

Abstract
Infantile neuronal ceroid lipofuscinosis (INCL), a lethal hereditary neurodegenerative lysosomal storage disorder, affects mostly children. It is caused by inactivating mutations in the palmitoyl-protein thioesterase-1 (PPT1) gene. Nonsense mutations in a gene generate premature termination codons producing truncated, nonfunctional or deleterious proteins. PPT1 nonsense-mutations account for approximately 31% of INCL patients in the US. Currently, there is no effective treatment for this disease. While aminoglycosides such as gentamycin suppress nonsense mutations, inherent toxicity of aminoglycosides prohibits chronic use in patients. PTC124 is a non-toxic compound that induces ribosomal read-through of premature termination codons. We sought to determine whether PTC124-treatment of cultured cells from INCL patients carrying nonsense mutations in the PPT1 gene would correct PPT1 enzyme-deficiency with beneficial effects. Our results showed that PTC124-treatment of cultured cells from INCL patients carrying PPT1 nonsense-mutations induced PPT1 enzymatic activity in a dose- and time-dependent manner. This low level of PPT1 enzyme activity induced by PTC124 is virtually identical to that induced by gentamycin-treatment. Even though only a modest increase in PPT1 activity was achieved by PTC124-treatment of INCL cells, this treatment reduced the levels of thioester (constituent of ceroid) load. Our results suggest that PTC124-treatment induces PPT1 enzymatic activity in cultured cells from INCL patients carrying PPT1 nonsense-mutations, and this modest enzymatic activity has demonstrable beneficial effects on these cells. The clinical relevance of these effects may be tested in animal models of INCL carrying nonsense mutations in the PPT1 gene.
Keywords: Lysosomal storage disease, Neurodegeneration, Infantile neuronal ceroid lipofuscinosis, Batten disease, PTC124, Nonsense mutation
1. Introduction
Neuronal ceroid lipofuscinoses (NCLs), commonly known as Batten disease, represent a group of the most prevalent (1 in 12,500 births) neurodegenerative lysosomal storage disorders (LSDs) affecting both children and adults (1–6). The infantile form of NCL (INCL), caused by inactivating mutations in the CLN1 gene, encoding palmitoyl-protein thioesterase-1 (PPT1), is the second most lethal disease among all NCLs. Despite our knowledge that PPT1 gene mutations cause INCL (7), a clear pathogenic mechanism remains unclear and an effective treatment is currently unavailable.
It is estimated that nonsense mutations in genes account for 5–70% of the genetic disorders (8, 9). Nonsense mutations in the PPT1 gene have been reported in the majority of the INCL patients in the US (10). These mutations cause premature translational termination, which results in the production of truncated PPT1 protein that lack enzymatic activity causing INCL pathogenesis (10). Compelling evidence from diseases such as cystic fibrosis (CF) indicated that stimulating the production of specific gene products from <1% to as little as 5% of normal levels may greatly reduce the severity or eliminate the principal manifestations of the disease. It has also been reported that the aminoglycoside antibiotic, gentamycin, suppresses premature termination of protein synthesis caused by nonsense mutations in a gene (11–14). However, repeated use of high doses of aminoglycosides, required for such a therapy, has significant adverse effects. To address the need for a nontoxic drug, which is capable of suppressing premature termination, a recent report identified PTC124 (3-[5-(2-fluorophenyl)-1, 2, 4-oxadiazol-3-yl] benzoic acid), a novel and non-toxic chemical compound to accomplish this task (15). Current reports indicate that PTC124-treatment corrects nonsense mutations in the gene encoding cystic fibrosis transmembrane regulator (CFTR) (16). More recently, PTC124 both in cell culture and in vivo has been shown to mediate read-through of nonsense mutations causing Usher syndrome gene, USH1C (17). In a phase II clinical trial with PTC124 on patients with CF, it has been reported that oral administration of this compound reduces the epithelial electrophysiological abnormalities caused by the CFTR mutation (18). The safety and tolerability of PTC124 have been reported (19). This drug selectively induces ribosomal read through of premature stop codons and does not affect the normal termination codons.
In the present study, we sought to determine whether the PPT1 nonsense mutations found in INCL patients in the US can be suppressed by PTC124 and if any beneficial effects of this treatment could be assessed in cultured cells from these patients. Our results show that PTC124-treatment of cultured cells from INCL patients carrying PPT1 nonsense mutations induced PPT1 enzymatic activity in a dose and time-dependent manner. This level of induction is virtually identical to that induced by gentamycin, an aminoglycoside antibiotic reported to induce ribosomal read through of nonsense mutations. Remarkably, although PTC124-treatment yielded only a modest increase in PPT1 activity, this treatment reduced the levels of thioester (ceroid) load present in untreated cells. Moreover, PTC124-treatment suppressed apoptosis. Our results show that PTC124-treatment of cultured cells from INCL patients carrying PPT1 nonsense mutations can induce enzymatic activity that in turn produce biological effects.
2. Material and Methods
2.1 Cell culture
Fibroblast and lymphoblast cells isolated from INCL patients with nonsense mutation (Table 1) in the Ppt1 gene were used for this study. Fibroblasts were grown in DMEM with 10% heat inactivated FBS and penicillin and streptomycin. Lymphoblast were cultured with RPMI 1640 media supplemented with 16 % heat inactivated FBS and penicillin and streptomycin. For immunofluorescence study COS-1 cells were used and cultured in DMEM containing 10 % heat inactivated FBS and penicillin and streptomycin. All cell types were maintained at 37° C in humidified atmosphere with 5% CO2.
Table 1.
Stop codons in Ppt1 nonsense mutations in INCL patients
| Types of mutations | Stop codon(s) | |
|---|---|---|
| Fibroblasts | ||
| INCL patient-1: | Nonsense, exon 5 (R151X) | UGA |
| Nonsense, exon 5 (R151X) | UGA | |
| INCL patient-2: | Nonsense, exon 5 (R151X) | UGA |
| Nonsense, exon 5 (R151X) | UGA | |
| INCL patient-3: | Nonsense, exon 1 (L10X) | UAG |
| Nonsense, exon 5 (R151X) | UGA | |
| Lymphoblasts | ||
| INCL patient-4: | Nonsense, exon 5 (R151X) | UGA |
| Nonsense, exon 5 (R151X) | UGA | |
| INCL patient-5: | Nonsense, exon 5 (R164 X) | UGA |
| Nonsense, exon 5 (R164 X) | UGA | |
| INCL patient-6: | Nonsense, exon 5 (R164 X) | UGA |
| Nonsense, exon 9 (Q291X) | UAG |
2.2 MTT assay for cell viability
Cell viability was measured using MTT [3-(4, 5-Dimethylthiazol-2-yl)-2,S-diphenyltetrazolium Bromide] (Sigma) according to a previously described method (20). Briefly, the cells were incubated with MTT (0.5 mg/ml) after treatment with varying concentrations of PTC124 (0.3 –30 µg/ml) [Selleck Chemicals (Houston, TX)] or Gentamycin (0.25–10 mg/ml) (Invitrogen). The formazan crystals formed within live cells was dissolved in acidified isopropanol and absorbance of the solution was measured at 570 nm. Cell viability is expressed as percentage of untreated control.
2.3 PPT1 Enzyme Assay
PPT1 enzyme activity was measured as previously described method (21). To summarize briefly, after treatment with gentamycin PTC124 cells were homogenized by sonication in water containing protease inhibitor. Cell homogenates (~ 10 µg) were then incubated with the substrate mixture consisting of 0.64 mM 4-Methlyumbelliferyl-6-thiopalmitoyl-β-D-glucoside (Moscerdam Substrates, Netherlands), 15 mM dithiothreitol (DTT), 0.375% (w/v) Triton X-100, and 0.1 U β-glucosidase from almonds (Sigma) in McIlvain’s phosphate/citrate buffer, pH 4.0. The reaction mixture was then incubated for 1 h at 37° C. The reaction was stopped by the addition of 0.5 M Na2CO3/NaHCO3, pH 10.7, containing 0.025% Triton X-100 and the fluorescence of the product, 4-methylumbelliferone, was measured in a fluorimeter (Flexstation2 of Molecular Device). Enzyme activity was estimated as nmole of product formed per hour per mg of protein.
2.5 Immunofluorescence study
COS-1 cells, grown in 2-chamber slide (Lab-Tek), were transfected with PPT1-myc-FLAG construct bearing a nonsense mutation (R151X) using Lipofectamine 2000 (Invitrogen). Transfected cells were treated with PTC124 (5 µg/ml) for 72 h. After treatment cells were fixed with 4% paraformaldehyde for 10 min at room temperature, washed twice with PBS and then permeabilized with 0.25% triton X-100 in PBS for 5 min. Cells were then washed with PBS twice and blocked with 10% BSA in PBS for 30 min, probed with FLAG antibody (Sigma, 1:1000) for 2 h, followed by incubation with secondary antibody (Alexa fluor 488 conjugated anti-mouse, Invitrogen) for 1 h. All incubations were performed at 37° C. Cell nuclei were stained with DAPI (Sigma-Aldrich). Slides were examined using Zeiss LSM 510 Inverted Meta confocal microscope (Carl Zeiss), and the image obtained was processed by the LSM image software (Carl Zeiss).
2.6 Determination of thioester levels in cultured cells from INCL patients
After treatment with PTC124 (5 µg/ml), lymphoblast cells were labeled with [35S]-cysteine as described previously (22, 23) with minor modifications. Briefly, PTC124 treated cells were washed and incubated for 30 min in cysteine/cystine- and serum-free medium, followed by incubation in the same medium containing [35S]-cysteine (final concentration: 100 µCi/ml) for 6 h at 37 °C. After incubation, the cells were washed twice with 2 ml of ice-cold PBS and centrifuged at 2,250g at 4 °C for 5 min. Pellet thus obtained was resuspended in PBS and used for extraction of lipid thioesters as described previously (22, 23).
2.7 Thin Layer Chromatography
Lipid thioesters were then resolved by TLC as per the method described previously (22, 23). After extraction thioesterified lipid/peptides were dissolved in chloroform:methanol mixture (1:1), applied on TLC plate and resolved using solvent mixture chloroform, methanol and water (65:25:4). After that the TLC plate was dried and exposed to film.
2.8 Transmission electron microscopy
Tranmission electron microsocopy was performed according to previously published method (23, 24). Briefly, after treatment with PTC124 (5 µg/ml) for 7 days, cells were fixed with 2.5 % glutaraldehyde in sodium phosphate buffer and then washed with Millonig’s phosphate buffer once and kept in the same buffer at 4°C until final processing. Ultra-thin sections were then prepared and stained with lead citrate and uranyl acetate and examined with a LEO 912 electron microscope (JFE Enterprises).
2.9 Measurement of cellular apoptosis
Detection of apoptotic cells were performed using Guava Nexin assay kit (Millipore) which uses Annexin-V-PE for the identification of apoptotic cells and 7-AAD for the detection of late stage apoptotic or dead cells. Briefly, after being treated with or without PTC124 (5 µg/ml) INCL lymphoblast cells (2×104) were washed and incubated with Guava Nexin reagent at room temperature in dark for 20 min. Samples were then analyzed by flow cytometry using Guava EasyCyte Mini System (Millipore).
3. Results
3.1 PTC124-treatment of INCL fibroblasts yields small increase in PPT1 enzymatic activity
We first sought to determine whether PTC124-treatment induces PPT1 enzymatic activity in cells carrying PPT1 nonsense mutations. Accordingly, we used cultured INCL fibroblasts carrying C451T nonsense mutations in the PPT1 gene and treated these cells with increasing concentrations of either gentamycin (0.25 to 1mg/ml) (Fig. 1A) or PTC124 (0.15–15 µg/ml) (Fig. 1B). Since gentamycin has been previously shown to promote translational read-through in nonsense mutations we used this aminoglycoside as a positive control. We also used INCL fibroblasts with missense mutations as a negative control (data not shown) since neither PTC124 nor gentamycin is known to suppress missense mutations. Our results showed that PTC124 induces PPT1 enzymatic activity in cultured fibroblasts from INCL patients carrying C451T mutations in a dose- (Fig. 1B) and time-dependent manner (Supplemental Fig. 1). We found that PTC124 treatment increased PPT1 enzyme activity 1–1.3% of normal (Fig. 1B), which was virtually identical to that induced by gentamycin (Fig. 1A). In order to confirm these results, we used six additional cell lines from six different patients carrying PPT1 nonsense mutations and then treated with PTC124 (5µg/ml) for 48 hours. Because the type of stop codon usage is important for the efficiency of PTC124-mediated read through (15), we determined the stop codon usage in cultured cells from different INCL patients that we used for our experiments. In Table 1 the stop codon usage in the fibroblast and lymphoblast cell lines from different INCL patients are shown. The results of these experiments showed that while PPT1 enzymatic activity was increased in all three cell lines, fibroblasts from patient 1 yielded slightly higher PPT1 activity than that of patient 2 and 3 (Supplemental Fig. 2A). Treatment of PTC124 also increased PPT1 activity at similar level in all three lymphoblast cells (Supplemental Fig. 2B). At this point, we wanted to make sure that the observed effects of PTC124 are due to the read through of the nonsense mutations in the PPT1 gene and not due to interference of PTC124 with the fluorimetric assay used for determining PPT1 enzyme activity. Thus, we performed experiments in which cell extracts from both normal control as well as INCL fibroblasts with nonsense mutations were incubated with PTC124, DMSO (diluent of PTC124) or the assay buffer only before determining PPT1 activity. The results showed that neither PTC124 nor DMSO or the assay buffer interfered with the enzyme assay (Supplemental Fig. 3). Taken together, these results showed that PTC124 is capable of stimulating PPT1 enzymatic activity in cultured INCL cells carrying nonsense mutations.
Figure 1. Effects of gentamycin and PTC124 on PPT1 activity and viability in INCL cells.
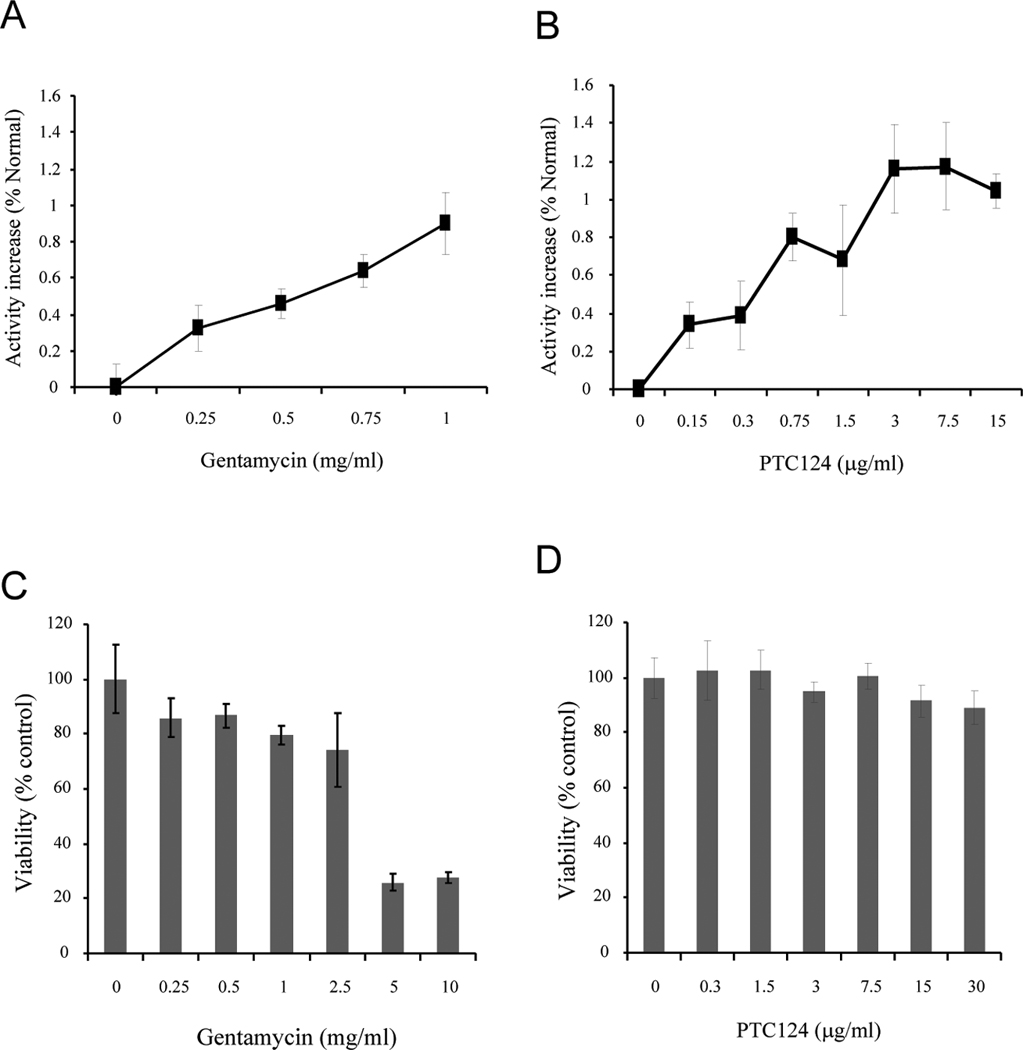
Fibroblasts from INCL patients with nonsense mutation (R151X) were treated with varying doses of (A) gentamycin (0.25–1mg/ml) or (B) PTC124 (0.3 to 30µg/ml) for 48hrs and then PPT1 enzymatic activity was assayed. Increase in PPT1 activity is plotted as percent of activity in normal cells. The Viability in cells treated with varying concentrations of gentamycin (C) or PTC124 (D) was determined and plotted as percent of untreated control. Note dramatic reduction in viability when the concentration of gentamycin was 5 mg/ml or above while virtually no adverse effects of PTC124 on cell viability were detected. The results are presented as the mean of at least 3 independent experiments ± SD.
3.2 PTC-124 show virtually no toxicity to cultured cells from INCL patients
We then sought to determine whether PTC124 treatment show any toxic effects compared to gentamycin on cultured cells from INCL patients. Accordingly, we used fibroblasts from an INCL patient carrying the most common PPT1 nonsense mutation (C451T) found in the US INCL patient population and tested the viability of these cells treated with varying doses of gentamycin (0.25–10 mg/ml) or PTC124 (0.3–30 µg/ml). Our results showed that viability of the cells was gradually reduced with increasing doses of gentamycin (0.25 to 2.5 mg/ml) and a drastic reduction in viability occurred when cells were treated with 5 and 10 mg/ml of this drug (Fig. 1C). In contrast to these results, treatment of the cells with PTC124 from 0.3–30 µg/ml showed virtually no alteration in viability (Fig. 1D). These results suggest that PTC124, unlike gentamycin, has no demonstrable toxicity on cultured cells from INCL patients, at the concentrations used.
3.3 PTC124-mediated production of full length PPT1
To determine whether PTC124-mediated translational read-through of the nonsense mutations in cultured INCL fibroblasts induced PPT1-protein, we first performed Western blot analysis of PTC124-treated and untreated cell lysates. However, the results showed no demonstrable PPT1 protein bands (data not shown). We then transfected COS-1 cells with nonsense PPT1-myc-FLAG construct (Fig 2A) and treated the transfected cells with PTC124 (5 µg/ml) for 72 h. FLAG and myc are attached at the C-terminal end of PPT1. A point mutation within PPT1 sequence is designed to produce stop codon (R151X) leading to generate truncated protein lacking FLAG or myc tags as shown (Fig. 2A). Thus, FLAG-tagged PPT1 can only be detected if it is read through during translation. We performed immunofluorescence analysis of PTC124-treated and untreated COS-1 cells transfected with nonsense PPT1-myc-FLAG construct using FLAG antibody to see if PTC124 induces production of full length PPT1-myc-FLAG fusion protein. The results showed that appreciable FLAG-immunoreactivity can be clearly observed in cells treated with PTC124 but not in the untreated control cells (Fig. 2B) suggesting translational read-through of PPT1 nonsense mutation by this compound.
Figure 2. PTC124 mediates translational read through of PPT1-nonsense mutations.
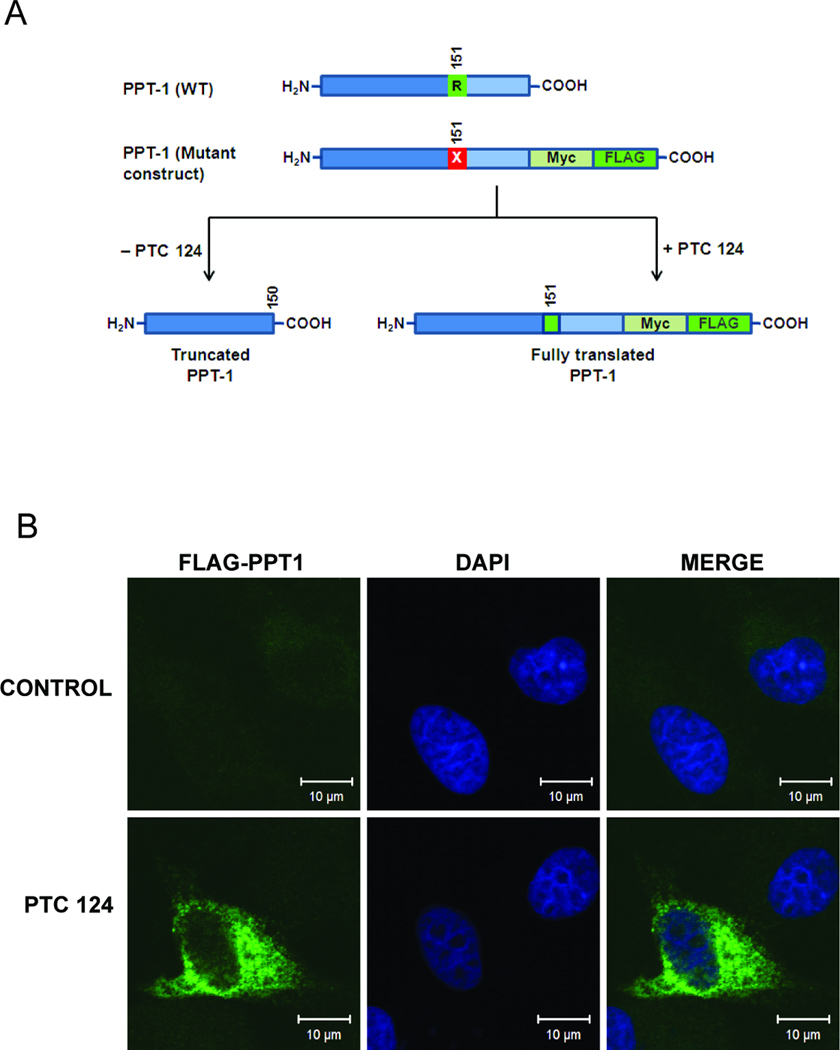
(A) PPT1-myc-FLAG construct used to transfect COS-1 cells is shown schematically. Myc and FLAG tags are incorporated at the C-terminus of PPT1. A point mutation is designed within the PPT1 sequence that causes generation of truncated PPT1-protein without FLAG or myc tags. However translational read through mediated by PTC124 can produce full length PPT1-protein with FLAG and myc tags. (B) Immunofluorescence analysis of PTC124 (5 µg/ml) treated and untreated COS-1 cells transfected with nonsense PPT1-myc-FLAG using FLAG antibody. Note that some PTC124-treated cells showed immunorectivity for FLAG, but no immunoreactivity for FLAG was observed in untreated cells.
3.4 Reduction in ceroid levels in INCL cells treated with PTC124
Since PPT1-deficiency causes abnormal accumulation of palmitoylated proteins (constituent of ceroid), we sought to determine whether PTC124-induced PPT1 enzyme activity observed in our experiments was effective in reducing the ceroid load in cultured INCL cells carrying nonsense mutations. Accordingly, we treated cultured lymphoblasts from patients (patients 4, 5 & 6) carrying nonsense mutations in the PPT1 gene with PTC124 for 48 hours and then labeled these cells with [35S]-cysteine, extracted the lipids and resolved the thioesters by high performance thin layer chromatography as previously reported (22). Labeled thioester containing bands were detected by autoradiography. The results showed that the densities of several lipid thioester containing bands in cells derived from three different patients were appreciably reduced in PTC124-treated cells compared with those of the untreated counterparts (Fig. 3, arrows). These results suggested that a modest increase in PPT1 enzyme activity induced after 48 hours of PTC124-treatment can lower lipid thioester load in these cells.
Figure 3. Reduced thioester load in PTC124-treated lymphoblast cells.
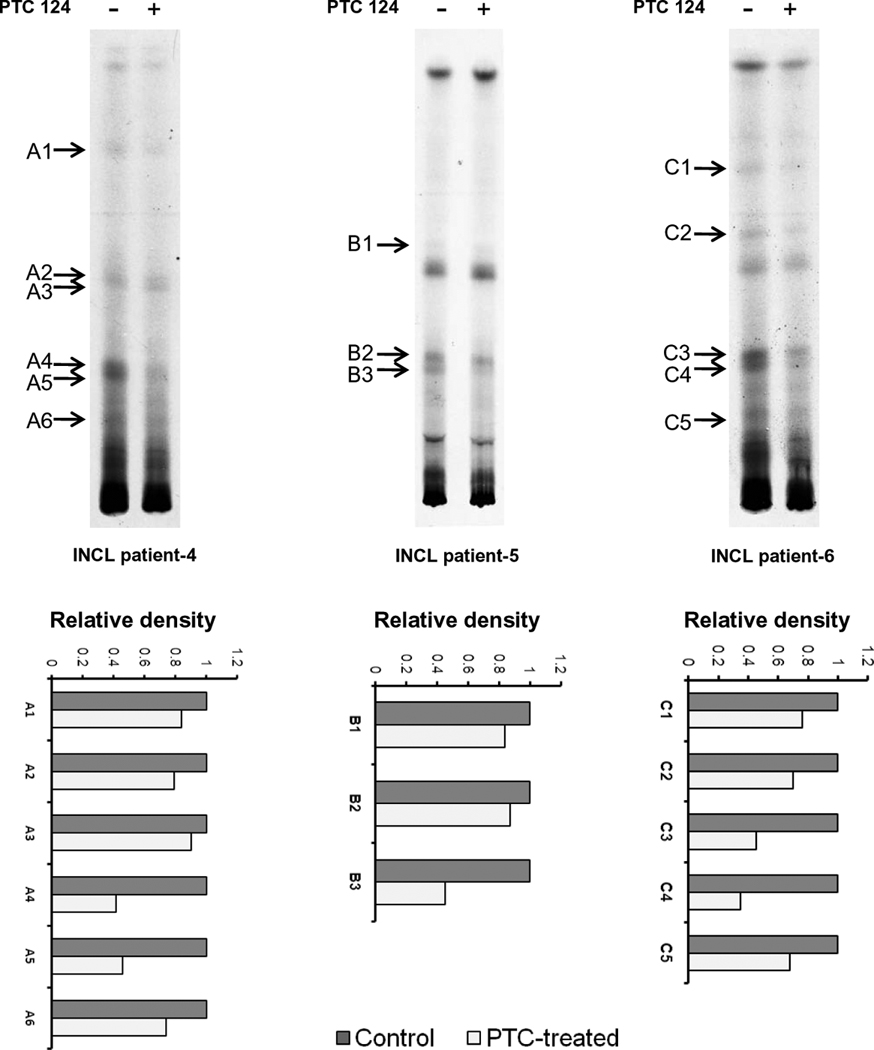
Lymphoblasts isolated from three different patients carrying PPT1 nonsense mutations were pretreated with PTC124 (5 µg/ml) for 48 h and then labeled with [35S] cysteine for 6h. Labeled thioesters were extracted and resolved by high performance thin layer chromatography. Arrows indicate thioester bands that are reduced in intensity following PTC124 (5 µg/ml)-treatment of the cells. Lower panel shows densitometric analysis of the thioester bands in PTC124-treated cells with respect to untreated control.
3.5 Low level induction of PPT1 activity causes moderately reduced level of GRODs
One of the pathological features of INCL is the presence of granular osmiophilic deposits (GRODs) when the cells or tissues are examined by transmission electron microscopy (TEM). It has been suggested that GRODs represent the undegraded s-acylated (palmitoylated) proteins due to the absence of PPT1 enzymatic activity. Since PTC-124 induced PPT1 activity and reduction in thioester load in INCL cells, we sought to determine whether there is any change in GROD level in PTC124-treated cells, which could be appreciated by transmission electron microscopic (TEM) analysis. Accordingly, we performed TEM of untreated and PTC124-treated (for 1 week) INCL lymphoblasts and analyzed the GRODs. The results showed that compared with the untreated cells (Fig. 4, upper panels) the PTC124-treated cells contained appreciably lower number of GRODs (Fig. 4, lower panels). These results suggest that even a small increase in PPT1 activity in PTC124-treated cells can reduce the level of lipid thioesters as well as the number of GRODs in INCL lymphoblasts carrying PPT1 nonsense mutations.
Figure 4. Level of GRODs in INCL lymphoblasts treated with PTC124.
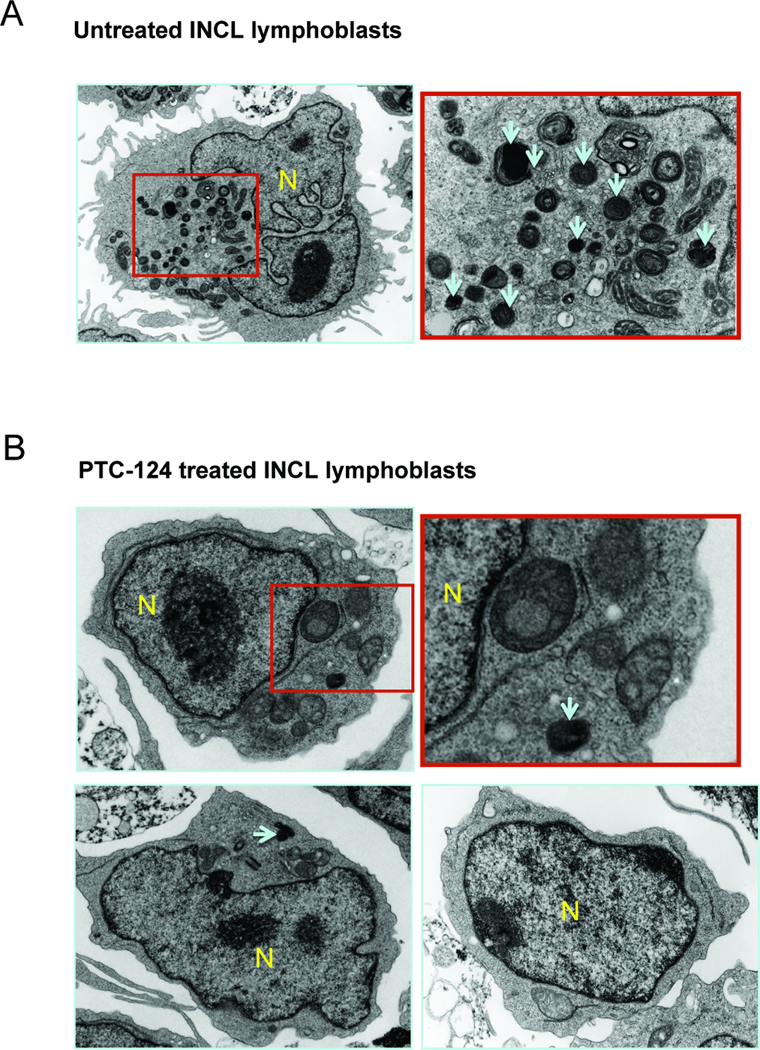
INCL lymphoblasts were cultured with or without PTC124 (5 µg/ml) for 1 week. Cells were then fixed, processed and analyzed by transmission electron microscopy. The upper panel represents untreated control cells and the lower panel represents PTC124-treated cells. Arrows indicate GRODs; N = nucleus.
3.6 PTC124 suppresses apoptosis in INCL cells carrying PPT1 nonsense mutations
Since it has been reported that CNS neurons in the brain of INCL patients (24) as well as those in Ppt1-KO mice that mimic INCL (25–31) and cultured lymphoblasts from INCL patients (23) undergo increased levels of apoptosis, we sought to determine whether PTC124 treatment suppressed apoptosis. Accordingly, we treated INCL lymphoblasts with PTC124 and compared the level of apoptosis with that of untreated lymphoblasts by FACS analysis using annexin-V, which labels the apoptotic cells. The results showed that within only one week of treatment with PTC124 there is an appreciable decrease in the level of apoptotic cells (Fig. 5 A–C).
Figure 5. PTC124 reduces apoptosis level in INCL cells.
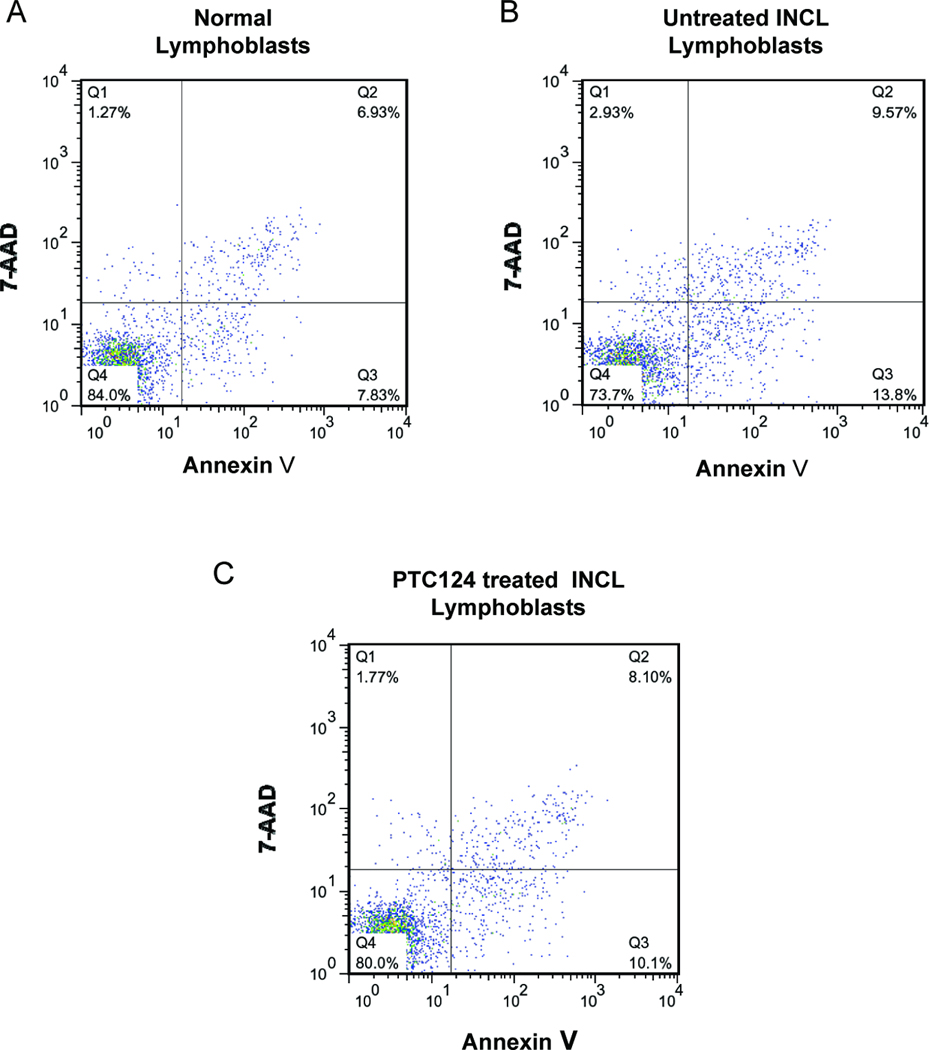
Normal and INCL lymphoblasts were cultured with (5 µg/ml) or without PTC124 for 1 week. Percentage of apoptotic cells in normal lymphoblasts (A), INCL lymphoblast treated without (B) or with PTC 124 (C) were determined by FACS analysis using annexin-V. Q4 represents the viable cells. Q2 + Q3 represent apoptotic cells. 7-AAD staining in Q1 represents dead cells.
4. Discussion
In the present study, we tested PTC124 on cultured cells from INCL patients carrying nonsense mutations in the PPT1 gene and demonstrated that this drug induces PPT1 enzymatic activity, albeit at a modest level compared with the activity in normal cells. Remarkably, even with this low level of PPT1 activity the PTC124-treated cells showed a modest reduction in thioester load in these cells and a lower level of GRODs characteristically found in this disease. We also noted that compared with untreated INCL cells PTC124-treatment caused an increase in cell viability (Supplemental Fig. 4). Taken together, these results show that PTC124 is non-toxic, and induction of modest PPT1 activity can mediate a reduction in ceroid levels, reduced GRODs and decreased level of apoptosis in INCL cells carrying nonsense PPT1 mutations.
It has been reported that premature termination codons (PTCs), which generate premature stop codons causing truncated protein production, account for approximately 33% of alleles that underlie various genetic disorders (reviewed in 8, 9, 11). PTCs may have a variety of origins including frame-shift deletions, insertions or nonsense mutations. Nonsense mutations generate truncated, nonfunctional, or proteins with harmful effects due to gain-of function or dominant negative functions. These mutations in the Ppt1 gene have been reported in the majority of the INCL patient population studied in the US (10). These mutations cause premature translational termination, which results in the lack of PPT1 activity leading to INCL pathogenesis.
In genetic diseases such as cystic fibrosis (CF) increased level of specific gene products from <1% to as little as 5% of normal may greatly reduce the severity or eliminate the principal manifestations of the disease (12, 13, reviewed in 32). In lysosomal storage diseases, even a small increase in enzyme activity can dramatically influence the clinical phenotype, especially if a drug can facilitate the production of some normal enzyme, such that a read-through approach can be a good therapeutic strategy (reviewed in 33). Previously, aminoglycosides have been identified to suppress nonsense alleles in Duchenne muscular dystrophy and cystic fibrosis (15, 16). However, these drugs have serious side effects at therapeutically relevant concentrations (11). PTC124 is not structurally unrelated to aminoglycosides or other drugs (15). Recently, it has been demonstrated that PTC124, which is a non-toxic compound, suppresses nonsense mutations in the gene encoding CFTR. It was found that oral administration of PTC124 reduces the epithelial electrophysiological abnormalities caused by the CFTR mutation (18). Moreover, the safety and tolerability of PTC124 have also been reported (19). Further, it has been shown that this drug selectively induces ribosomal read through of premature stop codons and does not affect the normal termination codons (15). Additionally, PTC124 has been reported to be efficacious in a mouse model of muscular dystrophy (mdx mice) and to some extent in clinical trials for Duchenne muscular dystrophy (DMD) (15). Indeed, these studies have been suggested to represent paradigm-forming approaches to personalized medicine with the potential to lead to life-changing treatments for hereditary diseases such as DMD (reviewed in 32). Our results demonstrated that PTC124 promotes read-through of INCL-related nonsense mutations in the PPT1 gene similar to gentamycin but it has virtually no toxicity.
As stated earlier, INCL is a neurodegenerative lysosomal storage disease and a therapeutic agent must be able to cross the blood-brain barrier (BBB). Remarkably, disruption of the BBB is a common manifestation of most neurodegenerative disorders (34), allowing blood-borne materials, including small molecules like PTC124 and immune cells to enter the brain. We recently discovered that in Ppt1-knockout (Ppt1-KO) mice (35), a reliable animal model of INCL (36), BBB is disrupted (unpublished results). If this is the case in INCL patients, small molecules such as PTC124 would readily cross the BBB. While during early stages of treatment PTC124 may enter the brain without hindrance, improvement of the BBB due to this treatment may prevent subsequent entrance of these molecules once the BBB may be impermeable with its improved status. However, PTC124 is a small molecule which is hydrophobic and may cross the BBB even when it is not disrupted. These are some of the important questions that need to be answered before we can adequately assess therapeutic potential of this small molecule for neurodegenerative storage disorders like INCL. Generation of mice carrying Ppt1 nonsense mutations may allow testing of PTC124 in vivo to determine its therapeutic potential. Our current efforts are directed towards generating a mouse model of INCL carrying nonsense mutations in the Ppt1 gene.
Supplementary Material
Acknowledgements
We thank S.W. Levin, J.Y. Chou and I. Owens for critical review of the manuscript and helpful suggestions. We also thank H.-S.Jun for his help and suggestions for conducting the FACS analyses. This research was supported in part by the Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health and by a grant from the Batten Disease Support and Research Association (BDSRA).
Abbreviations
- NCL
Neuronal ceroid lipofuscinosis
- INCL
Infantile neuronal ceroid lipofuscinosis
- PTC
Premature stop codon
- GRODS
Granular osmiophilic deposits
- CFTR
Cystic fibrosis transmembrane regulator
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
The authors declare no conflict of interest.
References
- 1.Goebel HH, Wisniewski KE. Current state of clinical and morphological features in human NCL. Brain Pathol. 2004;14:61–69. doi: 10.1111/j.1750-3639.2004.tb00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mole SE. The genetic spectrum of human neuronal ceroid-lipofuscinoses. Brain Pathol. 2004;14:70–76. doi: 10.1111/j.1750-3639.2004.tb00500.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Haltia M. The neuronal ceroid-lipofuscinoses: from past to present. Biochim Biophys Acta. 2006;1762:850–856. doi: 10.1016/j.bbadis.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 4.Jalanko A, Tyynelä JL, Peltonen From genes to systems: new global strategies for the characterization of NCL biology. Biochim Biophys Acta. 2006;1762:934–944. doi: 10.1016/j.bbadis.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 5.Hobert JA, Dawson G. Neuronal ceroid lipofuscinoses therapeutic strategies: past present and future. Biochim Biophys Acta. 2006;1762:945–953. doi: 10.1016/j.bbadis.2006.08.004. [DOI] [PubMed] [Google Scholar]
- 6.Cooper JD, Russell C, Mitchison HM. Progress towards understanding disease mechanisms in small vertebrate models of neuronal ceroid lipofuscinosis. Biochim Biophys Acta. 2006;1762:873–889. doi: 10.1016/j.bbadis.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 7.Vesa J, Hellsten E, Verkruyse LA, Camp LA, Rapola J, Santavuori P, Hofmann SL, Peltonen L. Mutations in the palmitoyl protein thioesterase gene causing infantile neuronal ceroid lipofuscinosis. Nature. 1995;376:584–587. doi: 10.1038/376584a0. [DOI] [PubMed] [Google Scholar]
- 8.Frischmeyer PA, Dietz HC. Nonsense-mediated mRNA decay in health and disease. Hum Mol Genet. 1999;8:1893–1900. doi: 10.1093/hmg/8.10.1893. [DOI] [PubMed] [Google Scholar]
- 9.Mendell JT, Dietz HC. When the message goes awry: disease-producing mutations that influence mRNA content and performance. Cell. 2001;107:411–414. doi: 10.1016/s0092-8674(01)00583-9. [DOI] [PubMed] [Google Scholar]
- 10.Das AK, Becerra CH, Yi W, Lu JY, Siakotos AN, Wisniewski KE, Hofmann SL. Molecular genetics of palmitoyl-protein thioesterase deficiency in the U.S. J Clin Invest. 1998;102:361–370. doi: 10.1172/JCI3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Linde L, Kerem B. Introducing sense into nonsense in treatments of human genetic diseases. Trends Genet. 2008;24:552–563. doi: 10.1016/j.tig.2008.08.010. [DOI] [PubMed] [Google Scholar]
- 12.Howard M, Frizzell RA, Bedwell DM. Aminoglycoside antibiotics restore CFTR function by overcoming premature stop mutations. Nat Med. 1996;2:467–469. doi: 10.1038/nm0496-467. [DOI] [PubMed] [Google Scholar]
- 13.Kerem E. Pharmacologic therapy for stop mutations: how much CFTR activity is enough? Curr Opin Pul Med. 2004;10:547–552. doi: 10.1097/01.mcp.0000141247.22078.46. [DOI] [PubMed] [Google Scholar]
- 14.Brooks DA, Muller VJ, Hopwood JJ. Stop-codon read-through for patients affected by a lysosomal storage disorder. Trends Mol Med. 2006;12:367–373. doi: 10.1016/j.molmed.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 15.Welch EM, Barton ER, Zhuo J, Tomizawa Y, Friesen WJ, Trifillis P, Paushkin S, Patel M, Trotta CR, Hwang S, et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature. 2007;447:87–91. doi: 10.1038/nature05756. [DOI] [PubMed] [Google Scholar]
- 16.Du M, Liu X, Welch EM, Hirawat S, Peltz SW, Bedwell DM. PTC124 is an orally bioavailable compound that promotes suppression of the human CFTR-G542X nonsense allele in a CF mouse model. Proc Natl Acad Sci USA. 2008;105:2064–2069. doi: 10.1073/pnas.0711795105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goldmann T, Overlack N, Wolfrum U, Nagel-Wolfrum K. PTC124-Mediated Translational Readthrough of a Nonsense Mutation Causing Usher Syndrome Type 1C. Hum. Gene. Ther. 2011 Jan 14; doi: 10.1089/hum.2010.067. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 18.Kerem E, Hirawat S, Armoni S, Yaakov Y, Shoseyov D, Cohen M, Nissim-Rafinia M, Blau H, Rivlin J, Aviram M, et al. Effectiveness of PTC124 treatment of cystic fibrosis caused by nonsense mutations: a prospective phase II trial. Lancet. 2008;372:719–727. doi: 10.1016/S0140-6736(08)61168-X. [DOI] [PubMed] [Google Scholar]
- 19.Hirawat S, Welch EM, Elfring GL, Northcutt VJ, Paushkin S, Hwang S, Leonard EM, Almstead NG, Ju W, Peltz SW, et al. Safety, tolerability and pharmacokinetics of PTC124, a nonaminoglycoside nonsense mutation suppressor, following single and multiple-dose administration to healthy male and female adult volunteers. J Clin Pharmacol. 2007;47:430–444. doi: 10.1177/0091270006297140. [DOI] [PubMed] [Google Scholar]
- 20.Mossman T. Rapid colorimetric assay for cellular growth and survival. J. Immunol. Method. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 21.van Diggelen OP, Keulemans JLM, Winchester B, Hofman IL, Vanhanen SL, Santavuori P, Voznyi YV. A rapid fluorogenic palmitoyl-protein thioesterase assay: pre- and postnatal diagnosis of INCL. Mol. Genet. Metab. 1999;66:240–244. doi: 10.1006/mgme.1999.2809. [DOI] [PubMed] [Google Scholar]
- 22.Lu JY, Verkruyse LA, Hofmann SL. Lipid thioesters derived from acylated proteins accumulate in infantile neuronal ceroid lipofuscinosis: correction of the defect in lymphoblasts by recombinant palmitoyl-protein thioesterase. Proc Natl Acad Sci U S A. 1996;93:10046–10050. doi: 10.1073/pnas.93.19.10046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Z, Butler JD, Levin SW, Wisniewski KE, Brooks SS, Mukherjee AB. Lysosomal ceroid depletion by drugs: therapeutic implications for a hereditary neurodegenerative disease of childhood. Nat Med. 2001;7:478–484. doi: 10.1038/86554. [DOI] [PubMed] [Google Scholar]
- 24.Riikonen R, Vanhanen SL, Tyynela J, Santavuori P, Turpeinen U. CSF insulin-like growth factor-1 in infantile neuronal ceroid lipofuscinosis. Neurology. 2000;54:1828–1832. doi: 10.1212/wnl.54.9.1828. [DOI] [PubMed] [Google Scholar]
- 25.Gupta P, Soyombo AA, Atashband A, Wisniewski KE, Shelton JM, Richardson JA, Hammer RE, Hofmann SL. Disruption of PPT1 or PPT2 causes neuronal ceroid lipofuscinosis in knockout mice. Proc Natl Acad Sci USA. 2001;98:13566–13571. doi: 10.1073/pnas.251485198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bible E, Gupta P, Hofmann SL, Cooper JD. Regional and cellular neuropathology in the palmitoyl protein thioesterase-1 null mutant mouse model of infantile neuronal ceroid lipofuscinosis. Neurobiol Dis. 2004;16:346–359. doi: 10.1016/j.nbd.2004.02.010. [DOI] [PubMed] [Google Scholar]
- 27.Zhang Z, Lee YC, Kim SJ, Choi MS, Tsai PC, Xu Y, Xiao YJ, Zhang P, Heffer A, Mukherjee AB. Palmitoyl-protein thioesterase-1 deficiency mediates the activation of the unfolded protein response and neuronal apoptosis in INCL. Hum. Mol. Genet. 2006;15:337–346. doi: 10.1093/hmg/ddi451. [DOI] [PubMed] [Google Scholar]
- 28.Kim SJ, Zhang Z, Lee YC, Mukherjee AB. Palmitoyl-protein thioesterase-1 deficiency leads to the activation of caspase-9 and contributes to rapid neurodegeneration in INCL. Hum. Mol. Genet. 2006;15:1580–1586. doi: 10.1093/hmg/ddl078. [DOI] [PubMed] [Google Scholar]
- 29.Kim SJ, Zhang Z, Hitomi E, Lee YC, Mukherjee AB. Endoplasmic reticulum stress-induced caspase-4 activation mediates apoptosis and neurodegeneration in INCL. Hum. Mol. Genet. 2006;15:1826–1834. doi: 10.1093/hmg/ddl105. [DOI] [PubMed] [Google Scholar]
- 30.Wei H, Kim SJ, Zhang Z, Tsai PC, Wisniewski KE, Mukherjee AB. ER and oxidative stresses are common mediators of apoptosis in both neurodegenerative and non-neurodegenerative lysosomal storage disorders and are alleviated by chemical chaperones. Hum. Mol. Genet. 2007;17:469–477. doi: 10.1093/hmg/ddm324. [DOI] [PubMed] [Google Scholar]
- 31.Wei H, Zhang Z, Saha A, Peng S, Chandra G, Quezado Z, Mukherjee AB. Disruption of adaptive energy metabolism and elevated ribosomal p-S6K1 levels contribute to INCL pathogenesis: partial rescue by resveratrol (2011) Hum Mol Genet. 2011 Jan 13; doi: 10.1093/hmg/ddq555. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nelson SF, Crosbie RH, Miceli MC, Spencer MJ. Emerging genetic therapies to treat Duchenne muscular dystrophy. Curr Opin Neurol. 2009;22:532–238. doi: 10.1097/WCO.0b013e32832fd487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wilschanski M, Miller LL, Shoseyov D, Blau H, Rivlin J, Aviram M, Cohen M, Armoni S, Yaakov Y, Pugatch T, Cohen-Cymberknoh M, Miller NL, Reha A, Northcutt VJ, Hirawat S, Donnelly K, Elfring GL, Ajayi T, Kerem E. Chronic ataluren (PTC124) treatment of nonsense mutation cystic fibrosis. E. Eur Respir J. 2011 Jan 13; doi: 10.1183/09031936.00120910. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 34.Zlokovic BV. B. V. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57:178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 35.Gupta P, Soyombo AA, Atashband A, Wisniewski KE, Shelton JM, Richardson JA, Hammer RE, Hofmann SL. Disruption of PPT1 or PPT2 causes neuronal ceroid lipofuscinoses in knockout mice. Proc. Natl. Acad. Sci. U S A. 2001;98:13566–13571. doi: 10.1073/pnas.251485198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bible E, Gupta P, Hofmann SL, Cooper JD. Regional and cellular neuropathology in the palmitoyl protein thioesterase-1 null mutant mouse model of infantile neuronal ceroid lipofuscinoses. Neurobiol Dis. 2004;16:346–359. doi: 10.1016/j.nbd.2004.02.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.