

Abstract
Immunological dysfunction has been described to occur in chronic idiopathic urticaria (CIU), most notably in association with an inflammatory process. Some pharmacological agents as statins – drugs used in hypercholesterolaemia – display a broad effect on the immune response and thus should be tested in vitro in CIU. Our main objectives were to evaluate the effects of statins on the innate and adaptive immune response in CIU. Simvastatin or lovastatin have markedly inhibited the peripheral blood mononuclear cells (PBMC) proliferative response induced by T and B cell mitogens, superantigen or recall antigen. Simvastatin arrested phytohaemaglutinin (PHA)-induced T cells at the G0/G1 phase, inhibiting T helper type 1 (Th1), Th2, interleukin (IL)-10 and IL-17A cytokine secretion in both patients and healthy control groups. Up-regulation of suppressor of cytokine signalling 3 (SOCS3) mRNA expression in PHA-stimulated PBMCs from CIU patients was not modified by simvastatin, in contrast to the enhancing effect in the control group. Statin exhibited a less efficient inhibition effect on cytokine production [IL-6 and macrophage inflammatory protein (MIP)-1α] induced by Toll-like receptor (TLR)-4, to which a statin preincubation step was required. Furthermore, statin did not affect the tumour necrosis factor (TNF)-α secretion by lipopolysaccharide (LPS)-stimulated PBMC or CD14+ cells in CIU patients. In addition, LPS-activated PBMC from CIU patients showed impaired indoleamine 2,3-dioxygenase (IDO) mRNA expression compared to healthy control, which remained at decreased levels with statin treatment. Statins exhibited a marked down-regulatory effect in T cell functions, but were not able to control TLR-4 activation in CIU patients. The unbalanced regulatory SOCS3 and IDO expressions in CIU may contribute to the pathogenesis of the disease.
Keywords: chronic idiopathic urticaria, cytokines, immunomodulation, proinflammatory factors, regulatory factors, statins
Introduction
Chronic urticaria (CU) is one of the most disabling types of urticaria and has great impact on quality of life [1,2]. One-third of the patients with CU have circulating autoantibodies against the α-chain of the high affinity receptor for immunoglobulin (Ig)E (FcεRI) or against the IgE molecule [3,4]. However, the majority of patients have no obvious cause and are considered to have chronic idiopathic urticaria (CIU) [5].
The pathogenic mechanism of CIU remains unknown, although some studies have demonstrated that patients may have intrinsic abnormalities in basophils or mast cells [6,7]. The inflammatory processes in CIU seem not to be limited to the consequences of mast/basophil degranulation [8]. Our previous data supported a theory that immunological dysfunction is the primary condition for CIU, which is sustained by the disturbed innate immune response as increased levels of proinflammatory cytokines and altered pattern of regulatory cytokine secretion found in serum of CIU patients, regardless of the result of the autologous serum skin test [9].
To understand the proinflammatory process in CIU or the possible autoimmune basis in some patients, it is essential to search for mechanisms that could be involved in immunoregulation. Indoleamine 2,3-dioxygenase (IDO) expressed by antigen-presenting cells (APC) is an important inhibitor of T cell function [10]. IDO, an enzyme that degrades the essential amino acid tryptophan, has a complex regulatory role and is involved in the promotion of T cell tolerance to cancer and allografts, protection of the allogeneic fetus and autoimmunity [11,12]. Among other factors that have crucial roles in the regulation of the immune response, there is the suppressor of cytokine signalling protein (SOCS). SOCS3 negatively regulates the c-Jun N-terminal kinase/signal transduction and activator of transcription (JAK/STAT) signal transducer pathway and interleukin (IL)-6 and IL-23 gene expression [13], inhibiting the generation of T helper type 17 (Th17). The retinoic acid-related orphan nuclear hormone receptor C (RORC) has a direct role in the secretion of IL-17 [14] in Th17 cells and is thought to be a key regulator of inflammation [15].
Pharmacological agents with immunomodulatory potential should be investigated for treatment of the inflammatory process in CIU. Statins, known inhibitors of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase, are well-established drugs for hypercholesterolaemia. Statins display a broad effect on the immune response, including induction of T cell hyporesponsiveness [16], regulatory T cell expansion [17], polarization towards an anti-inflammatory T cell phenotype [18] and down-regulation of APC function [19]. Conversely, statins can greatly increase lipopolysaccharide (LPS)-induced IL-1β secretion in monocytes [20].
We have investigated the effects of statins on the innate immune activation and T cell stimulation in CIU patients. Statins were clearly effective in inhibiting the T cell response, arresting the cell cycle in G0/G1 stage, while not interfering in the cytokine production triggered by Toll-like receptor (TLR)-4 activation. The altered expression in peripheral blood mononuclear cells (PBMCs) of the regulatory factors IDO and SOCS3 which, in turn, were not regulated negatively by the statins, may contribute to the inflammatory process in CIU.
Materials and methods
Patients
All patients with continuous CIU (n = 22; three males and 19 females; median age 49 ± 12·5 years; aged 22–67 years) were selected from the Dermatological Outpatient Clinic of the Hospital das Clínicas de São Paulo (HC-FMUSP). These patients had wheals daily or at least four times a week, for 6 weeks or longer. Patients with physical urticaria, urticarial vasculitis, food allergies or autoinflammatory diseases were excluded, as described previously [6,9]. None of the patients took corticosteroids or immunosuppressive drugs. In total, 41 blood donors, matched for age and sex, were enrolled as healthy controls (HC; 11 males and 30 females, median age 42 ± 10 years, aged 22–60 years). In blood donors, the presence of immune-mediated or other relevant disorders were assessed and excluded. All individuals gave written informed consent, and the study protocol was approved by the institutional ethics committee of the HC-FMUSP.
PBMC isolation
PBMCs were isolated using Ficoll-Hypaque solution (Amersham Pharmacia Biotech, NJ, USA), and cultured in RPMI-1640 with 10% human AB serum (RPMI-S) (Sigma-Aldrich, St Louis, MO, USA) at 37°C in 5% CO2. In some experiments, CD14+ cells were isolated by magnetic cell sorting using positive selection with magnetic microbeads (Miltenyi Biotech, Paris, France). Ninety-five per cent purity in cell populations was achieved by flow cytometry.
Proliferation assay
PBMCs (2 × 106/ml) were stimulated with phytohaemagglutinin (PHA, 2·5 µg/ml; Gibco, Carlsbad, CA, USA) for 3 days, pokeweed mitogen (PWM, 5 µg/ml; Sigma) and tetanus toxoid (0·04 Lf/ml; Instituto Butantã, São Paulo, Brazil) for 6 days, or Staphylococcus aureus enterotoxin A (SEA, 0·04 µg/ml; Sigma) for 3 days. The experiments were performed in the presence or absence of simvastatin or lovastatin (Calbiochem, La Jolla, CA, USA) at the following concentrations: 0·25, 1·0, 5·0 and 25 µm. Cells were pulsed with 1 µCi/well of [3H]-thymidine (New England Nuclear, Boston, MA, USA) and the incorporated radioactivity was measured in a Betaplate (1205 Betaplate; Wallac, Waltham, MA, USA).
Cell cycle
Cell cycle analysis was performed using bromodeoxyuridine incorporation (BrdU Flow Kit; Becton Dickinson Biosciences, San Jose, CA, USA). PBMCs (3·0 × 106/ml) were cultured in RPMI-S with PHA (2·5 µg/ml; Gibco) for 3 days or PWM (5 µg/ml; Sigma) for 6 days in the presence of 25 µm simvastatin or lovastatin (Calbiochem) at 37C and 5% CO2. BrdU (10 µm/ml) was added 18 h before the end of each culture period. The cells were fixed and permeabilized with buffer (BD Cytofix/Cytoperm), treated with DNAse (30 µg/ml), and incubated with anti-BrdU fluorescein isothiocyanate (FITC) in a solution of 7-amino-actinomycin D (7-AAD) phycoerythrin–cyanin 5 (PE-Cy5). Cells were analysed on a fluorescence activated cell sorter (FACS)Calibur (Becton Dickinson) using BD CellQuest Pro™.
Cytokine secretion
PBMCs or purified CD14+ cells were stimulated with PHA (2·5 µg/ml; Gibco) for 48 h or LPS (100 ng/ml, Sigma) for 24 h with simvastatin or lovastatin (25 µm; Calbiochem). In some experiments, cells were preincubated with the statin for 24 h, and then PBMCs were stimulated with LPS (100 ng/ml) for 24 h, whereas CD14+ cells were stimulated with LPS (1 µg/ml) for 2 h. Supernatants were collected, centrifuged and analysed for cytokine and chemokine secretion by enzyme-linked immunosorbent assay (ELISA) commercial kits (R&D Systems, Wiesbaden, Germany).
Real-time polymerase chain reaction (PCR)
Total RNA from PBMCs, CD4+ T cells or CD14+ cells (1 × 106 cells) was extracted using the RNeasy Mini Kit (Qiagen, Valencia, CA, USA), and reverse transcription was carried out with a Sensiscript Reverse Transcriptase kit (Qiagen). Real-time PCR was performed with the following primers: albumin (sense 5′-GCTGTCATCTCTTGTGGG CTGT-3′, anti-sense 5′-AAACTCATGGGAGCTGCTGGTT-3′); IDO (sense 5′-GGTCATGGAGATGTCCGTAA-3′, anti-sense 5′-ACCAATAGAGAGACCAGGAAG AA-3′); RORC-γτ (sense 5′-TGGAAGTGGTGCTGGTTAGGA-3′, anti-sense 5′-AAGGCTCGGAACAGCTCCAT-3′; and SOCS3 (sense 5′-AGGAATGTAGCAGCGATG GAA-3′, anti-sense 5′-GCCCTGTCCAGCCCAATAC-3′). PCR was carried out in an 7300 real-time PCR System (Applied Biosystems, Carlsbad, CA, USA) based on specific cytokine primers and general SYBR green fluorescence detection for 10 min at 94°C followed by 45 cycles of 15 s at 95°C, 30 s at 60°C and 30 s at 72°C. Relative quantitative expression was calculated as 2-ΔΔCT following the guidelines of Livak et al. [21].
Statistical analysis
A Mann–Whitney U-test or Wilcoxon matched-pairs test was used to compare variables between CIU and healthy subjects. The Kruskal–Wallis test with Dunn's post-test was used to compare non-paired data. P values less than 0·05 were considered significant.
Results
Immunomodulatory effect of statins on the lymphoproliferative response
The effect of increasing concentrations of simvastatin and lovastatin (0·25, 1·0, 5·0 and 25·0 µm) on lymphocyte proliferation was analysed in PBMCs from patients with CIU and HC subjects.
Both statin drugs inhibited proliferation of lymphocytes in HC and CIU subjects in a similar way and in a concentration-dependent manner for all the mitogens or antigens tested. Figure 1a shows that simvastatin induced a marked inhibition of the cellular proliferative response to PHA in both HC and CIU patients, reaching more than 75% inhibition at 5 µm and more than 95% at 25 µm. Similarly, high levels of inhibition were obtained with either 5 or 25 µm simvastatin or lovastatin on the B cell proliferative response to PWM stimulation (Fig. 1c and d) and to Staphylococcus aureus enterotoxin A (Fig. 1e), and to the antigen-specific proliferative response against the recall antigen tetanus toxoid (Fig. 1f). The highest concentration of simvastatin (25 µm) was chosen for all the following experiments.
Fig. 1.
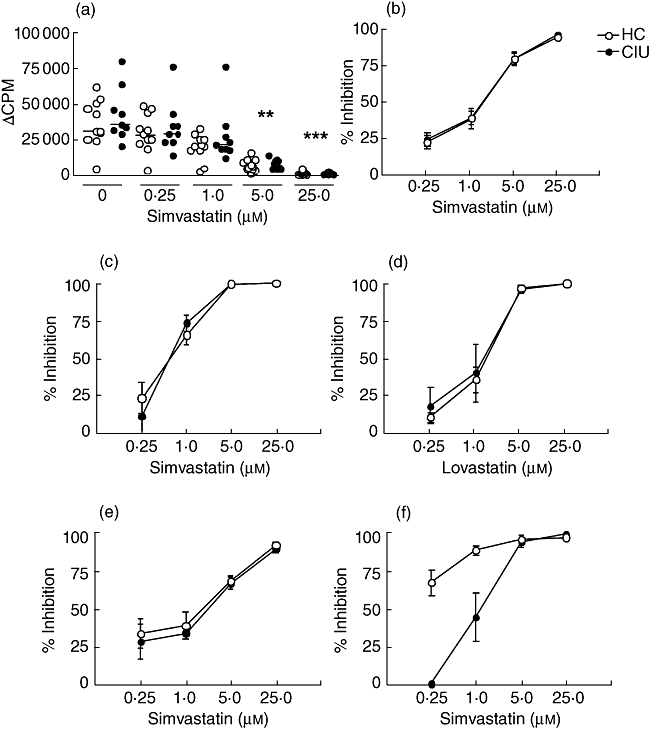
Inhibitory effect of simvastatin on the proliferative response of T cells from healthy controls (HC) and chronic idiopathic urticaria (CIU) patients. Peripheral blood mononuclear cells (PBMCs) from HC (n = 8–10) or CIU patients (n = 5–9) were cultured with (a) phytohaemagglutinin (PHA) (2·5 µg/ml) plus simvastatin (0·25–25 µm) for 3 days. The results are shown as Δcounts per minute (Δcpm) (stimulated cultures – basal) and the horizontal line represents the median, **P < 0·01, ***P < 0·001 compared to PHA response in the absence of simvastatin (0). (b–d) Inhibition percentage mediated by statin on the proliferative response induced by pokeweed mitogen (PWM) (5 µg/ml) with (c) simvastatin or (d) lovastatin (0·25–25 µm) for 6 days, or simvastatin with (e) enterotoxin A of Staphylococcus aureus (0·04 µg/ml) for 3 days or (f) tetanus toxoid for 6 days (0·04 Lf/ml), expressed by mean ± standard error of the mean.
We then evaluated whether the marked inhibition of proliferation was due to an increase in cellular apoptosis or to a possible interference in the cell cycle in response to PHA. Both simvastatin and lovastatin (25 µm) were able to interfere with the lymphocyte cell cycle progression, arresting cells in the G0/G1 phase (Fig. 2), while leading to a significant decrease in the percentage of cells in S and G2/M phases, in both CIU and HC groups. With regard to PBMC cultures from the HC group treated with lovastatin, there was an increase in the percentage of apoptosis compared to activated cultures not treated with lovastatin (Fig. 2b).
Fig. 2.
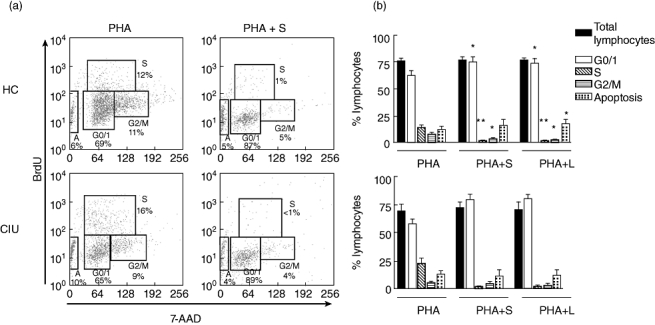
Statins arrest cell cycle at the G0/1 phase in response to phytohaemagglutinin (PHA) in healthy controls (HC) and chronic idiopathic urticaria (CIU) patients. Peripheral blood mononuclear cells (PBMCs) from HC (n = 5) or CIU patients (n = 4) were cultured with PHA (2·5 µg/ml) in presence of simvastatin (S, 25 µm) or lovastatin (L, 25 µm) for 3 days. Bromodeoxyuridine (BrdU) (10 µm/ml) was added 18 h before the end of the culture. (a) Illustrative histograms of cell cycle phases: G0/1, S, G/M, apoptosis of PBMC from HC or CIU patient cultured with PHA and S; (b) results represent median ± standard error of the mean of the percentage of total lymphocytes evaluated by flow cytometry. *P < 0·05; **P < 0·01; ***P < 0·001 when compared with the cell cycle response to PHA in the absence of statins.
Effect of simvastatin on cytokine secretion and expression of regulatory or proinflammatory factors induced by PHA
In view of the inhibitory effect of the statins on proliferative response, the modulatory effect of simvastatin on T cell cytokine secretion upon PHA stimulation was evaluated.
Figure 3 shows that the levels of interferon (IFN)-γ, IL-5, IL-10 and IL-17A induced by PBMC stimulated by PHA did not differ between HC or CIU patients and were decreased significantly by simvastatin. Cytokines such as IL-5 and IL-17A were inhibited to an undetectable level, regardless of the group analysed. These results showed that simvastatin is able to inhibit proinflammatory (IFN-γ and IL-17A) or regulatory (IL-10) cytokine secretion by T cells.
Fig. 3.
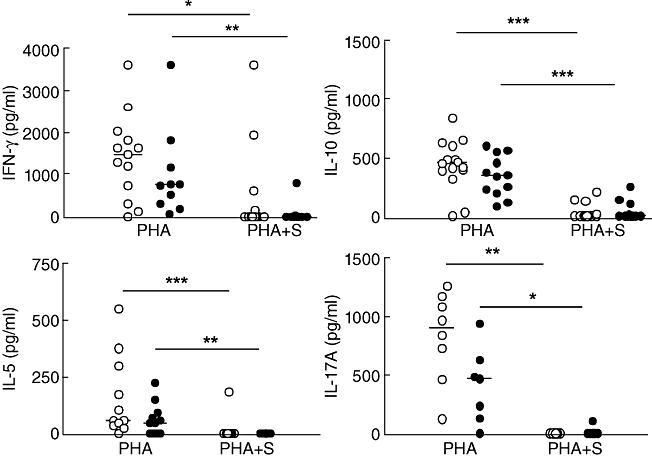
Effect of simvastatin on cytokine secretion induced by phytohaemagglutinin (PHA) in peripheral blood mononuclear cells (PBMCs) from healthy controls (HC) and chronic idiopathic urticaria (CIU) patients. PBMCs from HC (○, n = 8–12) and CIU patients (•, n = 8–10) were cultured with PHA (2·5µg/ml) in the presence of 25 µm simvastatin (S) for 48 h. Supernatants were assessed for interferon (IFN)-γ, interleukin (IL)-10, IL-5, and IL-17A secretion by enzyme-linked immunosorbent assay (ELISA). The horizontal line represents the median. *P < 0·05; **P < 0·01; ***P < 0·001.
The expression levels of the retinoic acid-related orphan nuclear hormone receptor C (RORCγt) gene, involved in IL-17 production, and the SOCS 3 gene, involved in the regulation of IL-6, IL-23 and IL-17 [22], were evaluated with or without PHA stimulation. The level of PHA-induced RORCγt mRNA was similar for CIU and HC PBMC cells in the presence or absence of statin (Fig. 4a).
Fig. 4.
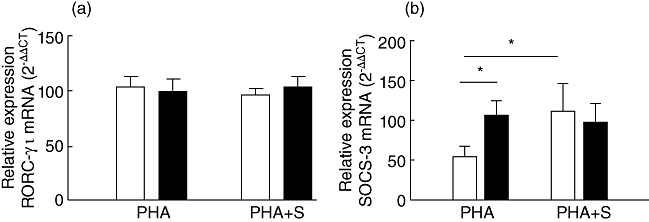
Effect of simvastatin on RORC-γτ and SOCS3 mRNA expression induced by phytohaemagglutinin (PHA) in cells from healthy controls (HC) and chronic idiopathic urticaria (CIU) patients. Peripheral blood mononuclear cells (PBMCs) from HC (n = 5–10, open bars) and CIU (n = 6–8, closed bars) were cultivated with PHA and simvastatin (S) for 48 h and assessed for retinoic acid-related orphan nuclear hormone receptor C (RORC)-γτ (a) and suppressor of cytokine signaling 3 (SOCS3) (b) mRNA expression by real-time polymerase chain reaction. Results are expressed as mean ± standard error of the mean. *P < 0·05; **P < 0·01.
Conversely, there was an increase in the expression level of the SOCS3 gene in PBMC from CIU patients stimulated with PHA compared to the HC counterparts (Fig. 4b). Simvastatin did not interfere in SOCS3 expression in CIU PBMCs, as it did in HC PBMCs (Fig. 4b). The level of PHA-induced SOCS3 expression was the same for CIU and HC when only T CD4+ cells were considered (data not shown).
Effect of simvastatin on cytokine secretion and expression of regulatory or proinflammatory factors induced by LPS
Stimulation involving the TLR-4 signalling pathway was evaluated by incubating simvastatin with LPS; however, the addition of LPS did not modulate cytokine secretion in PBMC (data not shown). We then assessed whether preincubation of PBMCs with simvastatin or lovastatin for 24 h would modulate cytokine secretion upon LPS stimulation. Figure 5a shows that tumour necrosis factor (TNF)-α secretion was inhibited by both statins only in the HC group. In contrast, the drugs were able to decrease IL-6 and macrophage inflammatory protein (MIP)-1α secretion significantly in both CIU patients and HC individuals (Fig. 5b and c).
Fig. 5.
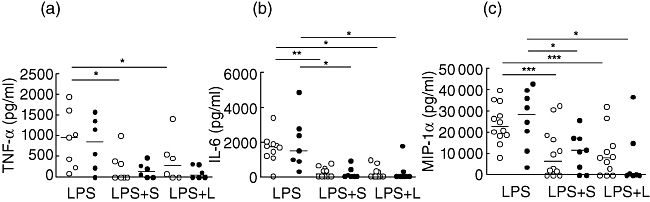
Effect of statin on cytokine secretion induced by lipopolysaccharide (LPS) in peripheral blood mononuclear cells (PBMCs) from healthy controls (HC) and chronic idiopathic urticaria (CIU) patients. PBMCs from HC (○, n = 7–9) and CIU patients (•, n = 6–7) were incubated with simvastatin (S) and lovastatin (L) (25 µm) for 24 h followed by LPS (100 ng/ml) stimulation for 24 h. Supernatant levels of tumour necrosis factor (TNF)-α, interleukin (IL)-6 and macrophage inflammatory protein (MIP)-1α were determined by enzyme-linked immunosorbent assay (ELISA). *P < 0·05, **P < 0·01, ***P < 0·001.
When cytokine secretion was analysed in LPS-stimulated monocytes, there was no difference in TNF-α secretion between CIU and HC subjects (Fig. 6a). This result corroborates the data obtained with pretreatment or co-cultivation of PBMCs with simvastatin stimulated with LPS, showing that this process was not effective at inhibiting TNF-α secretion from CIU cells (Fig. 5a). In contrast to PBMCs, monocytes from CIU patients produced increased amounts of MIP-1α compared to HC individuals (Fig. 6b).
Fig. 6.
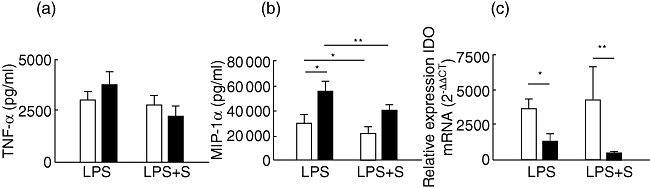
Effect of statin on indoleamine 2,3-dioxygenase (IDO) expression and cytokine secretion in cells from healthy controls (HC) and chronic idiopathic urticaria (CIU) patients. CD14+ cells (a,b) and peripheral blood mononuclear cells (PBMCs) (c) from HC (n = 7, open bars) and CIU patients (n = 4–7, closed bars) were cultured with 25 µm simvastatin (S) for 24 h and stimulated with lipopolysaccharide (LPS) for 24 h (PBMC) or 2 h (CD14+); CD14+ culture supernatant levels of tumour necrosis factor (TNF)-α (a) and macrophage inflammatory protein (MIP)-1α (b) were determined by enzyme-linked immunosorbent assay (ELISA). IDO mRNA expression was assessed by real-time polymerase chain reaction. Results are expressed as mean ± standard error of the mean. *P < 0·05; **P < 0·01.
To verify whether the expression of regulatory factor could be modulated by simvastatin, we assessed IDO mRNA expression in PBMC after statin treatment and LPS stimulation. The IDO expression upon LPS stimulation was reduced significantly in PBMC from CIU patients compared to HC, and this difference was more pronounced with statin treatment (Fig. 6c). This result showed that statin is less efficient to down-regulate TLR-4 signalling than in the activation of T cells in CIU.
Discussion
Although immune dysregulation is generally attributed to CIU, there is no clear evidence to date relating unbalanced immunoregulatory or proinflammatory factors with this disease. Therefore, the effects of immunomodulatory–pharmacological agents, such as statins, on the cell cycle proliferation, Th1, Th2 and proinflammatory cytokine production and on the regulatory factors expression were addressed.
Our findings showed that simvastatin or lovastatin induced a remarkable inhibition, up to 95%, in a concentration-dependent manner, on the proliferative response induced by T–B polyclonal stimuli, SEA and the antigen-specific proliferative response. Overall, the in vitro effects of statins on human PBMCs have been evaluated with polyclonal stimuli, and these agents were shown to be able to inhibit cellular proliferation in a concentration-dependent and statin-specific manner, simvastatin being the most potent, followed by lovastatin and mevastatin. This inhibitory effect has been demonstrated in autoimmune diseases such as multiple sclerosis [23].
The remarkable inhibition of the T cell proliferative response to PHA mediated by statin was a consequence of the cell cycle arrest at G0/G1 phase; this effect was more pronounced in PBMCs from HC individuals. Consequently, an increase in the percentage of G0/G1-arrested cells was observed along with a concomitant decrease in the number of cells in the S and G2/M phases, compared to the non-treated stimulated cells. Although an increased number of G0/G1-arrested cells was detected in both HC and CIU subjects, no difference was observed in the number of cells from other phases in the CIU patients; a possible explanation is that an early T cell activation may be unaffected by statins in CIU, and/or there may be an altered intracellular signalling pathway. In fact, the inhibitory effect of simvastatin on T cell proliferation involves the impaired geranylation of Rho, Rac and Ras GTPases, whereas the early T cell activation markers seem not to be affected by atorvastatin [24].
Furthermore, we demonstrated that lovastatin (25 µm) induced apoptosis based on 7AAD exclusion compared with healthy control cells; a longer cultivation period was probably required to evaluate apoptosis more effectively in patients' cells. Usually, apoptosis takes place by strong stimulation, such as PMA/ionomycin in CD4+ T cells, in the presence of atorvastatin (10 µm) [24] or fluvastatin (10 µm) in resting CD4+ T cells, but not in CD4+ T cells that are strongly activated with high concentrations of PMA/ionomycin [25].
The ability of statins to efficiently arrest cells at G0/G1 phase was indicated by the inhibition of cytokine production upon PHA stimulation, showing that simvastatin could have anti-inflammatory effects by inhibiting secretion of the proinflammatory cytokines Th1 and Th17 and also suppress the anti-inflammatory cytokine Th2. Th17 has been identified in a subset of CD4+ T cells that is critical for the development of autoimmune diseases [15], and down-modulation of this cytokine could be a target for treatment.
Although we found a clear inhibition of IL-17A secretion by statin in PHA-stimulated PBMCs, the up-regulated SOCS3 expression in PBMC was not affected upon statin treatment, contrary to the significant increase in HC samples. The increase of SOCS3 expression could be related to the cytokine inhibition by T cells. SOCS3 induction may have an inhibitory effect on multiple inflammatory cytokine signal transduction pathways that negatively regulate the JAK/STAT signal transduction pathway and IL-6 and IL-23 gene expression; SOCS3 may also inhibit the expression of RORC in CD4+ T cells [22]. In fact, simvastatin-treated monocytes used to treat activated allogenic CD4+ T cells significantly inhibited RORC transcription factor and IL-17A gene expression in CD4+ T cells in comparison to CD4+ T cells cultured with supernatants of untreated monocytes [22].
Conversely, simvastatin was less effective in controlling innate immune stimulation by LPS, as cell pretreatment with statins was required to produce inhibition of IL-6 and MIP-1α and yet not sufficient to interfere with TNF-α secretion by PBMCs in CIU patients. Similarly, statins did not interfere in TNF-α secretion by CD14+ cells stimulated with LPS from both HC and CIU patients, despite the maintenance of MIP-1α inhibition. It is possible that different statins exert differential effects on CD14+ cells with regard to their proinflammatory properties, as simvastatin does not affect TNF-α secretion by CD14+ cells. The concentration threshold for statin-induced proinflammatory response has been considered to be between 0·1 and 10 µm simvastatin in activated PBMCs [26]. Statins may also enhance secretion of proinflammatory cytokines such as IL-6, IL-18, IL-1, IL-12 and TNF-α from monocytes and dendritic cells (DCs) [27]. With regard to the present CIU and HC samples, we verified an increased level of MIP-1α secretion by CD14+ cells induced by LPS. This chemokine was already shown to be increased in CIU PBMCs stimulated with PHA plus IL-2 [28].
Furthermore, IDO, which exerts an essential role in the control of T cell responses, was decreased significantly in CIU PBMCs stimulated with LPS, reaching a 2·5-fold suppression compared to HC. Although we did not evaluate tryptophan catabolism, the altered enzyme expression may be critical for the immune dysfunction in CIU. Among some inhibitors of IDO, nitric oxide has been shown to interact directly with IDO [29], and IL-4, IL-13 and transforming growth factor (TGF)-β are known to be suppressors of IDO [30].
Several aspects of the negative regulation of IDO expression in CIU remain to be investigated. However, the present finding of SOCS3 up-regulation associated with the decreased expression of IDO may indicate a counter-regulatory mechanism. SOCS3 expression leads to the proteasomal degradation of IDO, which is involved in immunity rather than tolerogenesis [31]. Moreover, IDO overexpression by APCs may result in immunosuppression and reduced T cell responses [32].
In summary, the impaired expression of regulatory factors such as IDO and SOCS3, currently detected in the CIU, may play an important role in the pathogenesis of the disease. The balance between anti-inflammatory and proinflammatory cytokines is critical for a more or less favourable outcome of the chronic disease. Statins exert a broad inhibitory effect on T cell functions such as proliferative responsiveness and cytokine production. Statins also exert a different immunomodulatory effect on monocytes and are less efficacious at controlling innate immune stimulation. Altogether, statins displayed a more anti-inflammatory than proinflammatory effect, disclosing a role for these pharmacological agents particularly related to T cell activation.
Acknowledgments
We thank Dr Gabriela Ribeiro-dos-Santos for reviewing the manuscript. This research was supported by FAPESP (06/53601–8 and 06/57926–9), and LIM 56 HC-FMUSP.
Disclosure
The authors declare no financial conflict of interests.
References
- 1.O'Donnell BF, Lawlor F, Simpson J, Morgan M, Greaves MW. The impact of chronic urticaria on the quality of life. Br J Dermatol. 1997;136:197–201. [PubMed] [Google Scholar]
- 2.Grob JJ, Gaudy-Marqueste C. Urticaria and quality of life. Clin Rev Allergy Immunol. 2006;30:47–51. doi: 10.1385/CRIAI:30:1:047. [DOI] [PubMed] [Google Scholar]
- 3.Hide M, Francis DM, Grattan CE, Hakimi J, Kochan JP, Greaves MW. Autoantibodies against the high-affinity IgE receptor as a cause of histamine release in chronic urticaria. N Engl J Med. 1993;328:1599–604. doi: 10.1056/NEJM199306033282204. [DOI] [PubMed] [Google Scholar]
- 4.Grattan CE. Basophils in chronic urticaria. J Invest Dermatol Symp Proc. 2001;6:139–40. doi: 10.1046/j.0022-202x.2001.00027.x. [DOI] [PubMed] [Google Scholar]
- 5.Kaplan AP, Greaves M. Pathogenesis of chronic urticaria. Clin Exp Allergy. 2009;39:777–87. doi: 10.1111/j.1365-2222.2009.03256.x. [DOI] [PubMed] [Google Scholar]
- 6.Lourenco FD, Azor MH, Santos JC, et al. Activated status of basophils in chronic urticaria leads to interleukin-3 hyper-responsiveness and enhancement of histamine release induced by anti-IgE stimulus. Br J Dermatol. 2008;158:979–86. doi: 10.1111/j.1365-2133.2008.08499.x. [DOI] [PubMed] [Google Scholar]
- 7.Vonakis BM, Vasagar K, Gibbons SP, et al. Basophil FcepsilonRI histamine release parallels expression of Src-homology 2-containing inositol phosphatases in chronic idiopathic urticaria. J Allergy Clin Immunol. 2007;119:441–8. doi: 10.1016/j.jaci.2006.09.035. [DOI] [PubMed] [Google Scholar]
- 8.Tedeschi A, Asero R, Lorini M, Marzano AV, Cugno M. Plasma levels of matrix metalloproteinase-9 in chronic urticaria patients correlate with disease severity and C-reactive protein but not with circulating histamine-releasing factors. Clin Exp Allergy. 2010;40:875–81. doi: 10.1111/j.1365-2222.2010.03473.x. [DOI] [PubMed] [Google Scholar]
- 9.Dos Santos JC, Azor MH, Nojima VY, et al. Increased circulating pro-inflammatory cytokines and imbalanced regulatory T-cell cytokines production in chronic idiopathic urticaria. Int Immunopharmacol. 2008;8:1433–40. doi: 10.1016/j.intimp.2008.05.016. [DOI] [PubMed] [Google Scholar]
- 10.Mellor AL, Munn DH. IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol. 2004;4:762–74. doi: 10.1038/nri1457. [DOI] [PubMed] [Google Scholar]
- 11.Fallarino F, Grohmann U, Hwang KW, et al. Modulation of tryptophan catabolism by regulatory T cells. Nat Immunol. 2003;4:1206–12. doi: 10.1038/ni1003. [DOI] [PubMed] [Google Scholar]
- 12.Mellor AL, Munn DH. Tryptophan catabolism prevents maternal T cells from activating lethal anti-fetal immune responses. J Reprod Immunol. 2001;52:5–13. doi: 10.1016/s0165-0378(01)00118-8. [DOI] [PubMed] [Google Scholar]
- 13.Elliott J, Johnston JA. SOCS: role in inflammation, allergy and homeostasis. Trends Immunol. 2004;25:434–40. doi: 10.1016/j.it.2004.05.012. [DOI] [PubMed] [Google Scholar]
- 14.Weaver CT, Harrington LE, Mangan PR, Gavrieli M, Murphy KM. Th17: an effector CD4 T cell lineage with regulatory T cell ties. Immunity. 2006;24:677–88. doi: 10.1016/j.immuni.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 15.Chen Z, O'Shea JJ. Regulation of IL-17 production in human lymphocytes. Cytokine. 2008;41:71–8. doi: 10.1016/j.cyto.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zeiser R, Youssef S, Baker J, Kambham N, Steinman L, Preemptive NRS. HMG-CoA reductase inhibition provides graft-versus-host disease protection by Th-2 polarization while sparing graft-versus-leukemia activity. Blood. 2007;110:4588–98. doi: 10.1182/blood-2007-08-106005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mausner-Fainberg K, Luboshits G, Mor A, et al. The effect of HMG-CoA reductase inhibitors on naturally occurring CD4+CD25+ T cells. Atherosclerosis. 2008;197:829–39. doi: 10.1016/j.atherosclerosis.2007.07.031. [DOI] [PubMed] [Google Scholar]
- 18.Fehr T, Kahlert C, Fierz W, et al. Statin-induced immunomodulatory effects on human T cells in vivo. Atherosclerosis. 2004;175:83–90. doi: 10.1016/j.atherosclerosis.2004.02.016. [DOI] [PubMed] [Google Scholar]
- 19.Blanco-Colio LM, Martin-Ventura JL, Gomez-Hernandez A, Egido J. Statins, inflammation and atherosclerotic damage. Nefrologia. 2004;24:17–20. [PubMed] [Google Scholar]
- 20.Kuijk LM, Mandey SH, Schellens I, et al. Statin synergizes with LPS to induce IL-1beta release by THP-1 cells through activation of caspase-1. Mol Immunol. 2008;45:2158–65. doi: 10.1016/j.molimm.2007.12.008. [DOI] [PubMed] [Google Scholar]
- 21.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 22.Zhang X, Jin J, Peng X, Ramgolam VS, Markovic-Plese S. Simvastatin inhibits IL-17 secretion by targeting multiple IL-17-regulatory cytokines and by inhibiting the expression of IL-17 transcription factor RORC in CD4+ lymphocytes. J Immunol. 2008;180:6988–96. doi: 10.4049/jimmunol.180.10.6988. [DOI] [PubMed] [Google Scholar]
- 23.Neuhaus O, Strasser-Fuchs S, Fazekas F, et al. Statins as immunomodulators: comparison with interferon-beta 1b in MS. Neurology. 2002;59:990–7. doi: 10.1212/wnl.59.7.990. [DOI] [PubMed] [Google Scholar]
- 24.Brinkkoetter PT, Gottmann U, Schulte J, van der Woude FJ, Braun C, Yard BA. Atorvastatin interferes with activation of human CD4(+) T cells via inhibition of small guanosine triphosphatase (GTPase) activity and caspase-independent apoptosis. Clin Exp Immunol. 2006;146:524–32. doi: 10.1111/j.1365-2249.2006.03217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Samson KT, Minoguchi K, Tanaka A, et al. Effect of fluvastatin on apoptosis in human CD4+ T cells. Cell Immunol. 2005;235:136–44. doi: 10.1016/j.cellimm.2005.08.028. [DOI] [PubMed] [Google Scholar]
- 26.Coward W, Chow SC. Effect of atorvastatin on TH1 and TH2 cytokine secreting cells during T cell activation and differentiation. Atherosclerosis. 2006;186:302–9. doi: 10.1016/j.atherosclerosis.2005.08.013. [DOI] [PubMed] [Google Scholar]
- 27.Yilmaz A, Reiss C, Weng A, et al. Differential effects of statins on relevant functions of human monocyte-derived dendritic cells. J Leukoc Biol. 2006;79:529–38. doi: 10.1189/jlb.0205064. [DOI] [PubMed] [Google Scholar]
- 28.Piconi S, Trabattoni D, Iemoli E, et al. Immune profiles of patients with chronic idiopathic urticaria. Int Arch Allergy Immunol. 2002;128:59–66. doi: 10.1159/000058004. [DOI] [PubMed] [Google Scholar]
- 29.Thomas SR, Salahifar H, Mashima R, Hunt NH, Richardson DR, Stocker R. Antioxidants inhibit indoleamine 2,3-dioxygenase in IFN-gamma-activated human macrophages: posttranslational regulation by pyrrolidine dithiocarbamate. J Immunol. 2001;166:6332–40. doi: 10.4049/jimmunol.166.10.6332. [DOI] [PubMed] [Google Scholar]
- 30.Yuan W, Collado-Hidalgo A, Yufit T, Taylor M, Varga J. Modulation of cellular tryptophan metabolism in human fibroblasts by transforming growth factor-beta: selective inhibition of indoleamine 2,3-dioxygenase and tryptophanyl-tRNA synthetase gene expression. J Cell Physiol. 1998;177:174–86. doi: 10.1002/(SICI)1097-4652(199810)177:1<174::AID-JCP18>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 31.Orabona C, Pallotta MT, Volpi C, et al. SOCS3 drives proteasomal degradation of indoleamine 2,3-dioxygenase (IDO) and antagonizes IDO-dependent tolerogenesis. Proc Natl Acad Sci USA. 2008;105:20828–33. doi: 10.1073/pnas.0810278105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Terness P, Bauer TM, Rose L, et al. Inhibition of allogeneic T cell proliferation by indoleamine 2,3-dioxygenase-expressing dendritic cells: mediation of suppression by tryptophan metabolites. J Exp Med. 2002;196:447–57. doi: 10.1084/jem.20020052. [DOI] [PMC free article] [PubMed] [Google Scholar]