

Abstract
Proteases constitute a major class of drug targets. Endosomal compartments harbor several protease families whose attenuation may be beneficial to a number of biological processes, including inflammation, cancer metastasis, antigen presentation, and parasite clearance. As a step toward the goal of generalized but targeted protease inhibition in the endocytic pathway, we describe here the synthesis, characterization, and cellular application of a novel multifunctional protease inhibitor. We show that pepstatin A, a potent but virtually insoluble inhibitor of cathepsins D and E, can be conjugated to a single site on cystatin C, a potent inhibitor of the papain-like cysteine proteases (PLCP) and of asparagine endopeptidease (AEP), to create a highly soluble compound capable of suppressing the activity of all 3 principal protease families found in endosomes and lysosomes. We demonstrate that this cystatin–pepstatin inhibitor (CPI) can be taken up by cells to modulate protease activity and affect biological responses.
Protease inhibitors have emerged as a powerful drug class.(1) They include the inhibitors of angiotensin converting enzyme, inhibitors of HIV proteases, and proteasomal inhibitors such as Bortezomib (Velcade) used to treat multiple myeloma.(2)
The proteases of the endolysosomal pathway have frequently been proposed as therapeutic targets as they play important roles in the regulation of a wide variety of biological systems.(3) For example, lysosomal cysteine and aspartyl proteases are validated drug targets in several trypanosome species,(4) and the upregulation of certain endosomal proteases is associated with increased malignancy.(5) Asparagine endopeptidase (AEP or legumain) has also been implicated in the progression of malignant melanoma,(6) in the destruction of the therapeutic drug l-asparaginase,(7) and in neuroexitotoxity.(8) Down-regulation of cystatins, which are natural cysteine protease inhibitors, can lead to increased malignancy(9) and faulty immune responses.(10) High expression of cathepsin D (Cat D) in non-Hodgkin’s lymphoma has also been associated with increased malignancy(11) and is also associated with poor prognosis in breast cancer.(12) A further potential therapeutic application of endosomal protease inhibitors would be immune modulation since several recent studies demonstrate that the proteolytic activity in endosomes of antigen presenting cells may be too high, leading to antigen destruction and inefficient presentation to T-cells. Consequently, protease-resistant antigens often elicit more robust immune responses.13,14
Taken together, it seems that effective down-modulators of endo/lysosomal protease activity could be a valuable addition to the therapeutic armory. However, to date modulation of endo/lysosomal protease function has remained challenging, as there are multiple families of endosomal proteases with an extensive functional redundancy.(15) As an additional problem, there is evidence in the literature that the knock-down/inhibition of specific proteases leads to the upregulation of others.3,16,17
Most endosomal proteases belong to 3 distinct families. There are several papain-like cysteine proteases (PLCPs), including cathepsin L, S, B, C as well as several others.(18) Alongside these there are the aspartyl proteases related to pepsin: cathepsins D and E. Finally, there is an additional cysteine protease termed asparaginyl endopeptidase (AEP) or legumain that is more closely related to the caspases.(19) Each of these 3 classes can be inhibited by distinct and nonoverlapping small molecule inhibitors,20,21 but in vivo inhibition, or knockout, of these proteases frequently shows limited or no phenotype, most likely due to functional redundancy. We thus postulated that inhibiting all three families of endosomal proteases would provide a powerful tool for modulating endo/lysosomal function.
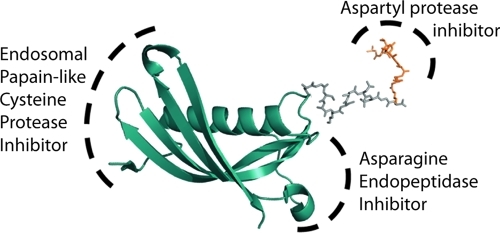
PLCPs and AEP are potently inhibited by a naturally occurring 14 kDa protein, cystatin C. The cystatins are a family of small proteins that inhibit PLCPs with subnanomolar affinity.(22) They are present in the bloodstream and are believed to play a role in the mopping up of proteases released during physiological and pathological responses. Importantly, cystatin C, as well as several family members, inhibit AEP via a distinct binding site with a Ki of 0.20 nM(23) (Figure 1). Cystatin C thus represents an excellent scaffold for the synthesis of a pan-endosomal protease inhibitor.
Figure 1.

Modeling of a cystatin–pepstatin inhibitor conjugate (CPI) as a potential inhibitor of all 3 major endosomal protease families: the papain-like cysteine proteases (PLCP), aspartyl proteases (Cat-D/E), and asparagine endopeptidase (AEP) are inhibited through 3 different motifs on the construct.
Cathepsin D and E, the endosomal aspartyl proteases, are inhibited with a Ki of 0.1 nM by pepstatin A,(24) an isopeptide first isolated from Actinomyces. Its major drawback is its virtual insolubility in aqueous media.(24) Nonetheless it is still widely used even in cell-based assays because more soluble alternatives are not readily available. Several attempts have been made to address this problem, such as conjugating pepstatin A to asialoglycoprotein(ASGP)(25) or to poly(ethylene glycol)(26) or more recently by mannosylating it, or conjugating it to mannosylated bovine serum albumin.27,28 PEG-ylation of pepstatin reduces its inhibitory potential 400-fold, and conjugating to mannosylated BSA reduces the Ki 10-fold, whereas conjugation to ASGP renders pepstatin inactive until the protein backbone is digested. Conjugation of pepstatin to peptides or fluorescent moieties did not significantly alter its inhibitory potential.29,30
In a novel approach, we decided to use cystatin C as the solubilizing agent for pepstatin A via a reducible peptide spacer linkage, thereby creating a highly soluble “Swiss army knife” protease inhibitor (figure 1) capable of suppressing the activity of all 3 major protease families that populate endosomes and lysosomes and that control their biological functions.(31)
Results and Discussion
We wanted tight control over the stoichiometry and localization of the introduced pepstatin, with no more than one pepstatin molecule per cystatin at a site away from the inhibitory domains of cystatin C (see Supplementary Figure S1a), and thus introduced a free cysteine into the protein backbone by site-directed mutagenesis,32,33 as it can be selectively modified in the presence of other nucleophilic residues. Issues associated with disulfide scrambling with the two existing disulfide bridges in cystatin C were avoided by using a mammalian expression system. Various mutants were tested (see Supplementary Figure 1a–c), and T102C was found to have the most favorable inhibitory properties. A C-terminal 6His-tag was also introduced, for purification and possible conjugation of the inhibitor to a solid phase carrier.
We chose to use methanethiosulfonate chemistry to introduce the pepstatin onto the free cysteine of cystatin C,33,34 due to its high selectivity for sulfhydryls and its facile introduction into the peptide backbone through a MTS-Boc-cysteine building block. Furthermore there is the potential for endosomal release of the pepstatin moiety by reduction of disulfides by the lysosomal thiol reductase GILT.(35)
Initial efforts focusing on an analogue with an ethyl spacer between pepstatin and the MTS moiety resulted in <5% protein recovery due to poor solubility of the resulting conjugate. As it has been reported that conjugation of the C-terminal end of pepstatin to lysine residues did not reduce its inhibitory potential significantly,29,30 we decided to introduce a charged peptide between the pepstatin and the MTS group (Figure 2) to increase solubility of the conjugate. It was found that a mild reduction (5 mM DTT) was needed prior to the modification reaction as the free cysteine had become modified during the expression and purification process (Supplementary Figure S2). This improved protein recovery levels to >95% with >90% modification as determined by mass spectrometry and SDS-PAGE analysis (Figure 2 and Supporting Information).
Figure 2.

Conjugation of pepstatin A to cystatin C T102C through a solubilizing peptide carrying a C-terminal MTS-cysteine residue. Conditions: (i) EDC/HOSu in DMF 96% yield; (ii) Fmoc-lysine/DMF, 89% yield; (iii) Fmoc-based solid phase peptide synthesis; (iv) PBS with 6% v/v DMSO, >90% conversion, 95% protein recovery.
Next, we used fluorescent substrates that are cleaved specifically by members of each of the three families of proteases to analyze the inhibitory capacity of our cystatin–pepstatin inhibitor (CPI; Figure 3a,b). When looking at the potential of CPI to inhibit recombinant enzymes, we found it inhibited the PLCP cathepsin L, AEP, and cathepsin D with a similar IC50 as wt-cystatin C and pepstatin A. Moreover, the CPI was also able to inhibit the 3 classes of protease activity present in dendritic cell lysates (Figure 3b). When the CPI conjugate was incubated with live mouse dendritic cells and their protease activity was determined postlysis, we found that the probe could simultaneously abolish cathepsin D/E activity as well as reduce PLCP and AEP activity by >90% (Supplementary Figure S3) with no cell death occurring (as determined by trypan blue assay).
Figure 3.

(a) Residual protease activity in fluorescence units per minute of recombinant cathepsin L, AEP, and cathepsin D in the presence of varying concentrations of CPI, cystatin, or pepstatin as measured using fluorescent substrates specific for each protease. (b) Residual activity of the same enzymes as measured in lysates derived from dendritic cells using the same fluorescent substrates as in panel a. Note that only CPI inhibits all 3 protease activities. (c) Fluorescence microscopy of quenched casein substrate (Enzcheck). Upon hydrolysis, the Enzcheck fluorophore is dequenched and emits green fluorescence, indicating protease activity. A20 antigen-presenting cells (APCs) were incubated with the Enzcheck substrate and treated in the absence or presence of CPI, cystatin, or pepstatin. Representative images of fixed cells immuno-stained for the late endosomal marker CD63 (red) are shown (scale bar, 10 μm). (d) Quantification of initial rates of Enzcheck fluorescence emergence in live A20 cells as measured on an Envision fluorimeter. Quenched Bodipy-casein (Enzcheck) was incubated with A20 cells in the presence or absence of CPI, cystatin C. Error bars represent SD based on a representative experiment performed in triplicate.
We next wanted to visualize the protease inhibition in live cells, rather than postlysis. For this we used a heavily BODIPY dye labeled casein construct (Enzcheck substrate). The high density of BODIPY groups results in the fluorescence being quenched. Upon proteolysis, however, the degradation of the protein backbone results in the dequenching of the BODIPY groups, resulting in the emergence of fluorescence.(36) First we had to demonstrate that this substrate was susceptible to cleavage by all three families of proteases, as some proteins are only cleaved (in vitro at least) by specific protease families.14,15 To test this we incubated the quenched fluorophore with mouse macrophage lysosomes, which are known to be rich in proteases from all three families. Indeed it was observed that the substrate was stabilized more effectively by CPI, relative to a PBS-control, than it was by cystatin C and pepstatin A alone (Supplementary Figure S4).
For the live cell assays we used the murine antigen presenting cell line A20, since it is resistant to relatively high levels of unconjugated pepstatin A needed for these experiments, which on the time scale for this experiment would be toxic to primary dendritic cell cultures.(37) It also possesses high levels of all three families of proteases. When the cells were treated with PBS, we observed the dequenching of the substrate by confocal microscopy (Figure 3c) in approximately 8% of all cells after 5 h. Longer timecourses resulted in extensive cell death in pepstatin-treated samples. When the component inhibitor inhibitor cystatin C or pepstatin A was added, this number was reduced to approximately 4%. When CPI was added together with the quenched casein <1% of cells became fluorescent. Proteolytic processing of BODIPY-casein could thus be stopped by the addition of the CPI. We also confirmed this observation by measuring the emergence of BODIPY fluorescence by fluorimetry. When A20 cells were incubated with or without the inhibitors and BODIPY fluorescence was measured at a the 5 h time point (Figure 3d), it was again observed that the BODIPY casein was dequenched more slowly in the presence of the CPI than it was in the presence of cystatin C or pepstatin A alone.
One of the potential therapeutic applications of the CPI is as a modulator of antigen processing. It has been reported that that protease-resistant antigens can make better immunogens and that reducing levels of proteases can boost antigen presentation. For example, in a recent study by Delamarre et al.(13) it was demonstrated that a heme-free form of horseradish peroxidase (apo-HRP) was more sensitive to lysosomal proteolysis than wild-type, heme-containing HRP (wt-HRP) in vitro. In vivo, apo-HRP induced much weaker immune responses than wt-HRP, leading the authors to suggest that apo-HRP was degraded too rapidly by the antigen processing machinery, preventing efficient loading of MHC complexes. We asked whether we could protect unstable apo-HRP from lysosomal degradation in vitro by adding the CPI. As a source of endolysosomal proteases we again used purified lysosomes from mouse macrophages. Indeed, after a 6 h incubation, apo-HRP was completely degraded (Figure 4a). In fact, more than 90% of the protein was degraded even after a 1 h incubation (data not shown). In contrast, a substantial fraction of wt-HRP remained intact after 6 h. Addition of cystatin C or pepstatin A individually produced only modest stabilization of apo-HRP (Figure 4a; left, lanes 3 and 5), indicating a functional redundancy between the lysosomal enzymes in these macrophages. However, addition of CPI stabilized apo-HRP dramatically such that after 6 h the amount remaining was similar to wt-HRP digested in the absence of CPI (Figure 4a; left, lane 4). These results suggest that incorporation of CPI into immunological adjuvants for unstable antigens may be worthwhile and is currently under investigation.
Figure 4.

(a) Protection of a destabilized variant of HRP (apo-HRP) and stable wild-type HRP (wt-HRP) from degradation by macrophage lysosomes (6 h incubation at 37 °C; pH 4.5). Inhibitors added as indicated at a final concentration of 5 μM. Only CPI protects the unstable variant apo-HRP (lane 4). Cystatin C (lane 3) and pepstatin A (lane 5) offer only partial protection against degradation of this destabilized protein. wt-HRP is largely stable in the presence of macrophage lysosomes (lanes 7–11). (b) CPI arrests EGF receptor downregulation and sustains signaling. COS7 cells were stimulated with EGF for up to 90 min following a preincubation in the presence or absence of CPI or cystatin C (15 μM final concentration). The downregulation of the EGF receptor (top panel) was assayed by Western blotting along with levels of phospho-Erk1/2 (p42/44 MAP kinase; bottom panel) and Rsk2 to assess total cellular protein loading (middle panel). (c) EGFR abundance throughout the treatment time course relative to t = 0 (left panel); phosphorylated p42/44 abundance throughout the treatment time course relative to t = 20 min (right panel).
We next assessed the capacity of CPI to inhibit endo/lysosomal proteases in live cells and whether it could modulate the biological functions of this compartmental system. One important role of the endocytic pathway is to degrade activated growth factor receptors following their ligand-stimulated endocytosis. For example, following ligand stimulation, the EGF receptor (EGFR) is ubiquitinated, clustered in clathrin coated pits, and delivered to the endosome system where it becomes sequestered on the internal vesicles of multivesicular bodies (MVBs) preventing recycling and shutting down its capacity to signal.38−40 MVBs then fuse with lysososomes, and the EGF receptor is degraded, although the specific lysosomal proteases that degrade EGF receptor remain to be fully defined, with the cysteine proteases all implicated.(11)
We preincubated the EGFR-positive kidney cell line COS-7 in the presence or absence of CPI or cystatin C (the insolubility of pepstatin A prevented this compound from giving meaningful data in this experiment) and then challenged with EGF. At different times following treatment the total cellular levels of EGFR remaining were monitored by Western blotting (Figure 4b). In control cells downregulation of EGFR was evident after 40 min and virtually complete after 90 min. By contrast, EGFR levels persisted in cells preincubated with CPI, demonstrating a block in receptor degradation (Figure 4c; left panel). Preincubation with cystatin C also suppressed EGFR downregulation, but not to the same extent as CPI, indicating that both cysteine and aspartyl proteases are involved in EGFR processing. The arrest in receptor degradation was not due to inhibitor toxicity since the MAP kinases Erk1/2 were activated normally in CPI and cystatin C treated cells (Figure 4b). In fact, sustained Erk activation was seen in the presence of CPI (Figure 4c; right panel), consistent with the prolonged half-life of EGFR observed in the same samples (left panel).
Perturbations in the proteolytic capacity of endosomes may not only impinge on protein turnover within but also affect progression of material along the endocytic subcompartments. To assess the effect(s) of CPI on endocytosis of cell surface receptors, we examined ligand-mediated trafficking of EGFR. HeLa cells expressing high endogenous levels of EGFR were pretreated in the presence or absence of CPI and stimulated with EGF for 5 and 90 min to allow observation of early and late events in receptor endocytosis, respectively (Figure 5). Following treatment, samples were fixed and immunostained for EGFR (green) and the transferrin receptor (TrfR, red). TrfR, also abundant in HeLa cells, is known to constitutively endocytose and recycle(41) but unlike EGFR does not travel downstream to the proteolytic late endosomes. It is therefore commonly used as a marker of early endosomes. As observed by confocal immuno-fluorescence microscopy, disappearance of EGFR following ligand treatment was substantially reduced in the presence of CPI relative to the control (Figure 5a, bottom panels), while the initial levels of internalized receptor appeared comparable (top panels). As stability of TrfR is unaffected by EGF stimulation, EGFR abundance in this assay was quantified relative to that of TrfR in the same cells (Figure 5b). These data further substantiate the CPI-dependent attenuation of EGFR turnover in the Western blot assay (Figure 4b), as the presence of EGFR on early endosomes and adequate induction of signaling shortly following ligand addition rule out the possibility that preincubation with CPI interferes with receptor internalization or alternatively abolishes receptor access to the plasma membrane.
Figure 5.

Confocal images of EGFR degradation. Inhibition of endolysosomal proteases slows EGFR downregulation and alters receptor trafficking. (a) Immunofluorescence microscopy of HeLa cells preincubated in the presence or absence of CPI and stimulated with EGF as indicated. Fixed samples were immunostained against endogenous EGFR (left panels, green) and transferrin receptor (TrfR, middle panels, red) with merges of the two channels shown (right panels). Representative images taken under the same magnification and laser settings are shown with 6x inset magnification and scalebar corresponding to 10 μm. (b) Quantification of EGFR abundance expressed as a fraction of TrfR in the same cells is represented relative to no inhibitor, t = 5 min sample. (c) Quantification of colocalization between the two receptors expressed as a fraction of EGFR overlapping TrfR. Error bars for quantification correspond to SD calculated on the basis of 20–25 cells for each conditions using Student’s t test.
Persistent localization of transitory membrane cargo to TrfR-positive endosomes can be associated with blocked trafficking of EGFR toward the lysosome.(42) Importantly, nearly half of the EGFR remaining in the presence of inhibitor at 90 min after stimulation with EGF still colocalized with TrfR, an extent similar to that observed at the 5 min time-point under either pretreatment condition (Figure 5a, bottom panel; Figure 5c). By contrast, in control cells only a small fraction of residual EGFR was found on TrfR-positive endosomes at this late time-point. Taken together with the Western blot data, these results allow us to tentatively hypothesize that CPI retards the maturation of the EGFR-positive endosomes, thereby affording prolonged ligand-induced signaling.
In summary, we have presented the construction of a single molecular entity that inhibits all three major families of endo/lysosomal proteases. This broad inhibition has been shown to attenuate destructive processing of labile proteins in vitro and to attenuate proteolytic events within the endo/lysosomal pathway in vivo. CPI and variants of it may be therapeutically useful not only because of its broad inhibitory range but because it can be readily modified to permit targeting to specific cell types, thus avoiding toxicity associated with systemically administered small molecular protease inhibitors. Moreover, the spectrum of lysosomal cysteine proteases targeted can be manipulated through mutation of the PLCP and AEP interacting domains.23,43
Methods
Synthesis of the Cystatin–Pepstatin Conjugate (3)
Dithiothreitol (DTT) was added to a solution of cystatin C-T102C-6His (1 mg mL–1 in PBS, 2.5 mL) to a final concentration of 5 mM. The mixture was gently shaken at RT for 10 min and buffer exchanged into phosphate buffered saline (PBS) by Sephadex G-25 resin (GE Healthcare). The reduced protein (50 μM 3.5 mL) was added to a solution of 2 in DMSO (2 mM, 200 μL), and the mixture was gently shaken at RT for 2 h, after which SDS-PAGE and LC–MS analysis showed the formation of a single product. The protein was purified after modification using nickel affinity chromatography, followed by dialysis into PBS (6000–8000 MWCO, 3 × 4 L).
Acknowledgments
We would like to thank S. Matthews for his help in providing mouse dendritic cells. S.I.v.K. was the recipient of a Wellcome Trust Sir Henry Wellcome Fellowship and a NWO Veni Fellowship; C.W. and J.D.C. were funded by a Wellcome Trust Programme Grant to C.W. H.O. and I.B. are funded by The Netherlands Foundation for Scientific Research (NWO).
Supporting Information Available
Characterization of cystatin C mutants, synthesis of the pepstatin analogue, optimization of the reduction conditions of CysCT102C, and general experimental procedures. This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- Leung D.; Abbenante G.; Fairlie D. P. (2000) Protease inhibitors: Current status and future prospects. J. Med. Chem. 43, 305–341. [DOI] [PubMed] [Google Scholar]
- Richardson P. G.; Sonneveld P.; Schuster M. W.; Irwin D.; Stadtmauer E. A.; Facon T.; Harousseau J. L.; Ben-Yehuda D.; Lonial S.; Goldschmidt H.; Reece D.; San-Miguel J. F.; Bladé J.; Boccadoro M.; Cavenagh J.; Dalton W. S.; Boral A. L.; Esseltine D. L.; Porter J. B.; Schenkein D.; Anderson K. C. (2005) Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. New Engl. J. Med. 352, 2487–2498. [DOI] [PubMed] [Google Scholar]
- Reiser J.; Adair B.; Reinheckel T. (2010) Specialized roles for cysteine cathepsins in health and disease. J. Clin. Invest 120, 3421–3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle P. S.; Sajid M.; O’Brien T.; DuBois K.; Engel J. C.; Mackey Z. B.; Reed S. (2008) Drugs targeting parasite lysosomes. Curr. Pharm. Design 14, 889–900. [DOI] [PubMed] [Google Scholar]
- Palermo C.; Joyce J. A. (2008) Cysteine cathepsin proteases as pharmacological targets in cancer. Trends Pharmacol. Sci. 29, 22–28. [DOI] [PubMed] [Google Scholar]
- Briggs J. J.; Haugen M. H.; Johansen H. T.; Riker A. I.; Abrahamson M.; Fodstad O.; Maelandsmo G. M.; Solberg R. (2010) Cystatin E/M suppresses legumain activity and invasion of human melanoma. BMC Cancer 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel N.; Krishnan S.; Offman M. N.; Krol M.; Moss C. X.; Leighton C.; van Delft F. W.; Holland M.; Liu J. Z.; Alexander S.; Dempsey C.; Ariffin H.; Essink M.; Eden T. O. B.; Watts C.; Bates P. A.; Saha V. (2009) A dyad of lymphoblastic lysosomal cysteine proteases degrades the antileukemic drug L-asparaginase. J. Clin. Invest 119, 1964–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z. X.; Jang S. W.; Liu X.; Cheng D. M.; Peng J.; Yepes M.; Li X. J.; Matthews S.; Watts C.; Asano M.; Hara-Nishimura I.; Luo H. R.; Ye K. Q. (2008) Neuroprotective actions of PIKE-L by inhibition of SET proteolytic degradation by asparagine endopeptidase. Mol. Cell 29, 665–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keppler D. (2006) Towards novel anti-cancer strategies based on cystatin function. Cancer Lett. 235, 159–176. [DOI] [PubMed] [Google Scholar]
- Henskens Y. M.; Veerman E. C.; Nieuw Amerongen A. V. (1996) Cystatins in health and disease. Biol. Chem. 377, 71–86. [DOI] [PubMed] [Google Scholar]
- Nicotra G.; Manfroi F.; Follo C.; Castino R.; Fusco N.; Peracchio C.; Kerim S.; Valente G.; Isidoro C. (2010) High expression of cathepsin D in non-Hodgkin’s lymphomas negatively impacts on clinical outcome. Dis. Markers 28, 167–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liaudet-Coopman E.; Beaujouin M.; Derocq D.; Garcia M.; Glondu-Lassis M.; Laurent-Matha V.; Prebois C.; Rochefort H.; Vignon F. (2006) Cathepsin D: newly discovered functions of a long-standing aspartic protease in cancer and apoptosis. Cancer Lett. 237, 167–179. [DOI] [PubMed] [Google Scholar]
- Delamarre L.; Couture R.; Mellman I.; Trombetta E. S. (2006) Enhancing immunogenicity by limiting susceptibility to lysosomal proteolysis. J. Exp. Med. 203, 2049–2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss C. X.; Villadangos J. A.; Watts C. (2005) Destructive potential of the aspartyl protease cathepsin D in MHC class II-restricted antigen processing. Eur. J. Immunol. 35, 3442–3451. [DOI] [PubMed] [Google Scholar]
- Matthews S. P.; Werber I.; Deussing J.; Peters C.; Reinheckel T.; Watts C. (2010) Distinct protease requirements for antigen presentation in vitro and in vivo. J. Immunol. 184, 2423–2431. [DOI] [PubMed] [Google Scholar]
- Dennemärker J.; Lohmüller T.; Müller S.; Aguilar S. V.; Tobin D. J.; Peters C.; Reinheckel T. (2010) Impaired turnover of autophagolysosomes in cathepsin L deficiency. Biol. Chem. 391, 913–922. [DOI] [PubMed] [Google Scholar]
- Bednarski E.; Ribak C. E.; Lynch G. (1997) Suppression of cathepsins B and L causes a proliferation of lysosomes and the formation of meganeurites in hippocampus. J. Neurosci. 17, 4006–4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turk B.; Turk D.; Turk V. (2000) Lysosomal cysteine proteases: More than scavengers. Biochim. Biophys. Act. 1477, 98–111. [DOI] [PubMed] [Google Scholar]
- Manoury B.; Hewitt E. W.; Morrice N.; Dando P. M.; Barrett A. J.; Watts C. (1998) An asparaginyl endopeptidase processes a microbial antigen for class II MHC presentation. Nature 396, 695–699. [DOI] [PubMed] [Google Scholar]
- Otto H.-H.; Schirmeister T. (1997) Cysteine proteases and their inhibitors. Chem. Rev. 97, 133–172. [DOI] [PubMed] [Google Scholar]
- Rozman-Pungercar J.; Kopitar-Jerala N.; Bogyo M.; Turk D.; Vasiljeva O.; Stefe I.; Vandenabeele P.; Bromme D.; Puizdar V.; Fonovi M.; Trstenjak-Prebanda M.; Dolenc I.; Turk V.; Turk B. (2003) Inhibition of papain-like cysteine proteases and legumain by caspase-specific inhibitors: when reaction mechanism is more important than specificity. Cell Death Differ. 10, 881–888. [DOI] [PubMed] [Google Scholar]
- Abrahamson M.; Alvarez-Fernandez M.; Nathanson C. M. (2003) Cystatins. Biochem. Soc. Symp. 179–199. [DOI] [PubMed] [Google Scholar]
- Alvarez-Fernandez M.; Barrett A. J.; Gerhartz B.; Dando P. M.; Ni J.; Abrahamson M. (1999) Inhibition of mammalian legumain by some cystatins is due to a novel second reactive site. J. Biol. Chem. 274, 19195–19203. [DOI] [PubMed] [Google Scholar]
- Umezawa H.; Aoyagi T.; Morishima H.; Matsuzaki M.; Hamada M. (1970) Pepstatin, a new pepsin inhibitor produced by Actinomycetes. J. Antibiot. 23, 259–262. [DOI] [PubMed] [Google Scholar]
- Furuno K.; Miwa N.; Kato K. (1983) Receptor-mediated introduction of pepstatin-asialofetuin conjugate into lysosomes of rat hepatocytes. J. Biochem. 93, 249–256. [DOI] [PubMed] [Google Scholar]
- Brygier J.; Vlncentelli J.; Nljs M.; Guermant C.; Paul C.; Baeyens-Volant D.; Looze Y. (1994) Preparation and preliminary characterization of poly(ethylene glycol)-pepstatin conjugate. Appl. Biochem. Biotechnol. 47, 1–10. [DOI] [PubMed] [Google Scholar]
- Free P.; Hurley C. A.; Kageyama T.; Chain B. M.; Tabor A. B. (2006) Mannose-pepstatin conjugates as targeted inhibitors of antigen processing. Org. Biomol. Chem. 4, 1817–1830. [DOI] [PubMed] [Google Scholar]
- Raiber E. A.; Tulone C.; Zhang Y.; Martinez-Pomares L.; Steed E.; Sponaas A. M.; Langhorne J.; Noursadeghi M.; Chain B. M.; Tabor A. B. (2010) Targeted delivery of antigen processing inhibitors to antigen presenting cells via mannose receptors. ACS Chem. Biol. 5, 461–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C. S.; Chen W. N. U.; Zhou M.; Arttamangkul S.; Haugland R. P. (2000) Probing the cathepsin D using a BODIPY FL-pepstatin A: Applications in fluorescence polarization and microscopy. J. Biochem. Biophys. Methods 42, 137–151. [DOI] [PubMed] [Google Scholar]
- Zaidi N.; Burster T.; Sommandas V.; Herrmann T.; Boehm B. O.; Driessen C.; Voelter W.; Kalbacher H. (2007) A novel cell penetrating aspartic protease inhibitor blocks processing and presentation of tetanus toxoid more efficiently than pepstatin A. Biochem. Biophys. Res. Commun. 364, 243–249. [DOI] [PubMed] [Google Scholar]
- Hsing L. C.; Rudensky A. Y. (2005) The lysosomal cysteine proteases in MHC class II antigen presentation. Immunol. Rev. 207, 229–241. [DOI] [PubMed] [Google Scholar]
- Chalker J. M.; Bernardes G. J. L.; Lin Y. A.; Davis B. G. (2009) Chemical modification of proteins at cysteine: Opportunities in chemistry and biology. Chem.—Asian J. 4, 630–640. [DOI] [PubMed] [Google Scholar]
- van Kasteren S. I., Garnier P. G., Davis B. G. (2007) Chemical methods for mimicking post-translational modification, in Protein Engineering, Springer Verlag, New York. [Google Scholar]
- Kenyon G. L.; Bruice T. W. (1977) Novel sulfhydryl reagents. Methods Enzymol. 47, 407–430. [DOI] [PubMed] [Google Scholar]
- Singh R.; Cresswell P. (2010) Defective cross-presentation of viral antigens in GILT-free mice. Science 328, 1394–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones L. J.; Upson R. H.; Haugland R. P.; PanchukVoloshina N.; Zhou M. J. (1997) Quenched BODIPY dye-labeled casein substrates for the assay of protease activity by direct fluorescence measurement. Anal. Biochem. 251, 144–152. [DOI] [PubMed] [Google Scholar]
- Moss C. X.; Villadangos J. A.; Watts C. (2005) Destructive potential of the aspartyl protease cathepsin D in MHC class II-restricted antigen processing. Eur. J. Immunol. 35, 3442–3451. [DOI] [PubMed] [Google Scholar]
- Vieira A. V.; Lamaze C.; Schmid S. L. (1996) Control of EGF receptor signaling by clathrin-mediated endocytosis. Science 274, 2086–2089. [DOI] [PubMed] [Google Scholar]
- Katzmann D. J.; Odorizzi G.; Emr S. D. (2002) Receptor downregulation and multivesicular-body sorting. Nat. Rev. Mol. Cell Biol. 3, 893–905. [DOI] [PubMed] [Google Scholar]
- Berlin I.; Higginbotham K. M.; Dise R. S.; Sierra M. I.; Nash P. D. (2010) The deubiquitinating enzyme USP8 promotes trafficking and degradation of the chemokine receptor 4 at the sorting endosome. J. Biol. Chem. 285, 37895–37908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciechanover A.; Schwartz A. L.; Dautry-Varsat A.; Lodish H. F. (1983) Kinetics of internalization and recycling of transferrin and the transferrin receptor in a human hepatoma cell line. Effect of lysosomotropic agents. J. Biol. Chem. 258, 9681–9689. [PubMed] [Google Scholar]
- Berlin I.; Schwartz H.; Nash P. D. (2010) Regulation of the epidermal growth factor receptor ubiquitination and trafficking by the USP8/STAM complex. J. Biol. Chem. 285, 4837895–37908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auerswald E. A.; Nagler D. K.; Assfalgmachleidt I.; Stubbs M. T.; Machleidt W.; Fritz H. (1995) Hairpin loop mutations of chicken cystatin have different effects on the inhibition of Cathepsin B, Cathepsin L and Papain. FEBS Lett. 361, 179–184. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.