

Background: HIV-RT uses Mg2+ for efficient nucleotide incorporation but is inhibited by other divalent cations.
Results: Zn2+ supports HIV-RT catalysis but with profoundly reduced kinetics.
Conclusion: Zn2+ inhibition is not due to catalysis blockage but to the formation of a highly stable, kinetically diminished complex.
Significance: HIV-RT is inhibited by low Zn2+ concentrations presenting an avenue for future drug research.
Keywords: Calcium, HIV, Manganese, Reverse Transcription, Zinc
Abstract
Several physiologically relevant cations including Ca2+, Mn2+, and Zn2+ have been shown to inhibit HIV reverse transcriptase (RT), presumably by competitively displacing one or more Mg2+ ions bound to RT. We analyzed the effects of Zn2+ on reverse transcription and compared them to Ca2+ and Mn2+. Using nucleotide extension efficiency as a readout, Zn2+ showed significant inhibition of reactions with 2 mm Mg2+, even when present at only ∼5 μm. Mn2+ and Ca2+ were also inhibitory but at higher concentrations. Both Mn2+ and Zn2+ (but not Ca2+) supported RT incorporation in the absence of Mg2+ with Mn2+ being much more efficient. The maximum extension rates with Zn2+, Mn2+, and Mg2+ were ∼0.1, 1, and 3.5 nucleotides per second, respectively. Zinc supported optimal RNase H activity at ∼25 μm, similar to the optimal for nucleotide addition in the presence of low dNTP concentrations. Surprisingly, processivity (average number of nucleotides incorporated in a single binding event with enzyme) during reverse transcription was comparable with Zn2+ and Mg2+, and single RT molecules were able to continue extension in the presence of Zn2+ for several hours on the same template. Consistent with this result, the half-life for RT-Zn2+-(primer-template) complexes was 220 ± 60 min and only 1.7 ± 1 min with Mg2+, indicating ∼130-fold more stable binding with Zn2+. Essentially, the presence of Zn2+ promotes the formation of a highly stable slowly progressing RT-(primer-template) complex.
Introduction
Human immunodeficiency virus reverse transcriptase (HIV-RT)2 like other RTs, possess both nucleotide polymerization and ribonuclease H (RNase H, activity that degrades RNA which is part of an RNA-DNA hybrid) capabilities. Both activities require a divalent cation as an integral cofactor in the mechanism of catalysis (1, 2). Under physiological conditions Mg2+ presumably functions as the cofactor, however other divalent cations (including Mn2+, Ni2+, and Cu2+) can also be used for nucleotide catalysis (3). Some divalent cations, such as Pd2+, Ca2+, Mn2+, and Zn2+, have been shown to inhibit Mg2+-dependent RT activity (3–7).
Magnesium binds to RT in the polymerase and RNase H domains where binding is coordinated by the acidic catalytic residues within the active sites. By analogy to other DNA polymerases, two Mg2+ ions participate in polymerization via a universal “two metal ion” mechanism (reviewed in Ref. 8). The number of Mg2+ ions bound at the RNase H active site is more controversial, as both one and two metal mechanisms have been proposed (reviewed in Ref. 9). In addition to the cation binding sites, other sites may also exist on the enzyme. For example, some reports suggest that as many as 21 Mg2+ ions are bound to a single Escherichia coli DNA polymerase I molecule (10). The role, if any, of these additional cations is unclear.
Many enzymes require cation cofactors that play an intricate role in the mechanism of catalysis. In humans these typically include magnesium, calcium, iron, manganese, and zinc, cobalt, copper, selenium, and molybdenum. Note that calcium, though required to stimulate the activity of several enzymes, typically acts as an allosteric regulator involved in cell signaling rather than a cofactor in catalysis. Most enzymes that carry out catalysis on nucleic acids have evolved to use Mg2+ as a primary cofactor. Although the majority of the ∼10 mm Mg2+ in cells is complexed with cellular components, the intracellular concentration of free available Mg2+, typically reported to be around 2 mm or less, is still much greater than other divalent cations (11–15). Unlike Mg2+, other cation cofactors are maintained at much lower levels in cells. The availability of these cations is carefully controlled by a combination of ion transporters, binding proteins that sequester the cations, and sequestration by cellular nucleotides and within specific cell organelles. For example, the concentration of free Zn2+ in cells is in the low nm range despite a total concentration (free and bound) of ∼0.1–0.5 mm and a plasma concentration of ∼15 μm (12, 16–20). The tight regulation is required due to the ability of some cations to alter transcription and inhibit or alter the activity of several enzymes. Zinc is particularly notable because it is important to many processes in cells and is a component of several proteins, particularly zinc finger containing proteins (16, 20, 21). In addition, Zn2+ has recently been shown to modulate signal transduction, and like Ca2+, serve a cellular second messenger role for specific processes (22, 23). Zinc is also a potent inhibitor of many enzymes, including several viral polymerases (5, 6, 18, 24–30). The mechanism of inhibition may take different forms including direct displacement of Mg2+ due to higher affinity of Zn2+ for the cation binding site, allosteric inhibition through binding at secondary sites on the enzyme, or more complex mechanisms that involve interactions with substrate or other cofactors (nucleotides or nucleic acids for example).
In this report we investigated the interaction of HIV-RT with Zn2+ (as well as Mn2+ and Ca2+) to determine how it affects RT activity. The results showed that Zn2+ supported both polymerase and RNase H activity at concentrations much lower than Mg2+ and was a potent inhibitor of RT in the presence of Mg2+. Surprisingly, the RT-Zn2+-(primer-template) complex was much more stable than the same complex formed with Mg2+ while rate of nucleotide incorporation was much slower. Overall Zn2+ inhibits RT by forming what is essentially a “dead-end complex” that can remain bound to the primer-template for hours but has very slow incorporation kinetics. The possibility of using Zn2+ or other cations as inhibitors of virus replication is discussed.
EXPERIMENTAL PROCEDURES
Materials
The following reagent was obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: pNL4–3 from Dr. Malcolm Martin (31). PCR primers and primers used to prime templates in reverse transcription assays were obtained from Integrated DNA Technologies, Inc. The HIV-RT clone was a generous gift from Dr. Michael Parniak (University of Pittsburg). HIV-RT was purified as described (32). Aliquots of HIV-RT were stored frozen at −80 °C and fresh aliquots were used for each experiment. RNase H minus (E478>Q) RT was a gift from Dr. Stuart Le Grice, HIV Drug Resistance Program, National Cancer Institute, Frederick, MD. Taq polymerase, T4 polynucleotide kinase, MuLV-RT, and restriction enzymes were from New England Biolabs. T7 and SP6 RNA polymerases, calf intestinal alkaline phosphatase (CIP), AMV-RT, and dNTPs were from Roche Diagnostics. DNase I-RNase-free and RNase H were from United States Biochemical Corporation. RNase inhibitor (RNasin) and the phiX174 HinfI digest DNA ladder was from Promega. Radiolabeled compounds were obtained from PerkinElmer. Sephadex G-25 spin columns were from Harvard Apparatus. QIAquick PCR Purification kits and RNeasy Mini kits for RNA cleanup were from Qiagen. All other chemicals were from Sigma or Fisher Scientific.
Generation of RNA Transcripts
Extension assays were carried out on a 425-nt RNA template derived from a 420-nt section of the HIV-1 gag-pol genome region (bases 1924–2343 on pNL43) and 5 additional non-HIV nts at the 5′-end. PCR amplification was performed using forward primer 5′-GATTTAGGTGACACTATAGTCAAACAGAAAGGCAATTTTAGGAAC-3′ and reverse primer 5′-TATCATCTGCTCCTGTATCT-3′. Underlined bases contain an SP6 promoter and italicized bases correspond to the 5 non-HIV nts. After amplification (94 °C/3 min followed by 25 cycles of: 94 °C/1 min, 50 °C/1 min, 72 °C/1 min, followed by a final extension at 72 °C for 5 min), template DNA was purified using the QIAquick PCR Purification kit and 8 μl was used in RNA transcription reactions with SP6 polymerase according to the manufacturer instructions. Reactions were allowed to proceed for 3 h, at which point they were treated with 0.4 units/μl of DNase I for 20 min at 37 °C. RNA was purified using the RNeasy Mini kit and quantified using UV spectroscopy. The 60-nt RNA for the RNase H assays was prepared by digesting 10 μg of pBSM13+ (Stratagene) plasmid with 20 units of HindIII in 20 μl using the manufacturer's protocol. Digested material was used in a transcription reaction with T7 RNA polymerase as described above. Transcription reactions were extracted with phenol:chloroform:isoamyl alcohol (25:24:1) and precipitated with ethanol. The RNA was gel purified on an 8% denaturing polyacrylamide gel, located by UV shadowing, and recovered by the crush and soak gel elution method (33). The amount of recovered RNA was determined spectrophotometrically from optical density. Gel-purified RNAs were dephosphorylated with CIP using the manufacturer's protocol and supplied buffer. The reactions were extracted and precipitated and the recovered RNA was end-labeled using T4 polynucleotide kinase as described below. The material was then run on a second denaturing polyacrylamide gel, located by autoradiography and recovered as described above. This material was hybridized to a DNA oligonucleotide as described below. The sequence of the recovered RNA was: 5′-GGGCGAAUUCGAGCUCGGUACCCGGGGAUCCUCUAGAGUCGACCUGCAGGCAUGCAAGCU-3′.
5′-End-labeling of DNA Primers for Reverse Transcription and Off-rate Determinations, and RNA Template for RNase H Assays
The reverse DNA primer (see above), DNA primer for the binding half-life determination assays (33 nt: 5′-TCCCCGGGTACCGAGCTCGAATTCGCCCTATAG-3′), and the RNA template for the RNase H assays (see above) were 5′ end-labeled using T4 polynucleotide kinase and [γ-32P]ATP using the manufacturer's protocol and supplied buffer. The material was run through a G-25 Sephadex column to remove excess ATP.
Nucleic Acid Hybridization
Hybrids were prepared by mixing RNA (425-nt product described above) and end-labeled DNA (reverse primer from above) for reverse transcription assays at an ∼1:1.5 ratio (template:primer) of 3′ termini in buffer containing 50 mm Tris-HCl (pH 8), 80 mm KCl, and 1 mm DTT. For binding half-life determination assays the end-labeled 33-nt primer described above was hybridized to a 50-nt DNA (5′-TTGTAATACGACTCACTATAGGGCGAATTCGAGCTCGGTACCCGGGGATC-3′) and for RNase H assays, the end-labeled 60-nt RNA was hybridized to a 23-nt DNA (5′-AGGATCCCCGGGTACCGAGCTCG-3′), each at a 1:1 ratio. For all hybrids, the mixtures were heated to 65 °C for 5 min, and then cooled slowly to room temperature.
Reverse Transcriptase Primer Extension Assay
Hybridized primer-template was incubated in 21 μl of buffer containing 50 mm Tris-HCl (pH 8.0), 80 mm KCl, 1 mm DTT, 100 μm each dNTP (final concentration, unless otherwise noted), 0.2 units/μl RNase inhibitor, and varying concentrations of ZnCl2, MnCl2, CaCl2, or MgCl2 (as indicated in legends). Following a 3 min pre-incubation at 37 °C, 4 μl of RT (final concentration of 25 nm) was added to begin extension. The final template concentration was 5 nm in the 25-μl reactions. The reactions were stopped by addition of 2× loading buffer (90% formamide, 10 mm EDTA (pH 8.0), and 0.025% bromphenol blue and xylene cyanol) at the indicated times. Samples were separated on 6 or 8% denaturing urea gels (7 m urea, 19:1 acrylamide:bisacrylamide), dried, and imaged using a Fujifilm FLA-5100 or FLA-7000.
RNase H Assays
Hybridized RNA-DNA from above (25 nm final concentration) was incubated in 21 μl of buffer containing 50 mm Tris-HCl (pH 8.0), 80 mm KCl, 1 mm DTT, 0.2 units/μl RNase inhibitor, and various concentrations of MgCl2 or ZnCl2 (as indicated in the legend to Fig. 5). A 3-min preincubation at 37 °C was followed by addition of 4 μl of HIV-RT (10 nm final concentration), and incubation was continued for 5 min (unless otherwise indicated). Products were resolved on 10% denaturing gels as described below.
FIGURE 5.
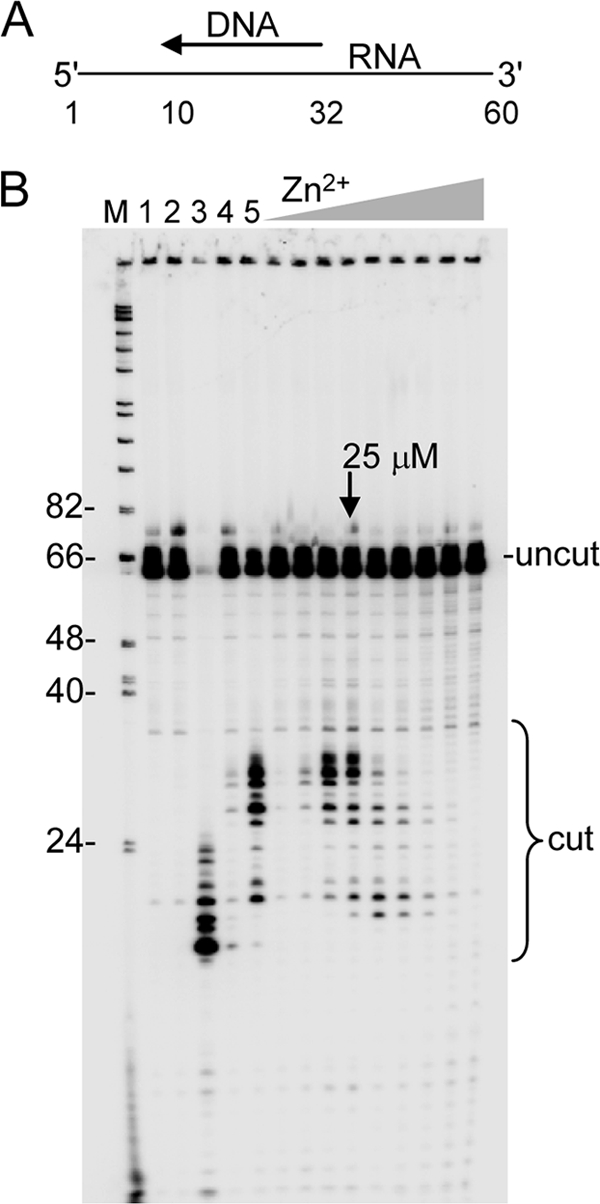
RNase H digestion with HIV-RT in the presence of varying concentrations of Zn2+. A, schematic diagram of the primer-template. A 5′-labeled 60 nt RNA is hybridized to a 23 nt DNA primer. B, in order, control lanes refer to: 1) no enzyme, 2) enzyme without additional divalent cation, and 3) E. coli RNase H digestion of substrate. Lanes labeled 4 and 5 contain 2 mm MgCl2 and no ZnCl2. Lane 4 was digested for 5 s, all other lanes are 5-min digests. Zn2+ concentrations were 2, 5, 10, 25, 100, 200, 400, 800, and 1200 μm. The concentration for optimal RNase H activity is noted with an arrow. Positions of uncut 60 nt RNA and cleavage products are indicated. Lane M, refer to Fig. 1B.
Assays to Determine RT-Substrate Binding Half-lives and Equilibrium Dissociation Constants (Kd)
Half-life and Kd determination assays for reactions with RT and ZnCl2, MnCl2, or MgCl2 were performed as described previously (34). A DNA-DNA primer-template was used: primer: 5′-TCCCCGGGTACCGAGCTCGAATTCCCCCTATAG-3′; template: 5′-TTGTAATACGACTCAGTATAGGGCGAATTCGAGCTCGGTACCCGGGGATC-3′.
Processivity Assay
Processivity assays were conducted using the conditions stated above under “Reverse Transcriptase Primer Extension Assay” with the following modifications: 1) the volume of the preincubation reaction was 50 μl; 2) divalent cations and dNTPs were not included in the preincubation step; 3) RT (50 nm final concentration) was included in the preincubation step; 4) reactions were initiated by the addition of a 10 μl solution containing a divalent cation (final concentration 400 μm or 2 mm for ZnCl2 and MgCl2, resp.); dNTPs (final concentration 100 μm each) and poly(rA)-oligo(dT) (final concentration 0.4 μg/ul) in 50 mm Tris-HCl (pH 8), 80 mm KCl, 1 mm DTT. 5-μl aliquots were removed at the indicated times (Fig. 6), and reactions were terminated with 5 μl of 2× loading buffer (see above). Samples were run on a 6% denaturing gel as stated above. The addition of poly(rA)-oligo(dT) limits synthesis to preformed complexes of RT-(primer-template) and further limits synthesis to a single processive cycle (35). Poly(rA)-oligo(dT) used in the reactions was prepared by mixing oligo (dT20) with poly(rA) at a 1:8 ratio (w/w) in 10 mm Tris-HCl (pH 7.5). The mixture was incubated for 10 min at 37 °C and slow cooled to room temperature.
FIGURE 6.

HIV-RT processivity with Mg2+ and Zn2+. Extension with 2 mm MgCl2 or 400 μm ZnCl2 was performed with the primer-template in Fig. 1A and in the presence of a non-radiolabeled poly(rA)-oligo(dT) trap which limits primer extension to a single binding event between RT and the primer-template. Lane 1, no enzyme; lane 2, trap control: enzyme was mixed with the trap then incubated with primer-template in standard reaction conditions for 16 min or 4 h for Mg2+ and Zn2+ reactions, resp.; lane 3, primer extension in the absence of trap for 16 min or 4 h for Mg2+ and Zn2+ reactions, respectively. Time points for trap reactions with MgCl2 were 30s, 1, 2, 4, 8, and 16 min, and for ZnCl2, 15 and 30 min, followed by 1, 2, 3, and 4 h. Refer to Fig. 1B for other markings.
Determination of Average Extension Rate
Average extension rates for reactions with 400 μm ZnCl2 or 2 mm MgCl2 were determined using results from the reverse transcription primer extension assays and the Fuji MultiGauge software (version 3.0) on the FLA-5100 or FLA-7000 and the molecular weight determination program. 30 and 1 min reaction times were used for ZnCl2 and MgCl2 reactions, respectively. At these time points, products had not reached the end of the template. The average extension rate was estimated by calculating the size in nts of each band on the gel (s) and subtracting the primer length (20 nt), then using the imager to determine the relative proportion (with the total being set to 1) of the total extended primers to which each band corresponded (y). The band's contribution to the average extension rate can be represented by the following equation: (s-20) × y. The average extension rate can then be calculated using the following expression, where t is the reaction time in seconds.
 |
As an example, if a 60-s reaction produced 4 extension product bands of sizes 30, 40, 60, and 80 nts that were of equal intensity then the average extension rate would be [(30–20) × 0.25 + (40 − 20) × 0.25 + (60 − 20) × 0.25 + (80 − 20) × 0.25]/60, which equals 32.5 nt/s.
Gel Electrophoresis
Denaturing polyacrylamide gels (19:1 acrylamide:bis-acrylamide, respectively), were prepared and subjected to electrophoresis as described (33).
RESULTS
Zn2+ Potently Inhibits HIV-RT Catalysis in the Presence of Mg2+
Although Zn2+ has been shown to inhibit HIV-RT activity, the mechanism and nature of the inhibition have not been investigated. To investigate this, primer-directed reverse transcription was performed using a 425-nt RNA template derived from the gag-pol region of the HIV genome primed with a 20-nt 5′ 32P end-labeled DNA primer (Fig. 1A). Full extension of the primer to the end of the template produced a 425 nt DNA product (as indicated in Fig. 1B). In these experiments, reactions were performed using 100 μm of each dNTP. Control reactions performed in the absence of Mg2+ with ZnCl2 (lanes 2 and 3, 1 and 30 min, resp.) and ZnSO4 (lanes 4 and 5, 1, and 30 min, resp.) at 400 μm each showed synthesis with Zn2+ under optimal conditions (see Fig. 3). Both ZnCl2 and ZnSO4 gave comparable results, and the former was used for all other experiments. Other reactions were performed for 1 or 30 min (as indicated) with 2 mm MgCl2 (optimal for extension rate (see Fig. 4) in the absence (Ø) or in the presence of increasing amounts of ZnCl2. Even very small amounts of Zn2+ (0.5–2 μm, first 3 lanes following the Zn2+-free reaction) had a detectable affect on the rate of nucleotide addition (based on results with 1 min reactions) to the primer and a more dramatic affect was observed at 5 μm, even thought the ratio of Mg2+:Zn2+ was 400:1 at this concentration. Severe inhibition was observed between 25 and 800 μm Zn2+. Both the maximum length of extension products and the extent of RT pausing were altered in the presence of Zn2+ with the former decreasing and the latter increasing. Reactions performed for 30 min indicated that the inhibition caused by low amounts of Zn2+ (0.5–25 μm) did not prevent the production of fully extended products although the quantity of those products was reduced at the expense of more intense pausing. The profile of synthesis in the presence of 2 mm Mg2+ plus 400 μm Zn2+ compared with 400 μm Zn2+ alone (lanes 3 and 5) were similar, suggesting that these reactions were being driven almost solely by Zn2+.
FIGURE 1.
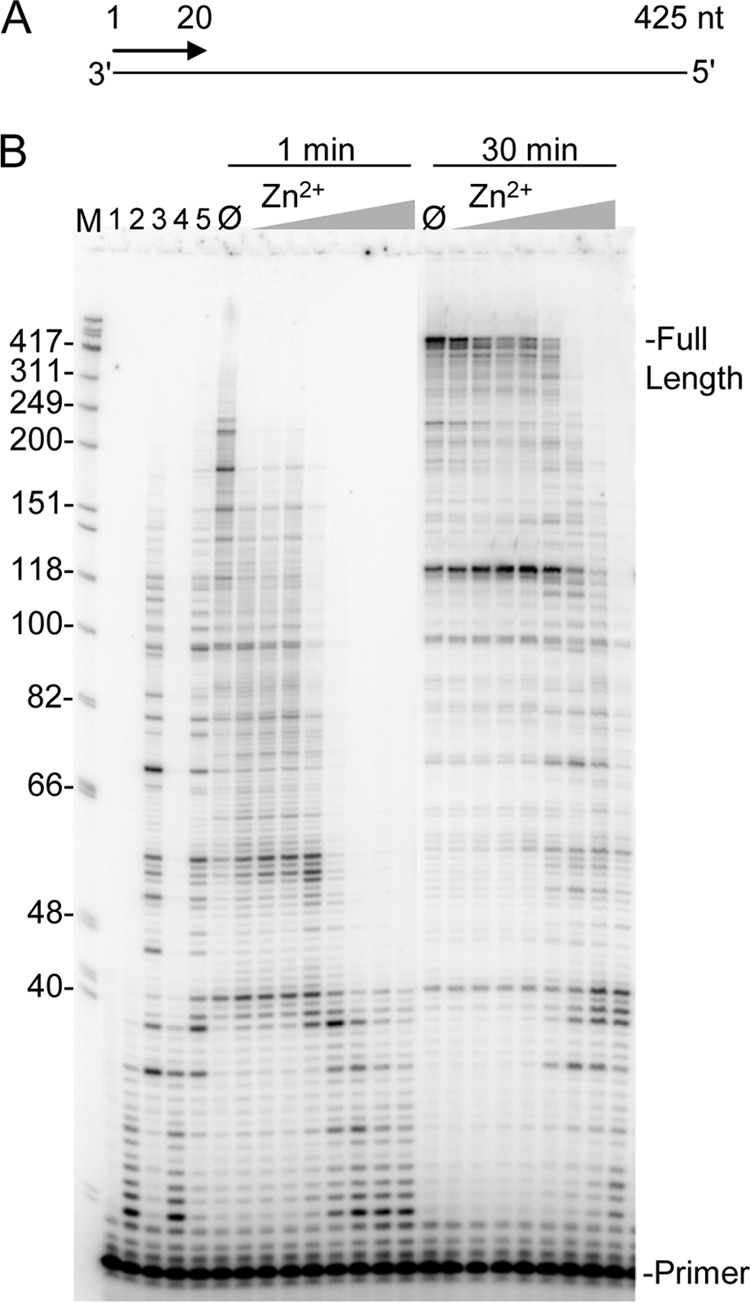
Reverse transcription with 2 mm Mg2+ and increasing Zn2+ concentrations. A, schematic representation of the primer-template used for assays. Numbers indicate size in nts. B, primers labeled at 5′-end were hybridized to the RNA template and reactions were performed in the presence of 2 mm MgCl2 and increasing concentrations of ZnCl2. Reactions were stopped after 1 min or 30 min as indicated and electrophoresed on a 8% denaturing gel. The concentration of ZnCl2 in reactions was 0.5, 1, 2, 5, 25, 100, 400, 800 μm. Lane M, size marker in nts as indicated; lane 1, no enzyme; lane 2 and 3, 400 μm ZnCl2 with no MgCl2 for 1 and 30 min, resp.; lanes 4 and 5, 400 μm ZnSO4 with no MgCl2 for 1 and 30 min, resp.; lane Ø, 2 mm MgCl2 with no ZnCl2. Positions of the 20 nt primer and full extension products are indicated.
FIGURE 3.

Reverse transcription with Zn2+ alone and various dNTP concentrations. Reactions were performed on the primer-template shown in Fig. 1A for 30 min with increasing concentrations of ZnCl2 as follows: 5, 10, 25, 100, 200, 400, 800, 1500, and 3000 μm. The concentration of each of the 4 equimolar dNTPs in the reactions is noted above the lanes. Lane M, refer to Fig. 1B; lane 1, 30 min reaction with 2 mm MgCl2 and 100 μm dNTPs; lane 2, reaction with no enzyme. Other markings are as indicated in Fig. 1B.
FIGURE 4.

Reverse transcription with various concentrations of Mg2+ and Mn2+. Reactions were performed on the primer-template shown in Fig. 1A for 3 min with 100 μm each dNTP and increasing concentrations of MgCl2 or MnCl2 as follows: 3.9, 7.8, 15.6, 31.3, 62.5, 125, 250, and 500 μm, followed by 1, 2, 4, 8, and 16 mm. Lane M, refer to Fig. 1B; lane 1, no enzyme; lane 2, enzyme without additional divalent cation. Lanes where optimal synthesis occurred are denoted on the figure with an arrow. Other markings are as indicated in Fig. 1B.
Zn2+ Is a More Potent Inhibitor of HIV-RT than Ca2+ and Mn2+
Both Ca2+ and Mn2+ have also been shown to inhibit HIV-RT (see Introduction). These physiologically relevant divalent cations were compared with Zn2+ in an inhibition assay in the presence of 2 mm Mg2+ as described above but in 2 min reactions (Fig. 2). The Zn2+, Ca2+, and Mn2+ were at 0, 7.8, 31, 125, 500, or 2000 μm. All three cations inhibited extension but Zn2+ was much more potent than the other two. The length of the longest extended products had decreased by ∼50% in the presence of just 7.8 μm Zn2+ while ∼500 μm Ca2+ or Mn2+ was required to observe the same inhibition. In general, inhibition of extension was accompanied by an increase in pausing rather than a decrease in the amount of total primers that were extended. At the higher concentrations some decrease in total primer extension was also observed. This was especially evident at 2000 μm in reactions with Zn2+ and Mn2+.
FIGURE 2.

Inhibition of reverse transcription with 2 mm Mg2+ by increasing concentrations of Zn2+, Ca2+, and Mn2+. Reactions were performed on the primer-template shown in Fig. 1A for 2 min with 2 mm MgCl2 and increasing concentrations of the indicated divalent cation (all chloride salts) as follows: 7.8, 31, 125, 500, and 2000 μm. Lanes and markings are as indicated in Fig. 1B with the exception of the -E lane that corresponds to no enzyme added.
Zn2+ Supports Nucleotide Addition by HIV-RT and the Optimal Concentration Is Dependent on dNTP Concentrations
Reactions performed in the absence of Mg2+ showed that Zn2+ supports RT synthesis at a highly reduced incorporation rate (Fig. 3). Even at 5 μm in these 30 min reactions some extension of the primer was observed at all dNTP concentrations, indicating that Zn2+ binds with high affinity to RT. The maximum rate of nucleotide incorporation was observed at 400 μm Zn2+ when 100 μm equimolar dNTPs (400 μm total) were used. An assay performed with a homologous DNA template yielded the same optimal concentration and similar profile with respect to extension rate versus the concentration of Zn2+ (data not shown). Because nucleotides can chelate divalent cations (36) and Zn2+ catalyzed synthesis peaks at a low concentration, the optimal determined with higher dNTP concentration is unlikely to reflect the actual affinity of Zn2+ for RT, as most Zn2+ would be complexed with nucleotides under these conditions. Reactions performed with 10 or 1 μm dNTP concentrations were optimal at ∼100 and ∼25 μm Zn2+, respectively. There was a decline in extension length with lower dNTP concentrations indicating that the Zn2+ catalyzed reactions require high nucleotide concentrations to proceed at a maximal rate. Overall the results with the lowest dNTP concentration (1 μm) where very little Zn2+ is sequestered indicated that HIV-RT has a very high affinity for Zn2+. For comparison purposes, a titration with Mg2+ and Mn2+ (Ca2+ showed no catalytic activity at any concentrations tested (data not shown)) at 100 μm dNTPs was conducted (3 min reactions in each case) (Fig. 4). Like Zn2+, Mn2+ showed optimal synthesis at a low concentration (∼250 μm) indicating a very high affinity for RT as most of the free Mn2+ would have been otherwise chelated under these conditions. This is consistent with previous results indicating that there is at least one high affinity binding site for Mn2+ on RT (4). In contrast, Mg2+ demonstrated a broad activity peak with an optimal at 2 mm, Some synthesis with Mn2+ was observed all the way down to 3.9 μm while synthesis with Mg2+ was first observed at 31 μm. At 2 mm Mg2+ and 400 μm total dNTPs the free concentration of Mg2+ in the reactions would be about ∼1.6 mm (36) indicating a much higher concentration optimum than Zn2+ or Mn2+. These results are consistent with Zn2+ showing strong inhibition of reactions with Mg2+ even when present at concentrations that were ∼100-fold lower than Mg2+ (Figs. 1 and 2). Inhibition by Mn2+ appeared less potent even though it apparently binds RT as tightly as Zn2+. This probably resulted from the much more efficient synthesis of HIV-RT in the presence of Mn2+ compared with Zn2+. For example, the maximum extension rate for reactions performed under optimal conditions (100 μm dNTPs with 2 mm, 400 μm, and 250 μm divalent cation, for Mg2+, Zn2+, and Mn2+, respectively) was determined by calculating the lengths of the longest products in reactions that had not proceeded to the end of the template, and dividing by the reaction time in seconds. Values of 3.5, 1, and 0.1 nt/s were determined for Mg2+, Mn2+, and Zn2+ respectively.
Interactions of divalent cations with the structured heteropolymeric template may have played a role in the differential catalysis rates. To test this, extension rates for Mg2+ and Zn2+ were also measured on an unstructured oligo(dT)-primed poly(rA) template (see supplemental data). The maximum rate of nucleotide incorporation was ∼25 nts/s and ∼1 nt/s, with 2 mm Mg2+ and 400 μm Zn2+, respectively. Both rates are much faster than with the structured heteropolymeric template, as expected. However, Zn2+ still leads to a much slower extension rate indicating that Zn2+ interactions with RT, not with secondary structures, are the likely cause of the slow incorporation.
An additional control was also performed in these assays and is shown in lane 2 of Fig. 4 and lane 2 of Fig. 5B. In these assays RT was incubated with the substrate but divalent cation was omitted from the reactions. Depending on the purification procedures, small amounts of divalent cation can co-purify with enzymes. This could lead to low levels of activity in reactions performed in the absence of additional divalent cation. No activity was observed when divalent cation was omitted from the reactions indicating that both polymerase (Fig. 4) and RNase H (Fig. 5B) activity are dependent on the addition of divalent cation.
Zn2+ Supports the RNase H Activity of HIV-RT at Concentrations Similar to Those Observed for Nucleotide Extension
An RNase H assay was performed for 5 min in the presence of various concentrations of Zn2+ (Fig. 5B). These results were compared with 5 s reactions with 2 mm Mg2+ (lane 4) to see initial cleavage events, and 5 min reactions (lane 5) to observe further processing. Optimal cleavage with Zn2+ occurred at ∼25 μm Zn2+, comparable to the optimal observed for dNTP incorporation with low dNTP concentrations (Fig. 3). Both Zn2+ and Mg2+ yielded a similar cleavage profile indicating that cleavage specificity and position are the same for both cations. At higher Zn2+ concentrations, cleavage was both inhibited and shifted toward smaller products. This may have resulted from a change in the cleavage specificity or more rapid processing of initial cleavage products at higher concentrations.
HIV-RT Forms a Very Stable Catalytically Competent Complex with Primer-Template in the Presence of Zn2+ That Traverses the Template Slowly but Can Remain Associated for Hours
Because the extension rate of RT was ∼35 times greater in the presence of Mg2+ compared with Zn2+ (see above), it was reasonable to assume that processivity (average numbers of nucleotides added to the primer in a single binding event between RT and the substrate) would be much greater with Mg2+. Surprisingly, this was not the case. Reactions to test processivity were performed on the primer-template shown in Fig. 1A by pre-binding RT to the preformed primer-template then adding dNTPs and divalent cation along with an excess of poly(rA)-oligo(dT) to sequester enzymes that were not bound to or had dissociated from the primer-template. This limits synthesis to a single binding even between the enzyme and substrate (37). Reactions with 2 mm Mg2+ (left panel, Fig. 6) showed the expected profile. Primers were extended at a maximum rate of ∼3.5 nt/s with some extension products reaching the end of the 425 nt template by 2 min and more by 4 min. The extension profile of reactions performed for 8 and 16 min looked very similar to those performed for 4 min indicating that the single round of enzyme extension was essentially complete in 4 min. In contrast, the first time point shown for the reactions with 400 μm Zn2+ was 15 min. In this case reactions proceeded at the expected ∼0.1 nt/s and some products reached the end of the template by about 1 h. Fully extended products continued to accumulate even up to the final 4 h time point, although there was just a small increase between 3 and 4 h and those profiles looked very similar. It was remarkable that single enzyme molecules could stay associated with a single template for hours. Although the maximum rate of synthesis was ∼0.1 nt/s, on average polymerases moved much more slowly, at a calculated rate of 0.022 ± 0.003 nt/s for Zn2+ as compared with 1.7 ± 0.4 nt/s for Mg2+ reactions (results are and average ± S.D. from 3 or more reactions using 30 min and 1 min time points for 400 μm Zn2+ and 2 mm Mg2+, respectively; see “Experimental Procedures” for calculation). Despite this, the final processivity on this template was only modestly higher for Mg2+.
To test the stability of the enzyme-substrate complexes further, the half-life (t½) of RT for binding to a DNA-DNA primer-template was determined in the presence of Zn2+, Mn2+, and Mg2+ (Fig. 7A). This experiment required the use of DNA-DNA since off-rates in the presence of divalent cation cannot be easily measured with RNA-DNA except with RNase H mutant enzymes. Remarkably, complexes with Zn2+ and Mn2+ were ∼130 (t½ = 220 ± 60 min) and ∼45 (t½ = 77 ± 1 min) times more stable than those with Mg2+ (t½ = 1.7 ± 1.0 min). This is consistent with the ability of single enzyme molecules to remain bound to the template for such a long time in the processivity experiments (Fig. 6). Equilibrium binding constants (Kd) in the presence of Zn2+ and Mg2+ were also measured on the same primer-template (Fig. 7B). Consistent with the slower off-rate (koff) for Zn2+-RT complexes, the Kd was smaller in the presence of Zn2+ versus Mg2+ (0.79 ± 0.13 versus 2.9 ± 0.1 nm, respectively), indicating higher affinity binding with Zn2+. However, the difference was only ∼3.5-fold as compared with the ∼130-fold difference for koff. This indicates that Zn2+ also lowers the on-rate (kon) for RT. It is important to note that the dramatic differences in t½ observed with DNA-DNA only provide a qualitative comparison between affinity differences that may occur during DNA synthesis on an RNA template or even a DNA template (see “Discussion”). Still the results clearly suggest that Zn2+ and Mn2+ lead to more stable RT-(primer-template) complexes in comparison to the natural divalent cation, Mg2+.
FIGURE 7.

A, representative plot of RT-(primer-template) complex binding stability in the presence of Mg2+, Mn2+, and Zn2+. Half-lives are indicated with their respective curves. Results are an average of 3 exp. ± standard deviations. B, representative plot of bound RT (nm, determined by extension) versus [HIV-RT] (nm) used to determine the Kd values in the presence of Zn2+ or Mg2+. Experiments were performed as described under “Experimental Procedures.”
Other Viral RTs from AMV and MuLV as Well as an RNase H− HIV Mutant (HIV-RTE478>Q) Can Also Use Zn2+ for Nucleotide Incorporation
Nucleotide incorporation was also tested with three other RTs including an RNase H minus form of HIV-RT (HIV-RTE478>Q) and two other viral RTs, AMV-, and MuLV-RT. Not only did all 3 RTs show activity, but peak activity occurred at∼400 μm Zn2+ in all cases (assay were with 400 μm total dNTPs). The similar results likely derive from common active site architecture in these closely related RTs.
DISCUSSION
Like Ca2+ and Mn2+, previous reports have also shown that Zn2+ inhibits HIV-RT nucleotide incorporation in the presence of Mg2+ (see Introduction). The mechanism of inhibition has been best studied using Mn2+. Bolton et al. (4) have proposed that Mn2+ inhibits TY1 and HIV-RT by binding with high affinity and displacing Mg2+ at one of two proposed divalent cation binding sites near the polymerase active site. The authors propose that the two sites do not equally affect catalysis and the site that Mn2+ binds tightly to is dominant. Therefore, even if the second site is occupied by Mg2+, RT reverts to a Mn2+ catalysis mode similar to what is observed in the presence of Mn2+ alone. This mode is much less efficient with respect to the rate of incorporation. The model is supported by mutational analysis as well as the observance of two divalent cations in the polymerase domain of HIV-RT in some crystals (38). Interestingly, TY1 retrotransposon mutants that could overcome Mn2+ inhibition mapped not to the polymerase but to the RNase H domain (39). This led the authors to propose a model where mutations in the RNase H domain could influence polymerization through allosteric effects. The fact that mutations in the polymerase domain conferring resistance were not found does not necessarily mean that this domain is uninvolved in the observed inhibition by Mn2+. It is possible that mutations that alter Zn2+ binding may have a severe impact on RT activity and are therefore not selected.
The model proposed for Mn2+ is consistent with what was observed for Zn2+ in the current experiments, although our findings do not necessarily support this model versus a simpler model where Zn2+ or Mn2+ (or even Ca2+) simply displace Mg2+ from one or more binding site in the polymerase domain that control catalysis. What was clear from the current results is that Zn2+ is a more potent inhibitor of Mg2+ catalysis than Mn2+ or Ca2+, as it is required at a lower concentration to inhibit extension (Fig. 2). This is probably not due to a higher affinity of Zn2+ for RT as the optimal concentrations for Zn2+ and Mn2+ catalysis in the absence of Mg2+ were comparable (∼400 μm and 250 μm for Zn2+ and Mn2+, resp., in the presence of 400 μm total dNTPs). Primer extension in the presence of Mn2+ was, however, much more efficient than with Zn2+ (Figs. 1 and 4, note time difference in reactions). The ability of polymerases to use Mn2+ in place of Mg2+ in vitro has been well documented and some polymerases function as well or even better with Mn2+, although it tends toward lower polymerase fidelity (8). Because experiments reported here essentially evaluated polymerization efficiency by the rate of extension and the length of the products that were produced, the greater inhibition of Mg2+ catalysis by Zn2+ probably resulted from Zn2+ producing a more catalytically deficient enzyme. In contrast to Zn2+ and Mn2+, no catalysis was observed with Ca2+, while the level of inhibition of Mg2+ catalysis was similar to Mn2+. The inability of RT to use Ca2+ for catalysis is not surprising as it is not typically a cofactor in catalysis for enzymes (see Introduction). Specific properties of Ca2+, which differ markedly from Mg2+, make it a poor candidate for nucleotide catalysis by polymerases (discussed in Ref. 40). Ca2+ is also known to inhibit other DNA polymerases (41) and has been used to help stabilize crystal structures of some polymerases by prevent catalysis during crystal formation. Some crystals made with Ca2+ show this ion bound at the pol active site suggesting that it could inhibit polymerization by displacing Mg2+. One polymerase, Sulfolobus solfataricus DNA polymerase, has been reported to be able to use Ca2+ for nucleotide polymerization, though less efficiently than Mg2+ (42).
Because RNase H catalyzed cleavage of the RNA-DNA hybrid was observed in the presence of Zn2+ alone, it was clear that this cation can bind to the RNase H active site and stimulate cleavage in a manner similar to Mn2+ (39). Also notable was the ∼10–25 μm Zn2+ optimal for cleavage (Fig. 5), which was very close to the optimal concentration of Zn2+ for polymerization in the presence of low (1 μm) concentrations of dNTPs (Fig. 3). This suggests that the affinity for Zn2+ at the catalytic binding sites in the polymerase and RNase H domains are similar, and that Zn2+ binds with greater affinity at these sites than Mg2+ which requires much higher concentrations for optimal activity (Fig. 4). Interestingly, all the RTs that were tested including HIV, HIVE478>Q, AMV, and MuLV showed similar optimal Zn2+ concentrations for nucleotide incorporation. This suggests that the active sites of these RTs have similar affinity for Zn2+.
HIV-RTE478>Q is a commonly used enzyme for studying polymerization properties in the absence of RNase H activity. The glutamic acid to glutamine mutation in the RNase H active site abrogates RNase H activity and also disrupts a divalent cation binding site in the RNase H domain. This enzyme has polymerization properties that essentially mimic wild type enzyme, although it does have altered binding characteristics to primer-template in the absence or at low concentrations of Mg2+ (43, 44). Polymerization with HIV-RTE478>Q occurred in the presence of Zn2+ with a concentration optimum and profile that was similar to wild type RT (Fig. 8). Inhibition of catalysis in the presence of Mg2+ by Zn2+ essentially mimicked that in the wild type enzyme (data not shown). Overall the results suggest that the mutation in the RNase H domain had no effect and an allosteric interaction of the type reported for TY1 RT (see above) was not observed. It is important to note that although the E478>Q mutation would presumably alter divalent cation binding in the RNase H domain, it is not clear how it would be changed and if the mutation would abrogate the binding of one or more cations. Therefore the results do not rule out potential allosteric interactions of the type proposed by others.
FIGURE 8.

Reverse transcription with various RTs in the presence of Zn2+. Extension was performed with the primer-template from Fig. 1A with the indicated enzyme. Reactions were for 30 min and ZnCl2 concentrations were as follows: 100, 200, 400, and 800 μm, followed by 1.5 and 3 mm. Lane M is as in Fig. 1B. Optimal concentrations for extension are noted for each enzyme with an arrow. Lane -E, no enzyme control; lane 1, extension with the indicated enzyme for 30 min with 2 mm MgCl2. Other markings are as indicated in Fig. 1B.
The dramatic increase (>100-fold) in the stability of RT-Zn2+-(primer-template) compared with RT-Mg2+-(primer-template) complexes was surprising. Importantly, the method for measuring dissociation did not directly reflect dissociation that occurs during active DNA synthesis. No dNTPs were present and the polymerase was not cycling thorough the various phases of nucleotide incorporation (primer-template binding, nucleotide binding, catalysis, and translocation, then dissociation). Each of these phases may have different dissociation rates. Reports have also shown that binding of RT to nucleotides can stabilize binding (45). Finally, DNA-DNA primer-templates were used in the off-rate experiments as RNA-DNA cannot be used since it is degraded by RNase H activity in the presence of divalent cation. Experiments have shown that in the presence of Mg2+, HIV-RT binds more stably to both RNA-DNA and DNA-DNA (43), so the results may under-represent the stability of Mg2+ complexes. Processivity measurements can be used as an indicator of dissociation during active synthesis although they cannot be used to directly calculate a dissociation rate. The processivity is dependent on several factors including the sequence/structure and type (RNA or DNA) of nucleic acid used, as well as the polymerase dissociation and incorporation rates. Processivity was greater in the presence of Mg2+ than with Zn2+ as the average length of extended products was clearly greater with Mg2+, but the difference was relatively small considering that RT incorporated nucleotides about 35 times more rapidly in the presence of Mg2+. This large difference in incorporation rate is compensated by very stable binding in the presence of Zn2+. With Zn2+, RT molecules were able to remain bound to the primer-template for hours even though nucleotides were being added to the primer at a maximal rate of ∼1 every 10 s and at an average rate that was much less than this (see “Results” and Fig. 7). This essentially means that polymerases stalled for long periods of time after each nucleotide addition but did not dissociate from the template. In contrast, extension with Mg2+ was essentially complete after 4 min as paused products remained stable and the level of fully extended primers did not change beyond this time point. Overall the processivity experiment supports the results from the off-rate experiments and indicates that the RT-Zn2+-(primer-template) complex is extremely stable. The reason for this high stability is unclear. Differing affects of the divalent cations on the template structure cannot be ignored as contributing factors and also the lower concentration of divalent cation that was used in the Zn2+ versus Mg2+ experiments may have had some effect. Still, these would not be expected to account for the greater than 100-fold difference in off-rates. Interestingly, RT-Mn2+-(primer-template) complexes were also more stable than those formed with Mg2+, though to a lesser extent (Fig. 7). Likewise, Mn2+ also reduced the rate of nucleotide incorporation, again to a lesser extent than Zn2+. This brings about the possibility that these cations may stabilize a particular confirmation of RT that is conducive to tight binding but not fast incorporation kinetics, or that they stabilize a tight binding confirmation and slower catalysis occurs because of relatively poor chemistry compared with Mg2+ (8). For example, they could promote the “productive” versus “nonproductive” RT confirmation postulated by Whörl et al. (46), the former of which is proposed to be more stable.
In conclusion, Zn2+ inhibits HIV-RT synthesis not by directly stopping catalysis, but by forming a highly stable complex that has very slow incorporation kinetics. The RT-Zn2+-(primer-template) complex can be thought of as a “dead-end complex” since it ties up RT- potentially for hours, in a complex that is from a kinetic perspective, minimally productive. HIV-RT apparently has higher affinity for Zn2+ (and Mn2+ and Ca2+ also) than Mg2+ at one or more of the divalent cation binding sites. Therefore, relatively low concentrations of Zn2+ can inhibit RT extension in the presence of much higher concentrations of Mg2+. Since Zn2+ has relatively low toxicity (47) and can inhibit RT extension at low concentrations (just a few μm was required to slow extension in the presence of 2 mm Mg2+, Fig. 1), it could, at least in theory, be useful for slowing viral replication and zinc-based compounds present an opportunity to exploit a weakness in HIV that might present a novel avenue for future drug development. However the possibly of using supplements or natural minerals including Zn2+ must be approached with caution. Even though Zn2+ can inhibit RT at low μm concentrations that are lower than the level of total Zn2+ in cells and plasma, they are still ∼2–3 orders of magnitude greater than the level of free available Zn2+ in cells (see Introduction). In addition, obtaining low μm levels inside cells would likely require much higher levels in the bloodstream unless there was a specific delivery mechanism. Finally, μm concentrations of free Zn2+ in cells would almost certainly have profound effects on the transcription of specific genes, the oxidation state of cells, and general heath (see Introduction and Ref. 47). Despite these hurdles, the work described in this report could help set the stage for further investigations with other cations or cation combinations that may be more specific for HIV-RT.
Supplementary Material
Acknowledgments
We thank Dr. Stuart LeGrice of the HIV Drug Resistance Program for HIV-RTE478>Q, Dr. Michael Parniak of the University of Pittsburg for the wild HIV-RT clone, and Dr. Malcolm Martin and the NIH AIDS Research and Reference Reagent Program for plasmid pNL4-3.
This work was supported by NIGM, National Institutes of Health Grant GM051140 (to J. J. D.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental materials.
- HIV-RT
- human immunodeficiency virus reverse transcriptase
- AMV
- avian myeloblastosis virus
- MuLV
- Moloney murine leukemia virus
- RNase H
- ribonuclease H
- CIP
- calf intestinal alkaline phosphatase
- nt
- nucleotide.
REFERENCES
- 1. Herschhorn A., Hizi A. (2010) Cell Mol. Life Sci. 67, 2717–2747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Goff S. P. (1990) J. Acquir. Immune Defic. Syndr. 3, 817–831 [PubMed] [Google Scholar]
- 3. Filler A. G., Lever A. M. (1997) AIDS Res. Hum. Retroviruses 13, 291–299 [DOI] [PubMed] [Google Scholar]
- 4. Bolton E. C., Mildvan A. S., Boeke J. D. (2002) Mol. Cell 9, 879–889 [DOI] [PubMed] [Google Scholar]
- 5. Levinson W., Faras A., Woodson B., Jackson J., Bishop J. M. (1973) Proc. Natl. Acad. Sci. U.S.A. 70, 164–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Palan P. R., Eidinoff M. L. (1978) Mol. Cell Biochem. 21, 67–69 [DOI] [PubMed] [Google Scholar]
- 7. Tan C. K., Zhang J., Li Z. Y., Tarpley W. G., Downey K. M., So A. G. (1991) Biochemistry 30, 2651–2655 [DOI] [PubMed] [Google Scholar]
- 8. Johnson K. A. (2010) Biochim. Biophys. Acta 1804, 1041–1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schultz S. J., Champoux J. J. (2008) Virus Res. 134, 86–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mullen G. P., Serpersu E. H., Ferrin L. J., Loeb L. A., Mildvan A. S. (1990) J. Biol. Chem. 265, 14327–14334 [PubMed] [Google Scholar]
- 11. Maguire M. E., Cowan J. A. (2002) Biometals 15, 203–210 [DOI] [PubMed] [Google Scholar]
- 12. Moomaw A. S., Maguire M. E. (2008) Physiology 23, 275–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Traut T. W. (1994) Mol. Cell Biochem. 140, 1–22 [DOI] [PubMed] [Google Scholar]
- 14. Delva P., Pastori C., Degan M., Montesi G., Lechi A. (2004) J. Membr. Biol. 199, 163–171 [DOI] [PubMed] [Google Scholar]
- 15. Wang S., McDonnell E. H., Sedor F. A., Toffaletti J. G. (2002) Arch. Pathol. Lab. Med. 126, 947–950 [DOI] [PubMed] [Google Scholar]
- 16. Cousins R. J., Liuzzi J. P., Lichten L. A. (2006) J. Biol. Chem. 281, 24085–24089 [DOI] [PubMed] [Google Scholar]
- 17. Dubben S., Hönscheid A., Winkler K., Rink L., Haase H. (2010) J. Leukoc. Biol. 87, 833–844 [DOI] [PubMed] [Google Scholar]
- 18. Honscheid A., Dubben S., Rink L., Haase H. (2011) J. Nutr. Biochem. doi: 10.1016/j.jnutbio.2010.10.007 [DOI] [PubMed] [Google Scholar]
- 19. Vallee B. L., Falchuk K. H. (1993) Physiol. Rev. 73, 79–118 [DOI] [PubMed] [Google Scholar]
- 20. Eide D. J. (2006) Biochim. Biophys. Acta 1763, 711–722 [DOI] [PubMed] [Google Scholar]
- 21. Coleman J. E. (1992) Annu. Rev. Biochem. 61, 897–946 [DOI] [PubMed] [Google Scholar]
- 22. Kitamura H., Morikawa H., Kamon H., Iguchi M., Hojyo S., Fukada T., Yamashita S., Kaisho T., Akira S., Murakami M., Hirano T. (2006) Nat. Immunol. 7, 971–977 [DOI] [PubMed] [Google Scholar]
- 23. Yamasaki S., Sakata-Sogawa K., Hasegawa A., Suzuki T., Kabu K., Sato E., Kurosaki T., Yamashita S., Tokunaga M., Nishida K., Hirano T. (2007) J. Cell Biol. 177, 637–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brewer G. J., Aster J. C., Knutsen C. A., Kruckeberg W. C. (1979) Am. J. Hematol. 7, 53–60 [DOI] [PubMed] [Google Scholar]
- 25. Fridlender B., Chejanovsky N., Becker Y. (1978) Virology 84, 551–554 [DOI] [PubMed] [Google Scholar]
- 26. Ilouz R., Kaidanovich O., Gurwitz D., Eldar-Finkelman H. (2002) Biochem. Biophys. Res. Commun. 295, 102–106 [DOI] [PubMed] [Google Scholar]
- 27. Siberry G. K., Ruff A. J., Black R. (2002) Nutritional Res. 22, 527–538 [Google Scholar]
- 28. Slaby I., Lind B., Holmgren A. (1984) Biochem. Biophys. Res. Commun. 122, 1410–1417 [DOI] [PubMed] [Google Scholar]
- 29. te Velthuis A. J., van den Worm S. H., Sims A. C., Baric R. S., Snijder E. J., van Hemert M. J. (2010) PLoS Pathog. 6, e1001176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Walther U. I., Wilhelm B., Walther S. C., Mückter H., Forth W. (2000) In Vitr. Mol. Toxicol. 13, 145–152 [DOI] [PubMed] [Google Scholar]
- 31. Adachi A., Gendelman H. E., Koenig S., Folks T., Willey R., Rabson A., Martin M. A. (1986) J. Virol. 59, 284–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hou E. W., Prasad R., Beard W. A., Wilson S. H. (2004) Protein. Expr. Purif. 34, 75–86 [DOI] [PubMed] [Google Scholar]
- 33. Sambrook J., Russell D. W. (2001) Molecular Cloning: A Laboratory Manual, 3rd Ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 34. Olimpo J. T., DeStefano J. J. (2010) Nucleic Acids Res. 38, 4426–4435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. DeStefano J. J., Buiser R. G., Mallaber L. M., Myers T. W., Bambara R. A., Fay P. J. (1991) J. Biol. Chem. 266, 7423–7431 [PubMed] [Google Scholar]
- 36. Goldschmidt V., Didierjean J., Ehresmann B., Ehresmann C., Isel C., Marquet R. (2006) Nucleic Acids Res. 34, 42–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. DeStefano J. J., Buiser R. G., Mallaber L. M., Fay P. J., Bambara R. A. (1992) Biochim. Biophys. Acta 1131, 270–280 [DOI] [PubMed] [Google Scholar]
- 38. Huang H., Chopra R., Verdine G. L., Harrison S. C. (1998) Science 282, 1669–1675 [DOI] [PubMed] [Google Scholar]
- 39. Yarrington R. M., Chen J., Bolton E. C., Boeke J. D. (2007) J. Virol. 81, 9004–9012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang M., Lee H. R., Konigsberg W. (2009) Biochemistry 48, 2075–2086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pelletier H., Sawaya M. R., Wolfle W., Wilson S. H., Kraut J. (1996) Biochemistry 35, 12762–12777 [DOI] [PubMed] [Google Scholar]
- 42. Irimia A., Zang H., Loukachevitch L. V., Eoff R. L., Guengerich F. P., Egli M. (2006) Biochemistry 45, 5949–5956 [DOI] [PubMed] [Google Scholar]
- 43. Cristofaro J. V., Rausch J. W., Le Grice S. F., DeStefano J. J. (2002) Biochemistry 41, 10968–10975 [DOI] [PubMed] [Google Scholar]
- 44. Schatz O., Cromme F. V., Grüninger-Leitch F., Le Grice S. F. (1989) FEBS Lett. 257, 311–314 [DOI] [PubMed] [Google Scholar]
- 45. Tong W., Lu C. D., Sharma S. K., Matsuura S., So A. G., Scott W. A. (1997) Biochemistry 36, 5749–5757 [DOI] [PubMed] [Google Scholar]
- 46. Wöhrl B. M., Krebs R., Goody R. S., Restle T. (1999) J. Mol. Biol. 292, 333–344 [DOI] [PubMed] [Google Scholar]
- 47. Plum L. M., Rink L., Haase H. (2010) Int. J. Environ. Res. Public Health 7, 1342–1365 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.