

Background: Oxidation sensitizes nociception by covalent cysteine modification of pain transduction transient receptor potential vanilloid 1 (TRPV1) channels.
Results: Cysteines within a unique C-terminal region are ligated to form intersubunit disulfide bonds to sensitize TRPV1.
Conclusion: Dimerization of adjacent C termini activates TRPV1.
Significance: This is potentially the first example of channel activation by physical approximation of two intrinsically unstructured peptide linkers.
Keywords: Ion Channels, Oxidative Stress, Protein Chemical Modification, Receptor Structure-Function, Signal Transduction, Capsaicin Receptor
Abstract
Covalent modification of the specific cysteine residue(s) by oxidative stress robustly potentiates transient receptor potential vanilloid 1 (TRPV1) and sensitizes nociception. Here we provide biochemical evidence of dimerization of TRPV1 subunits upon exposure to phenylarsine oxide and hydrogen peroxide (H2O2), two chemical surrogates of oxidative stress. A disulfide bond formed between apposing cysteines ligates two C termini, serving as the structural basis of channel sensitization by oxidative covalent C-terminal modification. Systematic cysteine scanning of the C terminus of a cysteineless TRPV1 channel revealed a critical region within which any cysteine introduced phenylarsine oxide activation to mutant TRPV1. Oxidative sensitization persisted even when this region is substituted with a random peptide linker containing a single cysteine. So did insertion of this region to TRPV3, a homolog lacking the corresponding region and resistant to oxidative challenge. These results suggest that the non-conserved linker in the TRPV1 C terminus senses environmental oxidative stress and adjusts channel activity during cumulative oxidative damage by lowering the activation threshold of gating elements shared by TRPV channels.
Introduction
The capsaicin receptor TRPV1 is a polymodal receptor that converts multiple noxious stimuli into electric signals (1). TRPV1 is subject to extensive modulation by neurotransmitters, inflammatory cytokines, growth factors, local hormones, and oxidative chemicals, thereby serving as an integration device for processing nociceptive information (2–8). As a substrate of many cellular signaling pathways, TRPV1 employs distinct cytoplasmic domains to translate various modulatory inputs into channel openings or closures, which ultimately adjust excitability of sensory afferents.
Oxidation is one of the most robust stimulators of TRPV1 activity (2, 9). It is conserved in both mammalian and avian TRPV1s, although birds and mammals have diverged during evolution to become differentially sensitive to capsaicin, the principal mammalian deterrent in chili peppers, and other endogenous lipid agonists (10). Although there might be a shared common gating mechanism for final channel opening as initially proposed, modulation of TRPV1 channel could arise from long-range allosteric interactions from distant domains. Diffusely distributed cytoplasmic modulatory domains couple to final channel opening differently, broadening the capacity of TRPV1 for signal integration during pain detection. The mammalian TRPV1 channel has distinct binding sites for small chemical agonists, including endogenous fatty acid or acylamides, vanilloids, 2-aminoethoxy-diphenyl borate, and purine nucleotides (ATP and GTP), as well as large macromolecular ligands such as calmodulin or inositide phospholipids (10–16). Ligand binding to each site either directly gates the channel or triggers conformation changes that secondarily modulate thresholds of channel opening by other modalities. Analysis of the gating mechanism of TRPV1 activation for even a single modality is hindered by incomplete understanding of coupling processes among different modes of channel activation. For instance, mutation in the extracellular proton acceptor (E600), which is not the capsaicin-binding site, nevertheless causes a pronounced shift of the dose-response curve for vanilloid ligands, complicating subsequent data interpretation from functional measurements (17). Mutational analysis directed toward critical residues required for oxidative TRPV1 channel modulation suffers from a similar problem. Unanticipated changes remote from regions containing mutations might alter the coupling of channel gates rather than purely perturbing local structures of the receptor critical for channel gating. We thus took advantage of functional conservation of oxidative sensitization of TRPV1 channels between avian and mammalian and selected the chicken receptor to simplify the mechanistic analysis of oxidative modulation because its insensitivity to chemical agonists capsaicin and 2-aminoethoxy-diphenyl borate helps to eliminate one gating mode, besides its better functional expression.
TRPV1 activity is heavily modulated by covalent modification, receptor trafficking and transcriptional or translational regulations (3, 18–21). To minimize the contribution of other signaling pathways that oxidative chemicals may indirectly induce in the whole cell context, we used the excised membrane patch, a reduced experimental system, to elucidate the mechanism of channel activation by direct oxidative covalent modification.
EXPERIMENTAL PROCEDURES
Cell Culture and Heterologous Expression
HEK293 cells were cultured with DMEM/F12 supplemented with 10% newborn calf serum and antibiotics. Cells were transfected with 400 ng wild-type or mutant receptors with or without 50 ng enhanced GFP plasmids using Turbofect reagents (Fermentas). Cells were plated on poly-d-lysine-coated coverslips and recorded 2 days following transfection.
Molecular Biology
Chimeric receptors were generated by the overlapped extension polymerase chain reaction. Single point mutants were constructed by overlapped extension PCR followed by restriction digestion and ligation or by QuikChange site-directed mutagenesis using Phusion (New England Biolab) or Pfu polymerase (Stratagene). For Western blotting, HA-tagged rat TRPV1 or 3× HA-tagged cysteineless chicken TRPV1 were constructed in a mammalian expression vector.
Non-reducing SDS-PAGE Analysis and Western Blotting
HEK293 cells transfected with cDNA-containing plasmids were grown in cell culture plates until 48 h after transfection. Cells were rinsed three times with PBS and then incubated in PBS containing 200 μm PAO2 for 1 h or 10 mm H2O2 for 30 min. The buffer solution was gently removed and cells were lysed in PBS containing 1% Triton X-100 and protease inhibitor mixtures (Thermo Scientific). To test the reversibility of covalent dithiaarsanane adducts, we included 10 mm mercaptoethanol in the lysis buffer. The lysate was resolved on 8% non-reducing SDS-polyacrylamide gel, which was transferred to an Immobilon membrane (Millipore). The membrane was incubated in blocking buffer (Tris-buffered saline with 0.05% Tween 20 with 5% nonfat milk) containing anti-HA antibody (at 1:1000 dilution, Covance, Inc.) at 4 °C overnight. TRPV1 proteins were visualized using a secondary goat-anti-rabbit HRP-conjugated antibody at 1:100,000 dilution (Thermo Scientific).
Electrophysiology
All electrophysiology experiments were conducted at room temperature (21 °C). The standard extracellular solution was composed of 10 mm HEPES, 145 mm CsCl, 1 mm MgCl2, and 2 mm CaCl2 (pH 7.4). The standard internal solution contained 10 mm HEPES, 145 mm Na gluconate, 1 mm Mg(gluconate)2, and 0.1 mm EGTA (pH 7.4). Access resistances of recording electrodes were 0.5 to 1 mΩ. Those of inside-out macropatch recordings were 0.2 mΩ. Pulse and PulseFit software (HEKA) was used for data acquisition and analysis. A voltage ramp from −120 to +80 mV given at 1 Hz was used to continuously record currents. Current amplitudes at +80mV were plotted against time.
Statistical Analysis
The differences of TRPV1 currents between maximal PAO stimulation and after BAL reversal were measured at +80mV. Data were rundown-corrected and presented as mean ± S.E. The sensitivity of each cysteine substitution to PAO application was determined using one-way analysis of variance. A p value less than 0.05 was considered to be statistically significant.
RESULTS
Cysteine Scanning Analysis Revealed That Numerous Positions in the C Terminus Are Permissive for Oxidative Activation of TRPV1
To study the positional specificity of disulfide-bond-evoked channel opening in chicken TRPV1, we introduced single cysteine residues individually back into a cysteineless mutant chicken TRPV1 (cys-cTRPV1) background at each position between amino acids 772 and 832. In most positions (56 of 59 mutants), cysteine substitutions yielded functional ion channels with discernible basal currents at a membrane potential of +80 mV under cell-attached recording conditions, provided that Cs+ was used as charge carriers in electrophysiological measurements. Excision of membrane patches into a buffer solution with simple electrolyte composition allowed us to quantify the effect of PAO, a cysteine-reactive chemical selective for vicinal thiols, on each mutant channel (Fig. 1, A and B). If the C terminus of TRPV1 folded into a discrete structural motif, covalent cysteine modification-induced channel activation should only be observed for cysteines introduced to a limited subset of positions: A pair of cysteines had to be in physical proximity with appropriate orientation to each other to form an intersubunit disulfide bond to activate the channel. Surprisingly, for a contiguous stretch of peptide between positions 772 and 809 of chicken TRPV1, the introduction of a cysteine conferred a response to PAO activation (Fig. 1C). No apparent periodicity was noted in this region, which suggests that the existence of cysteine, rather than a specific folded structure in this region, is sufficient for C terminus-mediated oxidative modulation. In contrast, introduction of cysteine residues into most positions of the more distal region of the C terminus (from Lys-810 to Asp-831) of cTRPV1 resulted in no significant oxidative sensitization (Fig. 1C).
FIGURE 1.

Single cysteine substitution mutants are activated by PAO. A, inside-out patch-clamp recording of the current of single cysteine mutant cys-cTRPV1 K777C activated by 10 μm PAO. The PAO-induced current can be reversed by 1 mm reducing agent BAL (all currents are measured at +80 mV). B, current-voltage relationships at time points before PAO (basal), maximal PAO stimulation, and full reversal by BAL are shown to demonstrate the characteristic rectification gating of TRPV1. C, statistical analysis of cysteine substitutions and averaged BAL-sensitive induced currents are displayed in the bar graph (n = 3–9). The results revealed the clustering of positions in C termini at which an introduced cysteine confers channel activation by PAO. Substitutions at positions 796, 799, and 814 had very low recordable currents, and their modulation were not further determined (n.d.).
PAO Reacted with Vicinal Thiols in Two Subunits to Form an Intersubunit Dimer
Given that a mutation to cysteine at almost position (33 of35 functional channels) along a continuous segment from Cys-772 to Glu-809 resulted in disulfide-mediated channel activation, it is unlikely that this region has a well folded structure with an interface that complements other domains of TRPV1. We hence considered an alternative hypothesis that this region is unstructured. Thermal motion might then expose the substituted cysteine, which can occasionally approximate the corresponding cysteine residue in an adjacent subunit and be driven to form a stable disulfide bond under oxidative conditions.
To determine whether such a disulfide bond is formed between subunits at positions permissive for oxidative activation, we used non-reducing SDS-PAGE to resolve TRPV1 monomers and oligomers in the lysates prepared from HEK293 cells transfected with TRPV1 cDNAs. No discrete bands of defined molecular sizes from detergent-solubilized cell lysates under non-reducing conditions were observed using wild-type rat TRPV1. We suspected that numerous cysteines in wild-type TRPV1 could have led to spurious disulfide bonds and introduced the anomalous mobility of channel proteins. Cysteineless cTRPV1 should not form high molecular weight homo-oligomers or complexes with other cellular proteins under non-reducing conditions. The cysteineless cTRPV1, as expected, migrated as a monomeric polypeptide on the non-reducing SDS gel. We thus focused on molecular weight analysis of single cysteine substitution mutants in this background (Fig. 2). We examined several single-cysteine substitution mutants (cysteine at positions 772, 774, 777, 784, 786, and 790) heterologously expressed in HEK cells. We treated live transfected cells with 200 μm PAO for 1 h to catalyze in situ cysteine modification. PAO treatment drove the dithiaarsanane adduct formation for channels in their native environments, provided that proximal thiols were present and covalent bonds formed were not broken by cellular antioxidants. A substantial fraction of channel proteins from these PAO-sensitive mutants had a shift of mobility to a range consistent with dimer formation in non-reducing SDS-PAGE. Dimeric adducts could be reversed to monomers by an excess of reducing agents (10 mm β-mercaptoethanol) (Fig. 2A). Interestingly, dimeric proteins cross-linked by PAO from each cysteine reversion or substitution mutants have slight but reproducible differences in their mobility, indicating their structural difference even under the denatured condition (Fig. 2B). The responsiveness in functional assays and reactivity to site-directed cysteine modification of each of these mutants to PAO provided supportive evidence that intersubunit dimerization from disulfide bonding causes channel activation.
FIGURE 2.
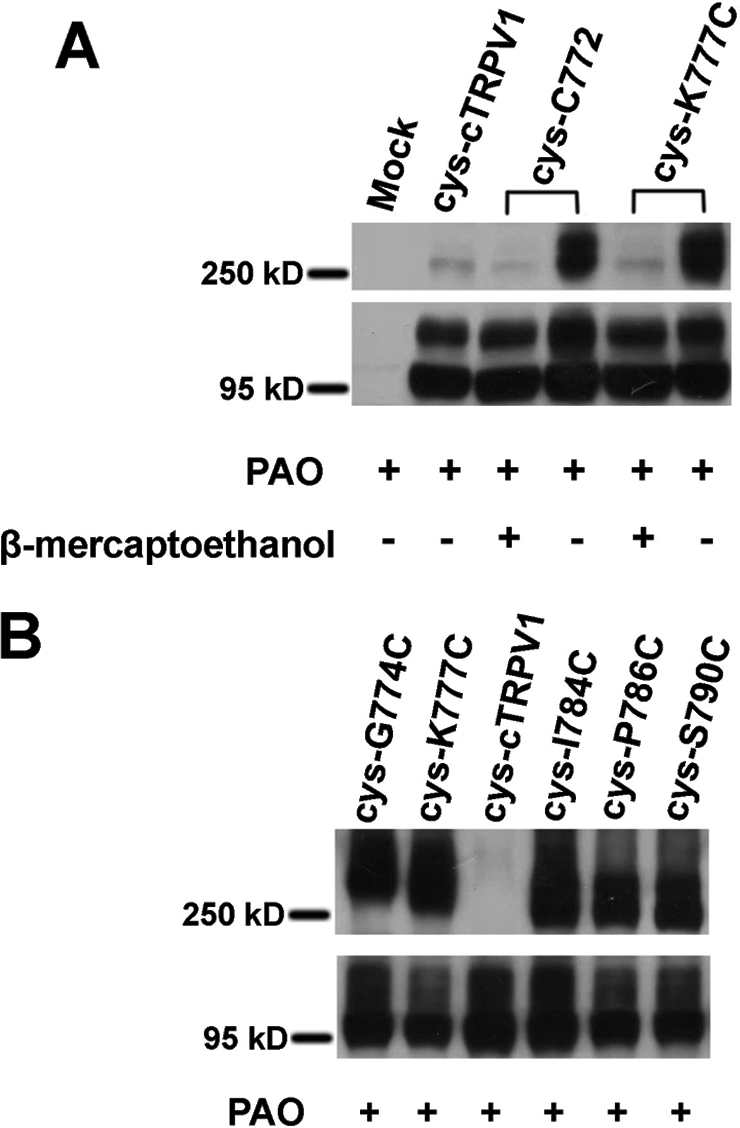
Non-reducing SDS-PAGE Western blot analysis of the PAO effect on HA-tagged cysteineless chicken TRPV1 and single-cysteine substitution mutants transiently expressed in HEK293 cells. A, under oxidative condition (200 μm PAO for 1 h), single-cysteine mutant cys-C772 and cys-K777C migrated as both monomeric (around 95 kDa) and dimeric (around 250 kDa) polypeptides, and the band for dimeric cys-C772 and cys-K777C reverted back to the sizes of monomers when the reducing agent β-mercaptoethanol was added to protein samples. The vast majority of cysteineless mutant migrated as a monomer even after PAO treatment. Mock-transfected cells did not have any proteins recognized by anti-HA antibodies in the corresponding positions for TRPV1 monomers or dimers. B, the cys-cTRPV1 mutants with single cysteine substitution at positions 774(cys-G774C), 777(cys-K777C), 784(cys-I784C), 786(cys-P786C), and 790(cys-S790C) migrated as both monomeric and dimeric polypeptides under oxidative conditions (200 μm PAO for 1 h).
Two Single Cysteine-containing Random Peptide Fragments Can Substitute the TRPV1 Linker Region to Introduce PAO Activation into cys-cTRPV1
Truncation of the very end of rat TRPV1 produced sensitized mutant channels with lowered thresholds for temperature- and voltage-dependent activation, suggestive of a tonic inhibitory role of the distal region of the C terminus of TRPV1 in channel gating (22, 23). An intersubunit disulfide bond within the segment just proximal to this “inhibitory” region might sensitize the full-length channel by relieving the inhibitory effect mediated by this distal C-terminal segment. This model predicts that distal C-terminal truncation of any PAO-sensitive single-cysteine substituted mutant chicken TRPV1 would be constitutively activated or sensitized even without oxidative stress and would result in no further activation by PAO. We thus created two versions of truncation mutants (Δ32 and Δ43) in a Cys-772 or 777C background to test this hypothesis (Fig. 3A). Both truncation mutants remained robustly activated by 10 μm PAO, which was also reversed by dithiol reducing agent 2,3-dimercaptopropanol (BAL) (PAO-induced currents = 2.0 ± 0.9 nA, n = 5 and = 0.89 ± 0.34 nA, n = 4 for Δ32 and Δ43, respectively, Fig. 3B). Clearly, cross-linking two neighboring C termini did not cause channel activation by removing autoinhibition by the C-terminal tail in TRPV1 gating.
FIGURE 3.

The PAO-induced channel activation does not require a linker of any specific primary amino acid sequence. A, nomenclatures and corresponding amino acid sequences of various cys-cTRPV1 mutants are shown. The cysteine introduced in these mutants was at the Lys-777 position of the cysteineless TRPV1. Representative traces of PAO activation and the reversal by the reducing agent BAL of truncation mutants cys-K777CΔ32 and cys-K777CΔ43 (B) and of random peptide linker mutants cys-cTRPV1(Δ773–787)CAP and cys-polyGS(773–787)Cys777 (C) are displayed in each panel.
The prevalence of permissive positions for introducing a single-cysteine substitution to confer oxidative activation to TRPV1 raised several mechanistic questions. First, is this region required, or is the presence of any cysteine residue within this region sufficient for oxidative activation of channels? Secondly, if this region is essential for oxidant-induced channel activation, does it simply serve as a spacer or, rather, obligate a certain primary sequence to exert its stimulatory function? To address these questions, we created the mutant cys-cTRPV1(Δ773–787)CAP by replacing the amino acids between Ser-772 and Arg-788 with a short tripeptide of Cys-Ala-Pro sequence (Fig. 3A). This mutant shortens the distance between the TRP domain and the distal C-terminal inhibitory region but contains a single cysteine residue. We recorded from this mutant and found that it is sensitized by PAO comparably to any single-cysteine substitution mutants (PAO-induced currents = 2.5 ± 1.3 nA, n = 6, Fig. 3C). We further replaced the Cys-Ala-Pro (CAP) tripeptide of cys-cTRPV1(Δ773–787)CAP with a random 15-amino acid-long poly-glycine-serine linker containing a single cysteine insertion, denoted cys-polyGS(773–787)Cys-777 (Fig. 3A). This channel was well expressed and showed pronounced PAO sensitization (PAO-induced currents = 1.6 ± 0.8 nA, n = 6, Fig. 3C).
Hydrogen Peroxide Activated the Single Cysteine Substitution Mutants and the Random Peptide Linker Mutant cys-polyGS(773–787)Cys-777
PAO and the more common oxidative stress H2O2 share the ability to join vicinal cysteines to form disulfide adducts. However, the disulfide bond formed by PAO treatment is slightly different, with an additional arsenic atom bridging two sulfurs. We therefore examined whether H2O2 could catalyze simple disulfide bond formation and what the functional consequences of such modification are. Oxidation of single-cysteine substitution mutants was detected as a shift of mobility of TRPV1 mutant polypeptides (Fig. 4A) on the non-reducing SDS-PAGE gel. We then assessed the ability of 10 mm H2O2 to activate mutant channels with a single cysteine per subunit. Channel activation by H2O2 was as robust as that arising from PAO application, although H2O2 activation and recovery kinetics were slower than PAO, and the reversal of activation required a higher concentration (10 mm) of the reducing agent BAL (Fig. 4, B and C). This may imply the stability of simple disulfides that sensitize TRPV1 in the oxidative process, which is consistent with a long-lasting sensitization of nociception in the animal model (9).
FIGURE 4.

Single-cysteine substitution mutants or the random peptide linker mutant is activated by H2O2. A, non-reducing SDS-PAGE Western blot analysis of the H2O2 effect on HA-tagged cTRPV1 mutants transiently expressed in HEK293 cells. After incubating in PBS with 10 mm H2O2 for 30 min, single-cysteine mutants cys-C772, cys-G774C, cys-K777C, cys-I784C, cys-P796C, and cys-S790C from whole cell lysates migrated as both monomeric and dimeric polypeptides, whereas the cysteineless cTRPV1 mutant only migrated as a monomer. B, inside-out patch-clamp recording revealed that the current from mutant cys-cTRPV1K777C was activated by 10 mm H2O2, which could be reversed by 10 mm BAL (measured at +80 mV). C, statistical analysis of H2O2-induced currents in mutants is displayed in the bar graph (n = 5–7). D, current-voltage relationships of cys-K777C at time points before H2O2 (basal), maximal H2O2 stimulation, and full reversal by BAL are overlaid.
Basic gating properties of TRPV1 channels, including strong voltage-dependent rectification (Fig. 4D), activation by an acidic pH, and direct modulation of current amplitude by Cs+ or Mg2+ (24, 25), are preserved in these mutants (data not shown). Oxidation primarily alters the energy barriers to reduce the activation threshold of TRPV1, thereby playing an important role in sensitization of this transduction channel.
The Short Peptide Linker Fragment from TRPV1 Transfers PAO Sensitivity to Oxidant-insensitive TRPV3
These results suggest that the critical segment in TRPV1 probably does not form a rigid structure to function as a gating module to regulate TRPV1 activity. Instead, it may operate as a flexible linker to connect distinct gating domains of TRPV1 to meter the environmental oxidative stress the cytoplasmic tails face. Clearly, the segment endowing oxidative modulation is dispensable for a functional channel. It is even not evolutionarily conserved across homologous channels in the same TRPV family. Alignment of multiple TRPV channel sequences revealed that the homologous TRPV3 channel lacks a corresponding segment (Fig. 5A). If these TRPV homologs have similar basic elements designed for gating movements facilitating channel opening, insertion of this extra sequence into TRPV3 might not disrupt other gating elements for TRPV3 channel function but successfully introduce oxidative modulation into TRPV3. We therefore transplanted this segment into analogous positions of oxidation-resistant TRPV3 and asked whether it confers oxidant sensitivity to the insertion mutant. Indeed, this minichimera transferred PAO-induced and reducing agent-reversible channel activation into TRPV3 (Fig. 5, B and C). The small insertion did not disrupt any critical structures for TRPV3 gating. However, C-terminal dimerization is sufficient to lower the activation threshold of this channel. This observation suggested that TRPV channels in the same subfamily may share a similar biophysical design to form ion-conducting devices and that TRPV1 may have acquired this additional sequence during evolution to adapt to its physiological function, the integration of multiple modulatory inputs into a signaling unit controlling sensory neuron excitability (Fig. 5D).
FIGURE 5.

Redox-insensitive TRPV3 can be converted into a PAO-activated ion channel by transferring the linker region unique to TRPV1 but missing in TRPV3. A, amino acid sequences of C termini of cTRPV1 (wild-type), human TRPV3, and the V3-V1-V3 minichimera are shown. The inserted linker sequence is marked in gray. Two cysteines in the native positions of cTRPV1 are highlighted in the alignment. B, the wild-type human TRPV3 channel exhibited basal channel activity in the excised inside-out membrane patch (top panel). Application of 10 μm PAO caused no significant increase in channel activity compared with spontaneously developed basal currents in the gluconate-based bath solution. 1 mm BAL slightly enhanced the basal current, which was reversible upon BAL washout (n = 5). i-V (current-voltage) relationships of hTRPV3 currents at the basal (a), PAO-treated (b), and BAL-treated (c) conditions were overlaid for comparison (bottom panel). All three traces showed characteristic rectification of TRPV3 channels. C, the V3-V1-V3 minichimera displayed similar basal activity increased spontaneously upon patch excision (top panel). In contrast to the wild-type channel, application of 10 μm PAO led to a rapid enhancement of the current amplitude. The slow increase of currents persisted after PAO washout. However, the current was rapidly suppressed by the reducing agent BAL. The small stimulation of BAL on the basal activity of this chimera was evidenced by a further reduction of current upon BAL washout (n = 6). i-V (current-voltage) relationships of the V3-V1-V3 minichimera were shown similarly in d, e, and f (bottom panel). The chimeric channel retained similar voltage-dependent gating to the wild type TRPV3. D, a summary alignment of amino acid sequences of C termini for TRPV1s and TRPV3s.
DISCUSSION
The cytoplasmic tails of TRPV1 contains many substrate sites for intracellular biochemical signaling pathways that alter activation thresholds of this nociceptive transduction channel. Protein phosphorylation, phospholipids, and calmodulin binding to the C terminus of mammalian TRPV1 exert substantial modulation on TRPV1 channel gating (26–28), presumably by triggering specific conformational change.
Our results highlight a novel mechanism by which TRPV1 is activated, where stable dimerization via covalent ligation between C termini of channel subunits is sufficient to open the channel. One unique feature of oxidative sensitization in contrast with other forms of TRPV1 modulation is that it does not require a specific local structure in which the disulfide bond forms. The stability of such a covalent bond serves as the molecular basis of sustained sensitization of TRPV1 following oxidative stress. Conceivably, the advantage of this form of modulation is to report the cumulative oxidative damage incurred under disease conditions.
The participation of channel C termini in TRPV1 gating is two-fold. Although some recent studies suggest that the proximal region of the TRPV1 C terminus directly interacts with phospholipids as the principal gating element (29), the linker region we identified seems to be unstructured and more involved in adjusting the activation threshold to trigger channel opening. Oxidation clearly sensitizes the channel so much as to exhibit substantial channel activities at room temperature. Covalent modification of cytoplasmic cysteines has recently been found to regulate human ether-a-go-go related channels (HERG) (30). Oxidative stress appears to be a general mechanism to alter electrical excitability.
Currently, there are no structural data available to allow visualization of the differences between the reduced and the oxidized forms of TRPV1. Although the C terminus of TRPV1 is responsible for integrating distinct modulations by multiple signaling pathways, the structural basis for such a small domain to carry out specific interactions with many signaling molecules remains elusive. The ability for the C termini to form disulfide-based dimers between two subunits within one TRPV1 complex is mostly likely from stochastic interaction between two adjacent C-terminal tails rather than requiring additional specific interaction partners from other regions of the channel. This is supported by the observation that simply inserting the linker peptide is sufficient to introduce oxidative activation into the PAO-insensitive wild-type TRPV3 channel, which has a different N terminus and bears more distant sequence homology than among TRPV1s themselves. There is also some sequence homology between C termini of cyclic nucleotide gated channels and of TRPV1 (31), but results from molecular modeling could not satisfactorily explain the widespread cysteine substitution in the C-terminal linker region for conferring the sensitivity to oxidation. Interestingly, swapping TRPV1 and TRPM8 C termini creates chimeric channels with switched temperature-dependent activation (32, 33). From the functional perspective, there are similar regulatory mechanisms of the C terminus-dependent gating in other tetrameric ion channels. KATP channels (Kir6.2), which exhibit modulation by multiple ligands, possess a C-terminal tail with distinct motifs to regulate their sensitivity to each ligand (34, 35). The Kir6.2 C terminus has an inhibitory role in channel gating. Limited C-terminal truncation generates a constitutively opened channel (36). However, truncated Kir6.2 can still be directly inhibited by ATP binding to a specific pocket located in the proximal part of the C terminus (36). Analogously, deletion of the distal region of the TRPV1 C terminus sensitizes the channel but does not occlude the stimulatory effect from dimerization of the more proximal part of the C termini. Such dimerization may trigger a more limited gating transition compared with full opening of TRPV1 channels in response to a chemical ligand like capsaicin. Although we have used a ligand-insensitive homolog to minimize the contribution of one gating mode to channel activation, dimerization-activated chicken TRPV1 is still subject to another gating mode. PAO-activated cTRPV1 exhibits strong voltage dependence comparable with the basal channel activity of human TRPV1 reported by Voets et al. (37). Recent studies of ligand-gated MthK potassium channels and cyclic nucleotide-gated channels suggest that four C termini may form a constricted ring to mobilize the more rigid hinge regions of these tetrameric ion channels during the gating process, and open their ion conduction pathways (38–40). Both MthK and CNG channels are activated by multiple gating mechanisms operating synergistically. In MthK, Ca2+ ions oligomerize the RCK domains to form the gating ring that is destabilized by protons (41). Although CNG channels exhibit unliganded basal channel activity and voltage-dependent channel activation, the binding of multiple cyclic nucleotide molecules to C-terminal ligand recognition domains also facilitates the major conformational change for maximal ion conduction (42, 43). Most importantly, each activation mechanism in these channels is mediated by distinct gating elements. Activation of TRPV1 by dimerization of C termini may as well trigger partial gating movements to facilitate further activation by other modalities, serving as the structural basis of receptor sensitization of this nociceptive transduction channel.
Acknowledgments
We thank Drs. Robert Oswald and Linda Nowak for helpful comments and stimulating discussion for experiments and manuscript preparation. We also thank Dr. D. E. Clapham for the generous gift of the human TRPV3 expression plasmid.
This work is supported by a Scientist Developmental Grant from the American Heart Association (to H. C.).
- PAO
- phenylarsine oxide.
REFERENCES
- 1. Tominaga M., Caterina M. J., Malmberg A. B., Rosen T. A., Gilbert H., Skinner K., Raumann B. E., Basbaum A. I., Julius D. (1998) Neuron 21, 531–543 [DOI] [PubMed] [Google Scholar]
- 2. Chuang H. H., Lin S. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 20097–20102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ji R. R., Samad T. A., Jin S. X., Schmoll R., Woolf C. J. (2002) Neuron 36, 57–68 [DOI] [PubMed] [Google Scholar]
- 4. Tominaga M., Wada M., Masu M. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 6951–6956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhang N., Inan S., Inan S., Cowan A., Sun R., Wang J. M., Rogers T. J., Caterina M., Oppenheim J. J. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 4536–4541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang X., Huang J., McNaughton P. A. (2005) EMBO J. 24, 4211–4223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhuang Z. Y., Xu H., Clapham D. E., Ji R. R. (2004) J. Neurosci. 24, 8300–8309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zygmunt P. M., Petersson J., Andersson D. A., Chuang H., Sørgård M., Di Marzo V., Julius D., Högestätt E. D. (1999) Nature 400, 452–457 [DOI] [PubMed] [Google Scholar]
- 9. Keeble J. E., Bodkin J. V., Liang L., Wodarski R., Davies M., Fernandes E. S., Coelho Cde F., Russell F., Graepel R., Muscara M. N., Malcangio M., Brain S. D. (2009) Pain 141, 135–142 [DOI] [PubMed] [Google Scholar]
- 10. Jordt S. E., Julius D. (2002) Cell 108, 421–430 [DOI] [PubMed] [Google Scholar]
- 11. Hu H. Z., Gu Q., Wang C., Colton C. K., Tang J., Kinoshita-Kawada M., Lee L. Y., Wood J. D., Zhu M. X. (2004) J. Biol. Chem. 279, 35741–35748 [DOI] [PubMed] [Google Scholar]
- 12. Kwak J., Wang M. H., Hwang S. W., Kim T. Y., Lee S. Y., Oh U. (2000) J. Neurosci. 20, 8298–8304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lishko P. V., Procko E., Jin X., Phelps C. B., Gaudet R. (2007) Neuron 54, 905–918 [DOI] [PubMed] [Google Scholar]
- 14. Numazaki M., Tominaga T., Takeuchi K., Murayama N., Toyooka H., Tominaga M. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 8002–8006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Prescott E. D., Julius D. (2003) Science 300, 1284–1288 [DOI] [PubMed] [Google Scholar]
- 16. Rosenbaum T., Gordon-Shaag A., Munari M., Gordon S. E. (2004) J. Gen. Physiol. 123, 53–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jordt S. E., Tominaga M., Julius D. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 8134–8139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bhave G., Zhu W., Wang H., Brasier D. J., Oxford G. S., Gereau R. W., 4th (2002) Neuron 35, 721–731 [DOI] [PubMed] [Google Scholar]
- 19. Salazar H., Llorente I., Jara-Oseguera A., García-Villegas R., Munari M., Gordon S. E., Islas L. D., Rosenbaum T. (2008) Nat. Neurosci. 11, 255–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xue Q., Jong B., Chen T., Schumacher M. A. (2007) J. Neurochem. 101, 212–222 [DOI] [PubMed] [Google Scholar]
- 21. Yoshida T., Inoue R., Morii T., Takahashi N., Yamamoto S., Hara Y., Tominaga M., Shimizu S., Sato Y., Mori Y. (2006) Nat. Chem. Biol. 2, 596–607 [DOI] [PubMed] [Google Scholar]
- 22. Liu B., Ma W., Ryu S., Qin F. (2004) J. Physiol. 560, 627–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vlachová V., Teisinger J., Susánková K., Lyfenko A., Ettrich R., Vyklický L. (2003) J. Neurosci. 23, 1340–1350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ahern G. P., Brooks I. M., Miyares R. L., Wang X. B. (2005) J. Neurosci. 25, 5109–5116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang S., Poon K., Oswald R. E., Chuang H. H. J. Biol. Chem. 285, 11547–11556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bhave G., Hu H. J., Glauner K. S., Zhu W., Wang H., Brasier D. J., Oxford G. S., Gereau R. W., 4th (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 12480–12485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jung J., Shin J. S., Lee S. Y., Hwang S. W., Koo J., Cho H., Oh U. (2004) J. Biol. Chem. 279, 7048–7054 [DOI] [PubMed] [Google Scholar]
- 28. Premkumar L. S., Ahern G. P. (2000) Nature 408, 985–990 [DOI] [PubMed] [Google Scholar]
- 29. Ufret-Vincenty C. A., Klein R. M., Hua L., Angueyra J., Gordon S. E. J. Biol. Chem. 286, 9688–9698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kolbe K., Schonherr R., Gessner G., Sahoo N., Hoshi T., Heinemann S. H. (2010) J. Physiol. 588, 2999–3009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. García-Sanz N., Fernández-Carvajal A., Morenilla-Palao C., Planells-Cases R., Fajardo-Sánchez E., Fernández-Ballester G., Ferrer-Montiel A. (2004) J. Neurosci. 24, 5307–5314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Brauchi S., Orta G., Mascayano C., Salazar M., Raddatz N., Urbina H., Rosenmann E., Gonzalez-Nilo F., Latorre R. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 10246–10251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Brauchi S., Orta G., Salazar M., Rosenmann E., Latorre R. (2006) J. Neurosci. 26, 4835–4840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Drain P., Li L., Wang J. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 13953–13958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Takano M., Xie L. H., Otani H., Horie M. (1998) J. Physiol. 512 ( Pt 2), 395–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tucker S. J., Gribble F. M., Zhao C., Trapp S., Ashcroft F. M. (1997) Nature 387, 179–183 [DOI] [PubMed] [Google Scholar]
- 37. Voets T., Droogmans G., Wissenbach U., Janssens A., Flockerzi V., Nilius B. (2004) Nature 430, 748–754 [DOI] [PubMed] [Google Scholar]
- 38. Jiang Y., Lee A., Chen J., Cadene M., Chait B. T., MacKinnon R. (2002) Nature 417, 515–522 [DOI] [PubMed] [Google Scholar]
- 39. Johnson J. P., Jr., Zagotta W. N. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 2742–2747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zagotta W. N., Olivier N. B., Black K. D., Young E. C., Olson R., Gouaux E. (2003) Nature 425, 200–205 [DOI] [PubMed] [Google Scholar]
- 41. Ye S., Li Y., Chen L., Jiang Y. (2006) Cell 126, 1161–1173 [DOI] [PubMed] [Google Scholar]
- 42. Craven K. B., Zagotta W. N. (2006) Annu. Rev. Physiol. 68, 375–401 [DOI] [PubMed] [Google Scholar]
- 43. Taraska J. W., Zagotta W. N. (2007) Nat. Struct. Mol. Biol. 14, 854–860 [DOI] [PubMed] [Google Scholar]