2011;41:22–9. doi: 10.1111/j.1872-034X.2010.00744.x. [DOI] [PubMed] [Google Scholar]
- 13.Hammerich L, Heymann F, Tacke F. Role of IL-17 and Th17 cells in liver diseases. Clin Dev Immunol. 2011 doi: 10.1155/2011/345803. Article ID 345803, 12 pages. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ma X, Hua J, Mohamood AR, Hamad AR, Ravi R, Li Z. A high-fat diet and regulatory T cells influence susceptibility to endotoxin-induced liver injury. Hepatology. 2007;46:1519–29. doi: 10.1002/hep.21823. [DOI] [PubMed] [Google Scholar]
- 15.Yamaguchi K, Yang L, McCall S, et al. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology. 2007;45:1366–74. doi: 10.1002/hep.21655. [DOI] [PubMed] [Google Scholar]
- 16.Guebre-Xabier M, Yang S, Lin HZ, Schwenk R, Krzych U, Diehl AM. Altered hepatic lymphocyte subpopulations in obesity-related murine fatty livers: potential mechanism for sensitization to liver damage. Hepatology. 2000;31:633–40. doi: 10.1002/hep.510310313. [DOI] [PubMed] [Google Scholar]
- 17.Vick B, Weber A, Urbanik T, et al. Knockout of myeloid cell leukemia-1 induces liver damage and increases apoptosis susceptibility of murine hepatocytes. Hepatology. 2009;49:627–36. doi: 10.1002/hep.22664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Z, Soloski MJ, Diehl AM. Dietary factors alter hepatic innate immune system in mice with nonalcoholic fatty liver disease. Hepatology. 2005;42:880–5. doi: 10.1002/hep.20826. [DOI] [PubMed] [Google Scholar]
- 19.Feldstein AE, Canbay A, Guicciardi ME, Higuchi H, Bronk SF, Gores GJ. Diet associated hepatic steatosis sensitizes to Fas mediated liver injury in mice. J Hepatol. 2003;39:978–83. doi: 10.1016/s0168-8278(03)00460-4. [DOI] [PubMed] [Google Scholar]
- 20.Zhou L, Ivanov II, Spolski R, et al. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. 2007;8:967–74. doi: 10.1038/ni1488. [DOI] [PubMed] [Google Scholar]
- 21.Mas E, Danjoux M, Garcia V, Carpentier S, Segui B, Levade T. IL-6 deficiency attenuates murine diet-induced non-alcoholic steatohepatitis. PLoS ONE. 2009;4:e7929. doi: 10.1371/journal.pone.0007929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Streetz KL, Luedde T, Manns MP, Trautwein C. Interleukin 6 and liver regeneration. Gut. 2000;47:309–12. doi: 10.1136/gut.47.2.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maher JJ, Leon P, Ryan JC. Beyond insulin resistance: innate immunity in nonalcoholic steatohepatitis. Hepatology. 2008;48:670–8. doi: 10.1002/hep.22399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ma X, Li Z. Pathogenesis of nonalcoholic steatohepatitis (NASH) Chin J Dig Dis. 2006;7:7–11. doi: 10.1111/j.1443-9573.2006.00237.x. [DOI] [PubMed] [Google Scholar]
- 25.Yang L, Anderson DE, Baecher-Allan C, et al. IL-21 and TGF-beta are required for differentiation of human T(H)17 cells. Nature. 2008;454:350–2. doi: 10.1038/nature07021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Manel N, Unutmaz D, Littman DR. The differentiation of human T(H)-17 cells requires transforming growth factor-beta and induction of the nuclear receptor RORgammat. Nat Immunol. 2008;9:641–9. doi: 10.1038/ni.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miossec P, Korn T, Kuchroo VK. Interleukin-17 and type 17 helper T cells. N Engl J Med. 2009;361:888–98. doi: 10.1056/NEJMra0707449. [DOI] [PubMed] [Google Scholar]
- 28.Wieckowska A, Papouchado BG, Li Z, Lopez R, Zein NN, Feldstein AE. Increased hepatic and circulating interleukin-6 levels in human nonalcoholic steatohepatitis. Am J Gastroenterol. 2008;103:1372–9. doi: 10.1111/j.1572-0241.2007.01774.x. [DOI] [PubMed] [Google Scholar]
- 29.Bettelli E, Carrier Y, Gao W, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–8. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 30.Korn T, Bettelli E, Gao W, et al. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature. 2007;448:484–7. doi: 10.1038/nature05970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mangan PR, Harrington LE, O'Quinn DB, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–4. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 32.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–89. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 33.Lan RY, Salunga TL, Tsuneyama K, et al. Hepatic IL-17 responses in human and murine primary biliary cirrhosis. J Autoimmun. 2009;32:43–51. doi: 10.1016/j.jaut.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rong G, Zhou Y, Xiong Y, et al. Imbalance between T helper type 17 and T regulatory cells in patients with primary biliary cirrhosis: the serum cytokine profile and peripheral cell population. Clin Exp Immunol. 2009;156:217–25. doi: 10.1111/j.1365-2249.2009.03898.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang JY, Zhang Z, Lin F, et al. Interleukin-17-producing CD4(+) T cells increase with severity of liver damage in patients with chronic hepatitis B. Hepatology. 2010;51:81–91. doi: 10.1002/hep.23273. [DOI] [PubMed] [Google Scholar]
- 36.Lemmers A, Moreno C, Gustot T, et al. The interleukin-17 pathway is involved in human alcoholic liver disease. Hepatology. 2009;49:646–57. doi: 10.1002/hep.22680. [DOI] [PubMed] [Google Scholar]
- 37.Kuang DM, Peng C, Zhao Q, Wu Y, Chen MS, Zheng L. Activated monocytes in peritumoral stroma of hepatocellular carcinoma promote expansion of memory T helper 17 cells. Hepatology. 2010;51:154–64. doi: 10.1002/hep.23291. [DOI] [PubMed] [Google Scholar]
- 38.Zhou L, Lopes JE, Chong MM, et al. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature. 2008;453:236–40. doi: 10.1038/nature06878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ichiyama K, Yoshida H, Wakabayashi Y, et al. Foxp3 inhibits RORgammat-mediated IL-17A mRNA transcription through direct interaction with RORgammat. J Biol Chem. 2008;283:17003–8. doi: 10.1074/jbc.M801286200. [DOI] [PubMed] [Google Scholar]