

Background: Mycophenolic acid sensitivity varies by 103-fold among IMP dehydrogenases (IMPDHs) from different sources even though the drug-binding site is completely conserved.
Results: Resistant IMPDHs have a different kinetic mechanism compared with sensitive enzymes.
Conclusion: Resistance results from a failure to accumulate the drug-sensitive intermediate.
Significance: This is a novel mechanism of drug resistance.
Keywords: Dehydrogenase, Drug Resistance, Enzyme Inhibitors, Enzyme Mechanisms, Fungi, Nucleoside/Nucleotide Biosynthesis, IMP Dehydrogenase (IMPDH)
Abstract
The filamentous fungus Penicillium brevicompactum produces the immunosuppressive drug mycophenolic acid (MPA), which is a potent inhibitor of eukaryotic IMP dehydrogenases (IMPDHs). IMPDH catalyzes the conversion of IMP to XMP via a covalent enzyme intermediate, E-XMP*; MPA inhibits by trapping E-XMP*. P. brevicompactum (Pb) contains two MPA-resistant IMPDHs, PbIMPDH-A and PbIMPDH-B, which are 17- and 103-fold more resistant to MPA than typically observed. Surprisingly, the active sites of these resistant enzymes are essentially identical to those of MPA-sensitive enzymes, so the mechanistic basis of resistance is not apparent. Here, we show that, unlike MPA-sensitive IMPDHs, formation of E-XMP* is rate-limiting for both PbIMPDH-A and PbIMPDH-B. Therefore, MPA resistance derives from the failure to accumulate the drug-sensitive intermediate.
Introduction
Penicillium brevicompactum produces a toxic small molecule called mycophenolic acid (MPA),4 which is used as an immunosuppressive drug (see Fig. 1A) (1). MPA is a potent inhibitor of eukaryotic IMP dehydrogenases (IMPDHs) and thus has antifungal activity (2). P. brevicompactum contains two genes for IMPDH, PbIMPDH-A and PbIMPDH-B (3). The PbIMPDH-B gene is located within the cluster that encodes the MPA biosynthetic proteins. Both PbIMPDH-A and PbIMPDH-B are more resistant to MPA compared with IMPDHs from fungi that do not produce the drug (e.g. Aspergillus nidulans IMPDH, AnImdA) (4, 5). However, PbIMPDH-B is remarkably resistant to MPA, with an ∼1000-fold greater IC50 than that of typical eukaryotic IMPDHs. Curiously, the active site of PbIMPDH-B is essentially identical to MPA-sensitive IMPDHs (see Fig. 1B), so the mechanistic basis of drug resistance is not understood.
FIGURE 1.

IMPDH reaction. A, mechanism of MPA inhibition. R5P, 1-β-d-ribofuranosyl 5-phosphate. B, the alignment of IMPDH-A and IMPDH-B from P. brevicompactum 23078 and Penicillium chrysogenum, IMPDH-A from A. nidulans, and IMPDH2 from Chinese hamster was mapped onto the x-ray crystal structure of the MPA complex of Chinese hamster IMPDH (Protein Data Bank code 1JR1 (8)). Dark magenta, 100% conserved; tan, 58% conserved; dark cyan, 17% conserved. MPA is shown in blue ball and stick, the IMP intermediate in gray ball and stick, K+ in purple, and residues within 4 Å of MPA and IMP in stick. Ala-249 is also shown because mutations at this residue confer MPA resistance.
IMPDH catalyzes the conversion of IMP to XMP via two distinct chemical reactions (6). The first step is a hydride transfer reaction that produces NADH and the covalent intermediate E-XMP* (Fig. 1B). NADH dissociates, and the enzyme switches from the open conformation (E-XMP*open) to the closed conformation (E-XMP*closed), where a mobile flap binds in the vacant cofactor site. This conformational change brings the catalytic Arg-429 into the active site to activate water for the hydrolysis of E-XMP*. (Chinese hamster IMPDH2 numbering will be used throughout unless noted otherwise.) Hydrolysis is at least partially rate-limiting in all IMPDHs studied to date (6), so E-XMP* is the predominant enzyme form.
MPA is an uncompetitive inhibitor of IMPDH. MPA binds to E-XMP*open in the cofactor-binding site and directly competes with flap closure (7–9). MPA thereby traps IMPDH in a nonproductive E-XMP*-MPA complex that is unable to complete the hydrolysis reaction. MPA will also bind to E-IMP, but the affinity for this complex is >300-fold lower than that for E-XMP* (10). Thus, the accumulation of E-XMP*open will be an important determinant of MPA sensitivity. Here, we show that the MPA resistance of PbIMPDH-B is elegantly conferred by a change in kinetic mechanism that reduces the accumulation of the drug-sensitive complex E-XMP*open.
EXPERIMENTAL PROCEDURES
Materials
IMP, MPA, 3-acetylpyridine adenine dinucleotide (APAD+), ADP, Tris, and common chemicals were purchased from Sigma. NAD+ was purchased from Roche Applied Science. KCl and trichloroacetic acid were purchased from Fisher. D2O and DCl were purchased from Cambridge Isotope Laboratories, Inc. (Andover, MA). [2-2H]IMP was synthesized as described previously (11). [8-14C]IMP was purchased from Moravek Biochemicals, Inc. Tiazofurin was obtained from NCI, National Institutes of Health.
Plasmids
Constructs for the recombinant expression of N-terminally His-tagged AnImdA, PbIMPDH-A, and PbIMPDH-B in Escherichia coli were constructed as described previously (5). Site-directed mutagenesis of PbIMPDH-B R429A was performed using a method based on QuikChange mutagenesis (Stratagene, La Jolla, CA). (This is residue 440 in PbIMPDH-B numbering.) Briefly, a T7 forward primer and reverse primer (GTC GTT CTC AGA GAA GTA GGC GGA AGC GCC AGC) were used to amplify by PCR a portion of the PbIMPDH-B gene carrying the R429A mutation. This PCR product was used as the forward and reverse mutant primers to complete the remainder of the QuikChange procedure according to the manufacturer's directions. Plasmids carrying the desired R429A mutation were verified by sequencing (GENEWIZ, South Plainfield, NJ).
Expression and Purification of His-tagged Proteins
Enzymes were expressed and purified as described previously (5). Briefly, plasmids were expressed in a ΔguaB derivative of E. coli BL21(DE3) (12); purified using a HisTrap affinity column (GE Healthcare) on an ÄKTApurifierTM system (GE Healthcare); and dialyzed into buffer containing 50 mm Tris (pH 8.0), 100 mm KCl, 1 mm DTT, and 10% (v/v) glycerol. All enzymes were purified to >90% purity as determined by SDS-PAGE.
Enzyme Concentration Determination
Enzyme concentration was determined by the Bio-Rad assay according to the manufacturer's instructions using IgG as a standard. The Bio-Rad assay overestimates IMPDH concentration by a factor of 2.6 compared with the concentration determined by active site titration using the irreversible inactivator 5-ethynyl-1-β-d-ribofuranosylimidazole-4-carboximide 5′-monophosphate (13), so a correction factor of 2.6 was used.
Enzyme Kinetics
Standard IMPDH assay buffer consisted of 50 mm Tris (pH 8.0), 100 mm KCl, 1 mm DTT, and various concentrations of IMP and NAD+ or APAD+. Enzyme activity was measured by monitoring the absorbance increase at 340 nm corresponding to the production of NADH or at 363 nm corresponding to the production of reduced 3-acetylpyridine adenine dinucleotide (APADH) on a Cary Bio-100 UV-visible spectrophotometer at 25 °C. Initial rates were calculated using ϵ340 = 6.2 mm−1 cm−1 and ϵ363 = 9.1 mm−1 cm−1. Initial velocity data were fit to either the Michaelis-Menten equation (Equation 1) or an uncompetitive substrate inhibition equation (Equation 2) using SigmaPlot (Systat Software).
MPA inhibition assays were performed at saturating IMP and half-saturating NAD+ concentrations to avoid complications arising from NAD+ substrate inhibition. The concentrations of IMP and NAD+ used for each enzyme are listed in the figure and table legends. MPA inhibition data were fit to either an uncompetitive inhibitor equation (Equation 3) or a noncompetitive/mixed inhibitor equation (Equation 4) using SigmaPlot.
Tight-binding inhibitor treatment was used for AnImdA. Data were fit to an uncompetitive tight-binding inhibitor equation (Equation 5) using SigmaPlot,
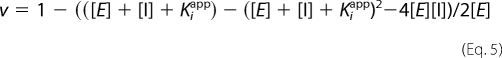 |
where Kiapp = Kii(1 + Km/[S]).
Multiple-inhibitor Experiment
Experiments with tiazofurin and ADP were performed in standard IMPDH assay buffer with constant concentrations of IMP and NAD+ for each enzyme as follows: AnImdA, 500 μm IMP and 250 μm NAD+; PbIMPDH-A, 1.5 mm IMP and 500 μm NAD+; and PbIMPDH-B, 3 mm IMP and 1 mm NAD+. Data were fit to an equation for multiple inhibition (Equation 6) using SigmaPlot,
where I and J are the inhibitors; v0 is the uninhibited initial velocity; Ki and Kj are the inhibition constants for I and J, respectively; and α is the interaction constant between the two inhibitors.
Primary Deuterium Isotope Effects
[2-2H]IMP was used as the substrate in standard IMPDH assay buffer. Activity was measured by holding IMP at a constant saturating concentration and varying the concentration of NAD+ or APAD+.
Solvent Deuterium Isotope Effects
Assay buffer was prepared in either H2O or D2O. pH meter readings were corrected to pD by adding 0.4 units. Activity was assayed by holding IMP at a constant saturating concentration and varying the concentration of NAD+ or APAD+.
Measurement of E-XMP*
Reaction mixtures contained 2 or 3 μm enzyme, 0.25 or 1 mm [8-14C]IMP (for AnImdA and PbIMPDH-A/B, respectively), and 5 mm NAD+ in standard IMPDH assay buffer at 25 °C. Enzyme, [8-14C]IMP, and NAD+ were mixed to initiate the reaction, and the reaction was quenched during steady state by precipitation with TCA to a final concentration of 10% (w/v). Enzyme samples were collected using HA-nitrocellulose filters (Bio-Rad) and washed with 10% (w/v) TCA. Radioactivity was measured using a scintillation counter. Control reactions in which enzyme was omitted were included for each experiment.
RESULTS
Mechanism of Inhibition
MPA is a potent inhibitor of AnImdA (IC50 = 26 nm), a moderate inhibitor of PbIMPDH-A (IC50 = 430 nm), and a poor inhibitor of PbIMPDH-B (IC50 = 27,000 nm) (5). If MPA binds exclusively to E-XMP*open, then uncompetitive inhibition should be observed versus both substrates for all three enzymes. However, if MPA binds to additional enzyme forms, noncompetitive inhibition will be observed.
We examined the effects of varying substrate concentration on MPA action to determine the mechanism of inhibition for all three fungal enzymes. MPA concentrations approximated enzyme concentration in experiments involving AnImdA, necessitating tight-binding inhibitor treatment (14). The data best fit uncompetitive inhibition for both IMP and NAD+ (Fig. 2 and Table 1), as expected if MPA binds selectively to E-XMP*open.
FIGURE 2.

Mechanism of MPA inhibition of fungal IMPDHs. A and B, AnImdA, [MPA] = 0 (●), 10 (○), 25 (▾), and 50 (▿) nm; [E] = 39 nm. A, versus NAD+, [IMP] = 500 μm, and Km = 10 μm. Data were fit to Equation 5. B, AnImdA versus IMP, [NAD+] = 500 μm, and Km = 170 μm. Data were fit to Equation 5. C and D, PbIMPDH-A, [MPA] = 0 (●), 200 (○), and 400 (▾) nm; [E] = 76 nm. C, versus NAD+, [IMP] = 2 mm, and Km = 130 μm. Data were fit to Equation 3. D, versus IMP, [NAD+] = 1 mm, and Km = 340 μm. Data were fit to Equation 4. E and F, PbIMPDH-B, [MPA] = 0 (●), 10 (○), and 25 (▾) μm; [E] = 71 nm. E, versus NAD+, [IMP] = 5 mm, and Km = 1.4 mm. Data were fit to Equation 3. F, versus IMP, [NAD+] = 5 mm, and Km = 0.79 mm. Data were fit to Equation 5. All fits were performed to nonlinear equations using SigmaPlot. Lineweaver-Burk plots are shown for inspection only.
TABLE 1.
Inhibition of fungal IMPDHs
| Inhibitor | AnImdA | PbIMPDH-A | PbIMPDH-B | PbIMPDH-B R429Aa |
|---|---|---|---|---|
| MPA IC50 (nm)b | 26 ± 2 | 430 ± 30 | (2.7 ± 0.9) × 104 | (3.5 ± 0.4) × 103 |
| MPA vs. IMP (nm) (mechanism) | Kii = 25 ± 3 (UC)c | Kii = 450 ± 150, Kis = 300 ± 100 (NC/mixed) | Kii = (2 ± 1) × 104, Kis = (2 ± 1) × 104 (NC/mixed) | NDd |
| MPA vs. NAD+ (nm) (mechanism) | Kii = 22 ± 2 (UC) | Kii = 500 ± 60 (UC) | Kii = (1.4 ± 0.1) × 104 (UC) | ND |
| Tiazofurin IC50 (mm)e | 0.24 ± 0.4 | 4 ± 0.4 | 3.5 ± 0.4 | ND |
| ADP IC50 (mm)e | 6 ± 2 | 10 ± 1 | 3.3 ± 0.3 | ND |
| αf | 0.8 ± 0.3 | 0.4 ± 0.1 | 0.8 ± 0.2 | ND |
a This is residue 449 in PbIMPDH-B numbering. The concentration of enzyme used for IC50 determination was 500 nm. The concentrations of substrates were 1 mm IMP and 3 mm NAD+.
b Values are from Ref. 5.
c UC, uncompetitive; NC, noncompetitive.
d ND, no data.
e IC50 values for tiazofurin and ADP are the apparent Ki values for tiazofurin and ADP obtained from the multiple-inhibitor experiment as described under “Experimental Procedures.” The concentrations of substrates were as follows: AnImdA, 250 μm IMP and 0.5 mm NAD+; PbIMPDH-A, 3 mm IMP and 500 μm NAD+; and PbIMPDH-B, 3 mm IMP and 1 mm NAD+.
f α = interaction constant between tiazofurin and ADP as described under “Experimental Procedures.”
MPA is also an uncompetitive inhibitor of PbIMPDH-A and PbIMPDH-B versus NAD+. However, MPA inhibition fits best to a noncompetitive/mixed pattern of inhibition versus IMP. This change in mechanism indicates that MPA no longer binds exclusively to E-XMP*. Perhaps the accumulation of E-XMP*open is so low that the association of MPA with other enzyme forms becomes significant.
MPA-resistant IMPDHs Have Less E-XMP*total
Whereas most IMPDHs accumulate E-XMP* in the steady state, the above observations suggested that less E-XMP* may accumulate in PbIMPDH-A and PbIMPDH-B. To test this hypothesis, the reactions were performed with radiolabeled IMP. The reactions were acid-quenched, and the protein-associated radioactivity was assessed by filter binding. This experiment measures all E-XMP* complexes (E-XMP*-NADH, E-XMP*open, and E-XMP*closed) and therefore provides an upper limit for the drug-sensitive E-XMP*open species. Whereas most IMPDHs contain ∼50% E-XMP* (6), 20 ± 1% of the active sites contain E-XMP* in PbIMPDH-A and only 7 ± 3% in PbIMPDH-B. These results suggest that MPA resistance may result from the failure to accumulate E-XMP*open.
More E-XMP*closed Cannot Account for Resistance
The above observations indicate that there is comparatively little E-XMP*total and, therefore, very little E-XMP*open during the reactions of PbIMPDH-A and PbIMPDH-B. The amount of E-XMP*open would be further reduced if the conformational equilibrium favored E-XMP*closed, as has been observed in other MPA-resistant IMPDHs (15, 16). We performed a multiple-inhibitor experiment to probe the open/closed equilibrium (6). Tiazofurin binds to the nicotinamide subsite of the NAD+-binding pocket, and ADP binds to the adenosine subsite. If the closed conformation is favored, one inhibitor will pull the equilibrium toward the open conformation, resulting in enhanced binding of the second inhibitor. This will be reflected in the interaction constant α, which will be <1 if such synergism is observed. In contrast, if E-XMP*open predominates, the two inhibitors will be independent, and α will be ∼1.
The value of α is ∼1 for both AnImdA and PbIMPDH-B (Table 1 and supplemental Fig. S1), indicating that the equilibrium favors the open form. In contrast, a synergistic interaction is observed for PbIMPDH-A (α = 0.4), which indicates that E-XMP*closed predominates. The value of α approximates the fraction of E-XMP*open. Because E-XMP*total = 20% (see above), then E-XMP*open can account for no more than 8% of enzyme in PbIMPDH-A.
Hydride Transfer Is Rate-limiting in Reactions of PbIMPDH-A and PbIMPDH-B
To further assess the accumulation of E-XMP*, we used isotope effects to identify the rate-limiting steps for each enzyme reaction. If hydrolysis is the rate-limiting step as typically seen, a solvent deuterium isotope effect will be observed. However, if hydride transfer is the slow step, there will be a primary deuterium isotope effect when [2-2H]IMP is used as the substrate.
No primary deuterium isotope effect is observed in the reaction of AnImdA, indicating that hydride transfer is fast, as observed for other IMPDHs (Table 2) (6). A small solvent deuterium isotope effect is observed on kcat, indicating that hydrolysis is partially rate-limiting step, also as expected. Therefore, E-XMP*open is the predominant enzyme form during the reaction of AnImdA, accounting for MPA sensitivity.
TABLE 2.
Isotope effects
| Enzyme | kcat | DV | DV/Km(NAD+) | D2OV | D2OV/Km(NAD+) | Rate-limiting step(s) |
|---|---|---|---|---|---|---|
| s−1 | ||||||
| Human IMPDH2 | 0.31 ± 0.01a | 1.0 ± 0.1a | 2.5 ± 0.2a | 1.8 ± 0.1a | NDb | Hydrolysis |
| AnImdA | 0.74 ± 0.06c | 1.0 ± 0.3 | 2.5 ± 0.6 | 1.6 ± 0.1 | 1.1 ± 0.1 | Hydrolysis |
| PbIMPDH-A | 0.70 ± 0.08c | 1.6 ± 0.2 | 1.6 ± 0.3 | 1.5 ± 0.1 | 1.2 ± 0.1 | Hydride transfer/hydrolysis |
| PbIMPDH-B | 0.41 ± 0.02c | 2.5 ± 0.3 | 3.4 ± 0.2 | 1.1 ± 0.1 | 0.9 ± 0.2 | Hydride transfer |
| PbIMPDH-B R429Ad | (4.0 ± 0.3) × 10−4 | ND | ND | 4.4 ± 0.4 | ND | Hydrolysis |
In contrast, PbIMPDH-A shows a primary deuterium isotope effect on kcat in addition to a solvent deuterium isotope effect, indicating that both steps are partially rate-limiting. This observation explains the relatively low amount of E-XMP*total that accumulates during the reaction of PbIMPDH-A.
PbIMPDH-B also shows a primary deuterium isotope effect on kcat but no solvent deuterium isotope effect, indicating that hydride transfer is completely rate-limiting in this reaction. E-IMP-NAD+ is therefore the predominant enzyme form for PbIMPDH-B. This kinetic mechanism minimizes the accumulation of E-XMP* in the steady state, rendering the enzyme resistant to MPA.
Increasing E-XMP* Makes PbIMPDH-A and PbIMPDH-B More MPA-sensitive
If the MPA resistance of PbIMPDH-A and PbIMPDH-B results from the low accumulation of E-XMP*open as proposed, then increasing the accumulation of E-XMP*open will make these enzymes more drug-sensitive. Reactions were performed in D2O to slow the hydrolysis of E-XMP*closed, which in turn will increase E-XMP*open. No change was observed in the IC50 for AnImdA in D2O (Table 3), as expected given that hydrolysis is already the rate-limiting step in this enzyme. In contrast, D2O makes the hydrolysis step fully rate-limiting in the PbIMPDH-A reaction, and a 6-fold decrease was observed in the IC50 of MPA. However, only a 2-fold decrease was observed in the IC50 of MPA for PbIMPDH-B because hydride transfer remains rate-limiting.
TABLE 3.
Effect of increasing E-XMP* on MPA inhibition
Conditions were as follows: AnImdA, 500 μm IMP and 500 μm NAD+; and PbIMPDH-A and PbIMPDH-B, 3 mm IMP and 1 mm NAD.
| Enzyme | Total E-XMP* | IC50(D2O)a | D2OIC50b | IC50(APAD+)a | AIC50c | IC50(D2O + APAD+)a | (D2O+A)IC50d |
|---|---|---|---|---|---|---|---|
| % | nm | nm | nm | ||||
| AnImdA | NDe | 21 ± 3 | 1.2 ± 0.2 | 13 ± 1 | 2.0 ± 0.2 | 15 ± 2 | 1.7 ± 0.3 |
| PbIMPDH-A | 20 ± 1 | 75 ± 10 | 5.7 ± 0.9 | 97 ± 5 | 4.4 ± 0.4 | 45 ± 3 | 9.4 ± 0.7 |
| PbIMPDH-B | 7 ± 3 | (1.2 ± 0.1) × 104 | 2.2 ± 0.8 | (1.0 ± 0.1) × 104 | 2.7 ± 0.9 | 3400 ± 200 | 8.0 ± 2.6 |
a Parentheses indicate the substitutions of D2O for H2O and/or APAD+ for NAD+.
b The ratio of IC50 to IC50(D2O).
c The ratio of IC50 to IC50(APAD+).
d The ratio of IC50 to IC50(D2O+APAD+).
e ND, no data.
The amount of E-XMP* can also be increased with the NAD+ analog APAD+. This dinucleotide has a greater reduction potential than NAD+ (E′0 = −0.258 for APAD+ −0.320 for NAD+) (17) and a lower affinity for IMPDH (6, 18). These properties will shift the reaction toward E-XMP*open, Importantly, because the reduced cofactor must dissociate before MPA can bind, the structure of E-XMP* formed in the reaction with APAD+ will be identical to the E-XMP* formed in the reaction with NAD+. As predicted, all of the enzymes became more MPA-sensitive by factors of 2–4 (Table 3). When APAD+ and D2O were combined, the effects were approximately additive for all three enzymes (Table 3).
Mutation R429A Increases MPA Sensitivity of PbIMPDH-B
Arg-429 is believed to act as a general base to activate water in the hydrolysis of E-XMP* (19). The mutation of the analogous Arg-419 to Ala decreases the hydrolysis step by a factor of 500 but has no effect on hydride transfer in Tritrichomonas foetus IMPDH (16). The R429A mutation caused a 1000-fold reduction in the value of kcat for PbIMPDH-B (Table 2). Hydrolysis is now completely rate-limiting as evidenced by a solvent isotope effect of 4.4. A burst of NADH production is observed in the pre-steady state, further confirming that hydrolysis is rate-limiting (supplemental Fig. S2). This mutant is 10-fold more sensitive to MPA than PbIMPDH-B.
DISCUSSION
The MPA resistance properties of PbIMPDH-A and PbIMPDH-B derive from changes in the kinetic mechanism. Unlike all other IMPDHs characterized to date (6, 21), hydride transfer is at least partially rate-limiting for both enzymes. This simple kinetic switch minimizes the accumulation of the drug-sensitive species E-XMP*open, allowing these enzymes to evade MPA inhibition.
The resistance of PbIMPDH-A can be almost completely reversed by increasing the accumulation of E-XMP*. The remaining 3-fold resistance can be attributed to the equilibrium between the open and closed conformations of E-XMP*, which favors the closed conformation in PbIMPDH-A. This shift in equilibrium may be due to the presence of Ser-249. MPA-sensitive IMPDHs contain an Ala at this position (Fig. 1B). Ser at this position accounts for the MPA resistance of IMD2 in Saccharomyces cerevisiae (20). The substitution of Thr causes a 3-fold increase in MPA resistance in Candida albicans IMPDH (15), shifting the equilibrium toward the closed conformation.
The resistance of PbIMPDH-B is only partially reversed by increasing E-XMP*. Like MPA-sensitive IMPDHs, PbIMPDH-B contains Ala-249. We have recently found that changes in residues 498–527 account for ∼7-fold of the 60-fold difference in MPA resistance between PbIMPDH-A and PbIMPDH-B; these residues form part of an allosteric site that binds K+ (8). However, many of the structural determinants of the MPA resistance of PbIMPDH-B remain a mystery.
Supplementary Material
Acknowledgments
Molecular graphics images were produced using the UCSF Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by National Institutes of Health Grant P41 RR001081).
This work was supported, in whole or in part, by National Institutes of Health Grant GM54403 (to L. H.). The work performed by B. G. H. was supported by Grants 09-064240 and 09-064967 from the Danish Council for Independent Research, Technology and Product Sciences.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1 and S2.
- MPA
- mycophenolic acid
- IMPDH
- IMP dehydrogenase
- PbIMPDH
- P. brevicompactum IMPDH
- AnImdA
- A. nidulans ImdA
- APAD+
- 3-acetylpyridine adenine dinucleotide.
REFERENCES
- 1. Bentley R. (2000) Chem. Rev. 100, 3801–3826 [DOI] [PubMed] [Google Scholar]
- 2. Noto T., Sawada M., Ando K., Koyama K. (1969) J. Antibiot. 22, 165–169 [DOI] [PubMed] [Google Scholar]
- 3. Regueira T. B., Kildegaard K. R., Hansen B. G., Mortensen U. H., Hertweck C., Nielsen J. (2011) Appl. Environ. Microbiol. 77, 3035–3043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hansen B. G., Genee H. J., Kaas C. S., Nielsen J. B., Regueira T. B., Mortensen U. H., Frisvad J. C., Patil K. R. (2011) BMC Microbiol. 11, 202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hansen B. G., Sun X. E., Genee H. J., Kaas C. S., Nielsen J. B., Mortensen U. H., Frisvad J. C., Hedstrom L. (2011) Biochem. J., in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hedstrom L. (2009) Chem. Rev. 109, 2903–2928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Link J. O., Straub K. (1996) J. Am. Chem. Soc. 118, 2091–2092 [Google Scholar]
- 8. Sintchak M. D., Fleming M. A., Futer O., Raybuck S. A., Chambers S. P., Caron P. R., Murcko M. A., Wilson K. P. (1996) Cell 85, 921–930 [DOI] [PubMed] [Google Scholar]
- 9. Gan L., Seyedsayamdost M. R., Shuto S., Matsuda A., Petsko G. A., Hedstrom L. (2003) Biochemistry 42, 857–863 [DOI] [PubMed] [Google Scholar]
- 10. Bruzzese F. J., Connelly P. R. (1997) Biochemistry 36, 10428–10438 [DOI] [PubMed] [Google Scholar]
- 11. Wang W., Hedstrom L. (1997) Biochemistry 36, 8479–8483 [DOI] [PubMed] [Google Scholar]
- 12. Macpherson I. S., Kirubakaran S., Gorla S. K., Riera T. V., D'Aquino J. A., Zhang M., Cuny G. D., Hedstrom L. (2010) J. Am. Chem. Soc. 132, 1230–1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang W., Papov V. V., Minakawa N., Matsuda A., Biemann K., Hedstrom L. (1996) Biochemistry 35, 95–101 [DOI] [PubMed] [Google Scholar]
- 14. Morrison J. F. (1969) Biochim. Biophys. Acta 185, 269–286 [DOI] [PubMed] [Google Scholar]
- 15. Köhler G. A., Gong X., Bentink S., Theiss S., Pagani G. M., Agabian N., Hedstrom L. (2005) J. Biol. Chem. 280, 11295–11302 [DOI] [PubMed] [Google Scholar]
- 16. Guillén Schlippe Y. V., Riera T. V., Seyedsayamdost M. R., Hedstrom L. (2004) Biochemistry 43, 4511–4521 [DOI] [PubMed] [Google Scholar]
- 17. Hermes J. D., Morrical S. W., O'Leary M. H., Cleland W. W. (1984) Biochemistry 23, 5479–5488 [DOI] [PubMed] [Google Scholar]
- 18. Digits J. A., Hedstrom L. (1999) Biochemistry 38, 2295–2306 [DOI] [PubMed] [Google Scholar]
- 19. Min D., Josephine H. R., Li H., Lakner C., MacPherson I. S., Naylor G. J., Swofford D., Hedstrom L., Yang W. (2008) PLoS Biol. 6, e206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jenks M. H., Reines D. (2005) Yeast 22, 1181–1190 [DOI] [PubMed] [Google Scholar]
- 21. Riera T. V., Wang W., Josephine H. R., Hedstrom L. (2008) Biochemistry 47, 8689–8696 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.