

Abstract
The pyridine nucleotide cycle is a network of salvage and recycling routes maintaining homeostasis of NAD(P) cofactor pool in the cell. Nicotinamide mononucleotide (NMN) deamidase (EC 3.5.1.42), one of the key enzymes of the bacterial pyridine nucleotide cycle, was originally described in Enterobacteria, but the corresponding gene eluded identification for over 30 years. A genomics-based reconstruction of NAD metabolism across hundreds of bacterial species suggested that NMN deamidase reaction is the only possible way of nicotinamide salvage in the marine bacterium Shewanella oneidensis. This prediction was verified via purification of native NMN deamidase from S. oneidensis followed by the identification of the respective gene, termed pncC. Enzymatic characterization of the PncC protein, as well as phenotype analysis of deletion mutants, confirmed its proposed biochemical and physiological function in S. oneidensis. Of the three PncC homologs present in Escherichia coli, NMN deamidase activity was confirmed only for the recombinant purified product of the ygaD gene. A comparative analysis at the level of sequence and three-dimensional structure, which is available for one of the PncC family member, shows no homology with any previously described amidohydrolases. Multiple alignment analysis of functional and nonfunctional PncC homologs, together with NMN docking experiments, allowed us to tentatively identify the active site area and conserved residues therein. An observed broad phylogenomic distribution of predicted functional PncCs in the bacterial kingdom is consistent with a possible role in detoxification of NMN, resulting from NAD utilization by DNA ligase.
Keywords: Enzymes, Genomics, NAD, NAD Biosynthesis, Vitamins and Cofactors
Introduction
The pyridine nucleotide cycle (PNC)3 is a network of biochemical transformations that allow cells to recycle the by-products of endogenous NAD consumption back to the coenzyme and to salvage the available pyridine bases, nucleosides, and nucleotides as NAD precursors. The importance of NAD regeneration through recycling pathways is emphasized by the occurrence of an intense nonredox NAD consumption as suggested by the rapid turnover of the coenzyme pool within the cell (1). In bacteria, the pyridine by-products of the NAD-consuming enzymes NMN and Nm can be recycled back to NAD through the PNC depicted in Fig. 1 (2, 3). Briefly, Nm can be converted to NAD through two different routes. The most commonly occurring pathway is initiated by Nm deamidation to Na, followed by Na conversion to NaMN, NaMN adenylation to NaAD, and NaAD amidation to NAD. The last three reactions comprise the so-called Preiss-Handler pathway (4, 5). The second Nm recycling route is a relatively rare, nondeamidated pathway, whereby Nm is directly phosphoribosylated to NMN and NMN is then adenylated to NAD. NMN can be recycled back to NAD through two pathways shown to be functional in Escherichia coli and Salmonella typhymurium (6): the predominant route, PNC IV, proceeds via NMN deamidation to NaMN, which is then converted to NAD by entering the Preiss-Handler pathway; the alternative route, PNC VI, comprises NMN hydrolysis to Nam followed by Nam conversion to NAD through the deamidated pathway. The same routes described for pyridine recycling can be used by the cell to salvage exogenous pyridines, e.g. Na and Nm. NmR and NMN can also be exogenous NAD precursors, the latter being converted to NmR prior to uptake (7). Once inside the cell via the PnuC transporter, NmR may be directly phosphorylated to NMN or degraded to the free pyridine base (2).
FIGURE 1.
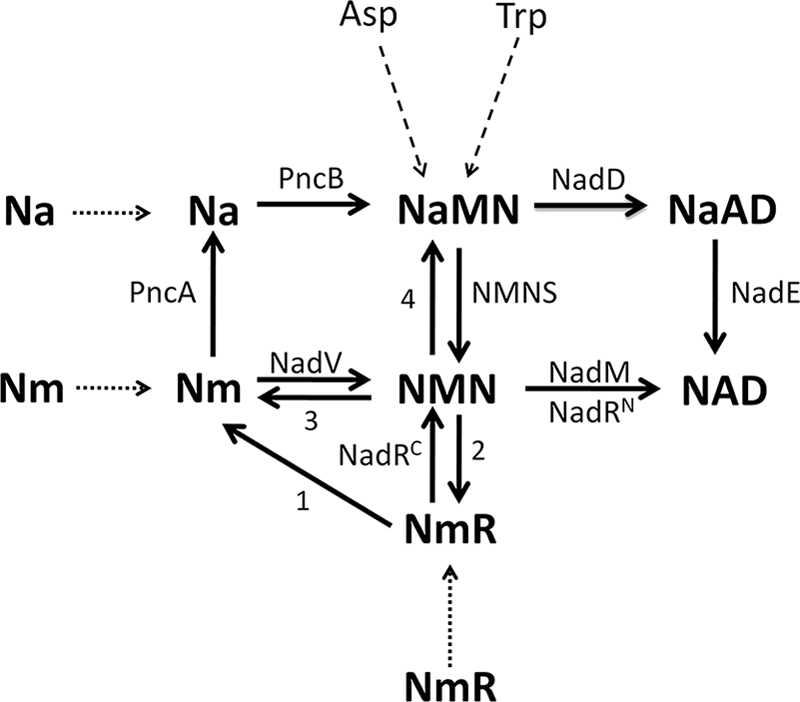
Pyridine nucleotide cycle in bacteria. The routes known to be functional across diverse bacterial species are shown by solid lines. The dotted and dashed lines relate to uptake and de novo NaMN synthesis, respectively. Enzymes are indicated with the acronism used to identify the corresponding gene locus: NadD, NaMN adenylyltransferase; NadE, NAD synthetase, NMNS, NMN synthetase; NadM, NMN adenyltransferase; NadRC, NmR kinase; NadRN, NMN adenylyltransferase; NadV, Nm phosphoribosyltransferase; PncA, Nm deamidase; PncB, Na phosphoribosyltransferase. The reactions whose responsible enzymes have not yet been identified and annotated are indicated with a number. Reaction 1 includes the reaction catalyzed in some species by a paralog of uridine phosphorylase, which may be considered a candidate for the role of bacterial NmR phosphorylase (2). Reaction 2 includes the reaction catalyzed by the periplasmic alkaline phosphatase AphA that is also endowed with NMN phosphatase activity (7). Reactions 3 and 4 are catalyzed by the “orphan enzymes” NMN glycohydrolase and NMN deamidase, respectively.
A comprehensive genomic reconstruction of the potential NAD biosynthetic machinery in the sequenced bacterial genomes reveals the occurrence of different combinations of the various PNCs, depending on the bacterial species. Indeed salvage and recycling pathways appear to be a subject of substantial variations even between closely related species. Although most of the enzymes involved in such routes have been characterized, some of them, like NMN deamidase (EC 3.5.1.42) and NMN glycohydrolase (EC 3.2.2.14), have not yet been assigned to any gene and still belong to the family of “orphan enzymes,” i.e. enzymes included in the ExPASy database for which no corresponding gene has been so far reported (8, 9). The existence of an enzyme endowed with NMN deamidase activity is supported by experimental evidence dating back to the early 1970s (10–13). In S. typhimurium and E. coli, it was suggested to be involved in NMN recycling through PNC IV (6, 14) and to prevent inhibition of bacterial NAD-dependent DNA ligase by accumulated NMN, a well known ligase inhibitor (1, 15, 16). In addition, physiological studies in S. typhymurium suggested that NMN deamidase might play a key role in salvaging of NmR via NMN; in fact, NMN would be deamidated to NaMN and thus enter the Preiss-Handler pathway rather than being directly adenylated to NAD (7, 17). Moreover, the product of the NMN deamidase-catalyzed reaction, e.g. NaMN, is used as the preferred phosphoribosyl donor by the enzyme CobT, which catalyzes a late step in adenosylcobalamin biosynthesis (18), thus conferring to NMN deamidase a role in the regulation of vitamin B12 biosynthesis.
The gene encoding NMN deamidase eluded identification for several years. In early genetic studies, a locus named pncC was proposed to code for the enzyme; however, it was found later to be involved in NMN uptake (7, 12, 19, 20). Here we describe the identification of the NMN deamidase encoding gene, following purification and partial sequencing of the enzyme from Shewanella oneidensis MR-1. We found that its sequence corresponds to a protein annotated as CinA and proposed to be involved in bacterial competence induction (21, 22). The availability of the three-dimensional structure of CinA from Agrobacterium tumefaciens, the only member of the CinA structural family as classified in the SCOPUS database, allowed us to annotate this fold as a novel amidohydrolase family.
EXPERIMENTAL PROCEDURES
Determination of NMN Deamidase Activity
The presence of the enzymatic activity in cell crude extracts was determined with an HPLC-based assay relying on direct quantitation of NaMN. Reaction mixtures containing 100 mm potassium phosphate buffer, pH 8.0, 10 mm sodium fluoride, 10 mm EDTA, 1 mm NMN, and appropriate amounts of extract were incubated for 10 min at 37 °C. The reactions were stopped with 0.6 m HClO4, and after 10 min on ice, the samples were centrifuged for 1 min at 12,000 × g. The supernatants were neutralized with 0.8 m K2CO3, kept on ice for 10 min, and centrifuged as described above. The supernatants were injected into an HPLC system equipped with a diode array detector. Nucleotide separation was performed on an ion-paired analytical Supelcosil LC18-S column (5 μm, 4.6 × 250 mm). Elution conditions were 4 min at 100% buffer A (100 mm potassium phosphate, pH 6.0, 8 mm tetrabutylammonium hydrogen sulfate, 12.5 min up to 15% buffer B (buffer A containing 30% methanol), and 23.5 min up to 90% buffer B, holding at 90% buffer B for 7 min, returning to 100% buffer A in 6 min and holding at 100% buffer A for 5 min. Flow rate was maintained at 1 ml/min, and temperature was fixed at 8 °C. In reaction mixtures containing purified enzyme preparations, phosphate buffer was substituted by 50 mm HEPES, pH 7.5, and EDTA was omitted. Elution conditions were modified as follows: 4 min at 100% buffer A, 6 min up to 7% buffer B, returning to 100% buffer A in 1 min, and holding at 100% buffer A for 5 min. In an alternative, spectrophotometric, continuous assay, NaMN release by the enzyme was coupled to the conversion of NaMN to NADH, in the presence of recombinant E. coli NadD (converting NaMN to NaAD) and NadE (amidating NaAD to NAD) and yeast alcohol deydrogenase. The coupled reaction was monitored by the increase of NADH absorbance at 340 nm at 37 °C. The assay mixture contained 50 mm HEPES buffer, pH 7.5, 0.5% ethanol, 14 mm semicarbazide, 11 mm MgCl2, 4.5 mm NH4Cl, 1.6 mm ATP, 0.1 mm NMN, 7 units of alcohol dehydrogenase, 0.02 unit of purified recombinant NadE and NadD, 0.56 mg/ml bovine serum albumin, and an appropriate amount of NMN deamidase. One unit of NMN deamidase activity is defined as the amount of enzyme catalyzing the formation of 1 μmol of NaMN/min at 37 °C.
Purification of S. oneidensis NMN Deamidase
S. oneidensis cells grown at 30 °C in LB medium (3 liters) to an A600 of 1.0 were harvested by centrifugation at 5,000 × g for 10 min and resuspended in 75 ml of lysis buffer (50 mm Tris/HCl, pH 7.5, 0.15 m NaCl, 1 mm DTT, 1 mm PMSF, and 0.002 mg/ml leupeptin, pepstatin, antipain, and chymostatin). The suspension was sonicated three times for 1 min, with 30-s intervals, and centrifuged at 10,000 × g for 10 min. To the supernatant, referred to as the crude extract, a 10% (w/v) solution of streptomycin sulfate was added dropwise to a final concentration of 1%. After 20 min of stirring, the sample was centrifuged at 10,000 × g for 10 min, and the pellet was discarded. To the supernatant, after dilution with 50 mm Tris/HCl, pH 7.5, to a protein concentration of ∼5 mg/ml, solid ammonium sulfate was added up to 50% saturation. The pH was maintained at 7.5 by dropwise addition of 1 m NH4OH. After stirring for 20 min, the precipitated proteins were collected by centrifugation at 3,000 × g for 40 min and resuspended in 75 ml 10 mm potassium phosphate buffer, pH 7.0 (buffer C), 3 m NaCl, 1 mm PMSF. The resulting suspension was centrifuged at 10,000 × g for 10 min, and the supernatant was loaded onto a phenyl-Sepharose column (2.5 × 12 cm) equilibrated with buffer C, containing 3 m NaCl. After washing with the same buffer, elution was performed with a linear gradient of NaCl from 3 to 0 m in buffer C. Active fractions were combined, concentrated by ultrafiltration through an YM-10 (Millipore) membrane, and diluted with buffer C to decrease the ionic strength to ∼13 mS/cm at 10 °C. The diluted pool was applied to a Reactive Red 120-agarose (Type 300; Sigma) column (1.5 × 13 cm) equilibrated with buffer C, containing 0.15 m NaCl. The column was washed with the same buffer and then eluted with a linear gradient of NaCl from 0.15 to 1.5 m in buffer C. Active fractions were combined, the pool was concentrated by ultrafiltration as described above and then diluted 10-fold with 1 mm potassium phosphate buffer, pH 7.0. The diluted pool was loaded onto a Resource Q FPLC (Pharmacia Biotech) column previously equilibrated with buffer C, containing 0.1 m NaCl. After washing with the same buffer, elution was performed with a discontinuous gradient, from 0.1 to 0.3 m NaCl in buffer D. Active fractions were tested for purity by SDS-PAGE, and homogeneous fractions were pooled and stored at 4 °C. Determination of protein concentration was evaluated according to Bradford (23), using bovine serum albumin as the standard.
Gel Filtration
Gel filtration of pure NMN deamidase from S. oneidensis and E. coli was carried out on a FPLC Superose 12 10/300 GL column (Amersham Biosciences) eluted with 10 mm potassium phosphate buffer, pH 7.0, 0.3 m NaCl, 1 mm DTT, at a flow rate of 0.5 ml/min. Bovine serum albumin (66 kDa), ovalbumin (45 kDa), and carbonic anhydrase (31 kDa) were used as the standards.
N-terminal Sequencing
After SDS-PAGE of the final enzyme preparation, NMN deamidase was electroblotted onto a polyvinylidene difluoride membrane. Transfer was performed at 4 °C for 3 h, in 10 mm N-cyclohexyl-3-aminopropanesulfonic, pH 11.0, containing 10% methanol. After membrane staining with Coomassie Brilliant Blue R-250, the enzyme band was excised and subjected to N-terminal sequencing by automated Edman degradation on a Procise model 491 sequencer (Applied Biosystem, Foster City, CA).
Cloning
Purified S. oneidensis MR-1 genomic DNA was amplified by PCR to generate DNA for cloning cinA into the BamHI and HindIII sites of pET-28c. The construct was sequence-verified for accuracy. The primers and plasmids used for cloning are listed in supplemental Table S1 and S2, respectively.
Proteins Expression and Purification
For S. oneidensis CinA (SO0272) expression, the construct was used to transform E. coli BL21(DE3) cells. Expression of E. coli YgaD, YfaY, and YdeJ proteins was obtained by growing clones from E. coli ASKA library encoding the proteins of interest (24). The cells were grown at 37 °C in Luria Bertani medium supplemented with the appropriate antibiotic. After reaching an A600 of 0.3, the cultures were shifted at 20 °C, and expression was induced with 1 mm isopropyl β-d-thiogalactopyranoside at an A600 of 0.6. After 12 h of induction, the cells were harvested by centrifugation at 5.000 × g for 10 min. Recombinant proteins were purified to homogeneity by chromatography on a HisTrap HP column (Amersham Biosciences).
Mutants Construction
In-frame deletion mutagenesis of nadA (SO2342), pncC (SO0272), and nadV (SO1981) was performed by two-stage homologous cross-over as described in Ref. 25 with minor modifications. Constructs for conducting mutagenesis were generated by PCR using primers 5-O with 5-I; 3-O with 3-I (supplemental Table S1) to generate DNA products that were subsequently joined and inserted in the SmaI site of pDS3.0. Double deletion mutants (nadA/pncC, nadA/nadV, and pncC/nadV) were generated by performing a second round of mutagenesis on previously generated single deletion mutants. Deletion of genomic DNA was validated by sequencing genomic PCR products generated by primers designated F-O and R-O (supplemental Table S1). The bacterial strains used are listed in supplemental Table S2.
Bioinformatics Tools and Resources
Functional annotations of genes involved in NAD metabolism in the selected set of ∼100 bacterial genomes were from the “NAD and NADP cofactor biosynthesis global” subsystem in The SEED genomic comparative genomic database (26). Multiple sequence alignments of protein sequences were produced by MUSCLE (27). The phylogenetic trees were constructed by the maximum likelihood method implemented in the PROML program of the PHYLIP package (28). The Protein Families database (Pfam) (29) was used to identify conserved functional domains.
Homology Modeling and Docking
An initial model of E. coli PncC structure was generated using the I-TASSER server (30). The structural stability of the model was tested by means of molecular dynamics simulation using the GROMACS simulation package v. 4.5.3 (31). The final model quality was evaluated using the JCSG Structure Validation Server, including the Protein Structure Quality Score Tool. Automated docking was performed with the program AutoDock 4.2.3 (32). For each simulation, the ligand top ranked conformation (minimal energy) in complex with the enzyme was subsequently refined by molecular dynamics simulations using the GROMACS 4.5.3 package, with the standard GROMOS96 force field (33). The 2A9S crystal structure was analyzed by using CASTp server to identify and measure surface-accessible pockets, as well as interior inaccessible cavities (34). All graphic manipulations and visualizations were performed with the Chimera program (35).
RESULTS
Bioinformatic Prediction of the NMN Deamidase Activity and Search for the Encoding Gene
In silico reconstruction of NAD biosynthetic pathway in bacterial sequenced genomes underscored a group of bacteria where salvage pathways leading to NMN operate without an adenylyltransferase of the NadM or NadR family, resulting in the inability of these species to directly adenylate NMN to NAD (supplemental Fig. S1). In this context, a NMN deamidase activity appears indispensable to feed NMN into the NadD catalyzed reaction via its conversion to NaMN. Based on the observation that genes involved in NAD biosynthesis very frequently show a strong tendency to form conserved operon-like clusters, we initially searched the bacterial genomes for the presence of a gene lying in a genomic context that would have suggested its function in NMN deamidation. However, the genomic context analysis did not allow us to predict a candidate gene for such a role. Therefore, to identify the NMN deamidase encoding gene, a classical biochemical approach consisting of native enzyme purification, partial protein sequencing, and genomic database search was pursued. Among the bacterial species where the metabolic gap has been identified is the γ-proteobacterium S. oneidensis, which was chosen as the organism to search for the gene based on its ability to synthesize NAD through a relatively limited number of routes, which is a desirable framework for in vivo functional studies.
Purification and Characterization of NMN Deamidase from S. oneidensis
The presence of NMN deamidase activity in the bacterial crude extract was demonstrated by the HPLC-based assay described under “Experimental Procedures.” HPLC analysis of the cell extract incubated in the presence of NMN revealed the appearance of a peak, absent in the same extract incubated without the nucleotide (Fig. 2A). Identity of the peak as genuine NaMN was confirmed by its coelution with an NaMN standard (Fig. 2A) and identical UV absorption spectra (not shown). The enzyme was purified to homogeneity from bacterial cell extract using a preliminary treatment with streptomycin sulfate and ammonium sulfate, followed by a combination of hydrophobic interaction, dye ligand, and ion exchange chromatography (Table 1). During all the chromatographic steps, NMN deamidase eluted as a single enzymatically active peak. In the final preparation, a single band of ∼47 kDa was observed upon SDS-PAGE analysis (Fig. 2B). Its correspondence with the NMN deamidase enzyme was confirmed by the strong correlation between the band intensity and the enzymatic activity in the fractions eluted in the last chromatographic step (not shown).
FIGURE 2.

Biochemical characterization of NMN deamidase from S. oneidensis. A, HPLC chromatogram of a reaction mixture containing an appropriate amount of bacterial cell extract incubated in the presence (continuous line) and in the absence (dashed line) of NMN. The dotted line represents the elution profile of NMN and NaMN standards. B, SDS-PAGE analysis of the various chromatographic steps of the enzyme purification: phenyl-Sepharose (lane b), RedA (lane c), and Mono Q (lane d). The arrow points to the pure protein. Lane a, molecular mass standards. C, kinetic analysis: activity versus substrate curve and Hill plot (inset). The data are the results of three independent experiments.
TABLE 1.
Purification of NMN Deamidase from S. oneidensis
| STEP | Proteins | Activity | Specific activity | Yield | Purification |
|---|---|---|---|---|---|
| mg | units | units/mg | % | fold | |
| Crude extract | 1117 | 3.192 | 0.003 | 100 | |
| Streptomycin sulfate | 887 | 3.306 | 0.004 | 104 | 1.3 |
| Ammonium sulfate | 404 | 2.337 | 0.006 | 73 | 2.0 |
| Phenyl-Sepharose | 68 | 1.071 | 0.016 | 34 | 5.3 |
| Dye ligand affinity Red A | 3.1 | 0.579 | 0.187 | 18 | 62.3 |
| Resource Mono Q | 0.075 | 0.135 | 1.800 | 4 | 600.0 |
Gel filtration experiments showed a native molecular mass of ∼80 kDa, which is consistent with a dimeric structure. The enzyme has no detectable deamidase activity toward NAD, NADP, Nm, and NmR. It does not require a divalent cation for the catalytic activity and is not affected by EDTA at concentrations up to 10 mm. It fully retains its activity in the presence of 0.2 mm iodoacetamide, suggesting that cysteine residues are not involved in catalysis. The enzyme shows a broad optimum pH, ranging from 5.5 to 9.0; 72 and 60% residual activity is observed at pH 10.0 and 5.0, respectively. Among several metabolites associated with NAD biosynthetic pathways (including NAD, NADP, NADH, NADPH, NaAD, Nm, Na, NmR, Qa, PRPP, and ADP-ribose), none exerted any effect on the enzyme activity. S. oneidensis NMN deamidase is an allosteric enzyme, as revealed by the sigmoid shape of the plot of the initial velocity of the enzyme-catalyzed reaction versus NMN concentration (Fig. 2C). From the Hill plot, a nH value of 2.6 was calculated, indicating a strong positive cooperativity. A S0.5 value of 16 μm and a kcat value of 3.0 s−1 were determined.
Identification of the S. oneidensis Gene Coding for NMN Deamidase
For the gene identification, the 47-kDa band of the final enzymatic preparation was electroblotted onto a PVDF membrane and subjected to N-terminal sequencing by automated Edman degradation. A single N-terminal sequence was revealed, MKLEMICTGEEVLS, that was identical to that of a S. oneidensis protein annotated as the competence/damage-inducible protein CinA, suggesting the identity of CinA with NMN deamidase. To verify this prediction, the S. oneidensis cinA gene was cloned, and the corresponding protein was overexpressed in E. coli (supplemental Fig. S2A). Significantly higher levels of NMN deamidase activity were found in the recombinant extracts in comparison with the controls prepared from cells harboring the nonrecombinant plasmid, confirming the identity of CinA as the S. oneidensis NMN deamidase (from now on referred as PncC). The recombinant enzyme, purified through nickel affinity chromatography, exhibited the same molecular and kinetic properties as the wild type enzyme (not shown).
In Vivo Functional Activity of S. oneidensis pncC Gene
The result of a comparative genomic reconstruction of NAD biosynthesis in S. oneidensis is illustrated in Fig. 3. S. oneidensis possesses readily detectable orthologs of nadA, nadB, nadC, and nadD genes involved in de novo NAD biosynthesis, as well as nadV gene coding for the enzyme Nm phosphoribosyltransferase that initiates the amidated salvage/recycling of Nm and is under ADP-ribose regulation (36). The lack of pncA and pncB orthologs, coding for the enzymes supporting conversion of Nm to NaMN via Na, indicates that the deamidated route is not operative in this bacterium. To confirm these bioinformatic predictions and the pncC role in the amidated route, S. oneidensis mutants were generated, and their growth phenotypes were compared with the wild type strain (Table 2 and supplemental Fig. S3). ΔpncC and ΔnadV mutants could grow on the defined medium like the wild type strain (not shown), whereas ΔnadA failed to grow, indicating that bacterium growth in the defined medium is sustained by a functional de novo NAD biosynthetic pathway. ΔnadA mutant could grow on the medium in the presence of added Nm or Qa, but not Na, confirming the presence of the amidated route and the absence of the deamidated one, as well as the bacterium capability to utilize exogenous Nm and Qa. Double mutants ΔnadA/ΔpncC and ΔnadA/ΔnadV allowed us to assess the physiological role of nadV and pncC genes in NAD biosynthesis. As expected, both double mutants lost the ability to grow on medium supplemented with Nm. These results clearly indicate the involvement of pncC and nadV genes in the Nm salvage/recycling amidated route. Notably, the ΔpncC single mutant failed to show any NMN deamidase activity (not shown), confirming that in S. oneidensis the pncC encoded product is the only deamidase that in vivo can exert the NMN deamidase function.
FIGURE 3.

Genomic reconstruction of NAD biosynthesis in S. oneidensis. Schematic representation of NAD biosynthetic routes as revealed by the in silico genomic reconstruction of NAD metabolism, integrated with the experimental results in Table 2.
TABLE 2.
Growth phenotypes of S. oneidensis knockout mutants
Growth was conducted at 30 °C for 24–48 h on defined medium (DM) or on defined medium supplemented with 200 μm nicotinamide (DM + Nm), quinolinate (DM + Qa), or nicotinic acid (DM +Na). ND, not determined.
| Strains | Media |
|||
|---|---|---|---|---|
| DM | DM + Nm | DM + Qa | DM + Na | |
| Wild type | + | + | + | + |
| ΔnadA | − | + | + | − |
| ΔnadA/ΔpncC | − | − | + | ND |
| ΔnadA/ΔnadV | − | − | + | ND |
PncC Domain Composition
S. oneidensis PncC is a 424-residue protein that is organized into two domains. The C-terminal domain (residues 252–407) is a domain of unknown function (PF02464, CinA domain), highly and widely conserved within bacteria. The N-terminal domain (residues 1–170) is also of unknown function (PF00994, probable molybdopterin binding domain, MocF domain) and exhibits some degree of homology with enzymes involved in the last step of molybdenum cofactor biosynthesis (37). Sequence similarity searches in the E. coli proteome using the S. oneidensis PncC as the query, showed the existence of three E. coli PncC homologs: YfaY, where a CinA domain is fused with a conserved MocF domain as in S. oneidensis, and two paralogs, YgaD and YdeJ, comprising only the CinA domain (Fig. 4). To assess whether the MocF or the CinA domain would be responsible for the NMN deamidase function, we overexpressed and purified YfaY, YgaD, and YdeJ (supplemental Fig. S2B) and tested them for NMN deamidase activity. The activity was confirmed only for YgaD (Fig. 4), indicating that YgaD is the only E. coli functional PncC. It consists of 165 residues, and its molecular mass, as determined by SDS-PAGE, is ∼20 kDa (supplemental Fig. S2B). The native molecular mass of ∼43 kDa, as calculated by gel filtration (not shown), is consistent with a dimeric structure. The catalytic properties of the E. coli enzyme are similar to those of the S. oneidensis ortholog, including the lack of pH dependence in a broad range of pH values and the strict specificity toward NMN. In contrast to S. oneidensis PncC, the E. coli enzyme follows the Michaelis-Menten kinetic behavior (not shown); however, it shares a high affinity for NMN (Km = 6 μm) and the same kcat value (3.3 s−1) with the S. oneidensis enzyme.
FIGURE 4.

Domain composition and enzymatic activity of S. oneidensis and E. coli proteins containing the CinA domain. Specific activity values refer to the pure recombinant proteins.
The finding that YgaD, comprising the CinA domain alone, is enzymatically active demonstrates that in S. oneidensis PncC, the NMN deamidase activity resides in the CinA domain, herein renamed PncC domain. The domain composition analysis was extended to a subset of 100 representative bacterial genomes by using bioinformatic tools. We identified 93 PncC domain-containing proteins that are evenly distributed in 86 analyzed genomes. A multiple alignment of the PncC domain of the most divergent sequences, including the functional and nonfunctional PncC domains of S. oneidensis and E. coli characterized in this work, is depicted in Fig. 5 (for the full alignment, see supplemental Fig. S4). Proteins containing the PncC domain might be divided into four subgroups, according to their domain composition and predicted enzymatic activity: (i) E. coli PncC homologs, i.e. single-domain proteins likely to be functional NMN deamidases, labeled PncC in Fig. 5; (ii) S. oneidensis PncC homologs, i.e. two-domain proteins, also predicted to be functional NMN deamidases, labeled MocF/PncC; (iii) E. coli YfaY homologs, i.e. two-domain nonfunctional proteins; and (iv) E. coli YdeJ homologs, i.e. single-domain nonfunctional proteins. It can be inferred that the lack of enzymatic activity in the YfaY subgroup is likely due to the occurrence of a PncC domain with multiple deletions and mutations. On the other hand, this argument does not explain the lack of activity in the YdeJ subgroup, where the PncC domain appears to be conserved. In this case, it might be assumed that subtle and critical changes within the active site might be the cause for the activity loss. Indeed, the structural analysis described below appears to support such predictions.
FIGURE 5.

Multiple alignment of the PncC domain of selected PncC proteins. Proteins are grouped and differentially colored according to the domain composition and predicted enzymatic activity. The domain composition of the different subgroups is shown on the bottom. Proteins experimentally characterized in this work are marked by a red star. Secondary structure elements according to the known three-dimensional structure of A. tumefaciens PncC are shown by arrows (α-helices) and zigzags (β-strands). Residues conserved in all functional proteins are marked with asterisks. The complete list of analyzed bacterial genomes and gene identifiers (locus tags) is provided in Fig. 7.
PncC Structural Analysis
The analysis was enabled by the availability of the high resolution (1.75 Å) crystal structure for the A. tumefaciens CinA (Protein Data Bank code 2A9S), as determined at the Midwest Center for Structural Genomics. Our assignment of the NMN deamidase function to this protein is based on its significant sequence homology with E. coli PncC (47% identity, 63% similarity). (Fig. 5). The 2A9S structure, which shows a dimeric organization, represents a unique, distinctive variant of the classic anticodon-binding domain fold of class II tRNA synthetases, which consists of a three layers α/β/α arrangement, comprising a central five mixed stranded β-sheet ordered β2-β1-β3-β4-β5, with β4 antiparallel to the other strands (38). Indeed, in the SCOP database, the structure is currently annotated as the only member of a CinA-like superfamily.
We used the 2A9S crystal structure as the template for the prediction of E. coli PncC structure through homology modeling. As expected, the deduced overall architecture of the E. coli enzyme model was essentially identical to the 2A9S structure (Fig. 6a). The root mean square deviation values calculated between the superimposed backbones of the E. coli enzyme model and the crystallographic template were all ∼1 Å. The peculiar fold of the enzyme consists of the classic β sheet at the center of the protein surrounded by two external layers comprising four (α1, α2, α3, and α7) and three (α4, α5, and α6) helices (Fig. 6a). Surface charge analysis was performed on the A. tumefaciens protein to predict the location of the putative active site (Fig. 6b). The most likely candidate was a small cleft (with an area of 283.3 Å2 and a volume of 381.1 Å3 (Fig. 6, b and c). Significantly, in this region of the structure, a number of amino acids are present, including Gly-46 and Ser-48 of chain A and Ser-31, Gly-34, Tyr-58, Gly-106, Ile-107, Ala-108, Gly-109, and Arg-145 of chain B, which are highly conserved in all bacterial NMN deamidases (Fig. 5 and supplemental Fig. S4), further supporting the identification of the active site. Computational NMN docking was carried out, indicating that the putative active pocket has an excellent space fit for NMN. Notably, 9 of the 13 residues predicted to be involved in NMN stabilization are strictly conserved among all single- and two-domain proteins likely to be functional NMN deamidases. In addition, Ser-31, Thr-105, and Gly-106 that interact with the NMN amide group, as well as Ser-48 hydrogen-bonded to the phosphate, are all missing in the PncC domain of the YfaY subgroup, in keeping with the lack of enzymatic activity in YfaY. Finally, the proteins in the YdeJ subgroup lack Ser-48 and Arg-145, both stabilizing the phosphate group, in agreement with the loss of NMN deamidase activity in YdeJ (Fig. 5 and supplemental Fig. S4).
FIGURE 6.

Structural analysis of NMN deamidase. a, ribbon representation of E. coli PncC (cyan) superposed to A. tumefaciens PncC monomer (Protein Data Bank code 2A9S; orange), showing the domain organization. b, electrostatic surface of the 2A9S dimer; positive charges are in blue, and negative charges are in red. A NMN molecule in the proposed binding site is shown in ball and stick representation. c, ribbon representation of the 2A9S dimer, rotated by 45 °C with respect to b, in complex with NMN. d, detailed view of the interactions between 2A9S and NMN. Hydrogen bonds are indicated as dotted red lines.
PncC Phylogenetic Distribution and Genomic Context Analysis
Proteins containing the PncC domain are widely distributed throughout the Eubacteria kingdom and are absent in Eukarya and Archaea. A phylogenetic tree of the selected species, with the distribution of the NAD biosynthetic enzymes, is shown in Fig. 7. The PncC domain is fused to the MocF domain in 40 proteins found in diverse taxonomic groups such as Firmicutes, Actinobacteria, Cyanobacteria, Thermotoga, and Bacteroidetes. In contrast, most Proteobacteria contain the single-domain proteins. In some taxonomic groups of Proteobacteria (like Enterobacteria and Vibrionales), we detected both the single-domain functional PncC and the two-domain nonfunctional protein. In addition, some Enterobacteria including the analyzed E. coli and Salmonella species have also the single domain nonfunctional protein. Notably, the enzyme is absent in those bacterial species lacking NadD, as well as in reduced genomes of obligate pathogens and symbionts. Intriguingly, the enzyme is absent or present as a nonfunctional protein in some species, like Staphylococcus aureus and Deinococcus radiodurans, even though a deamidated NAD biosynthetic route is operative. The neighbor-joining phylogenetic tree constructed for the 93 PncC domains is shown in supplemental Fig. S5. It contains a separate clade including most of the single-domain functional enzymes in Proteobacteria, and several mixed clades including both the single and two-domain enzymes. The branch of single-domain YdeJ paralogs from Enterobacteria is most closely located to PncC from γ-proteobacteria. In contrast, the two-domain YfaY paralogs from Enterobacteria and Vibrionales, as well as several other proteins of the YfaY subgroup, e.g. from Chloroflexus, Methylococcus, Pirellula, and Deinococcus, form a separate highly diverged clade. Intriguingly, the S. aureus protein of the YfaY subgroup clusters with the MocF/PncC enzymes from the Bacillales.
FIGURE 7.

Distribution of NMN deamidase and other NAD biosynthetic enzymes in bacteria. The phylogenetic tree of the representative set of 100 bacterial genomes was taken from the Genomic Encyclopedia of Bacteria and Archaea project (43). Genomic identifiers (locus tags) of genes encoding PncC proteins are listed in the first column. The color code reflects domain composition and enzymatic activity as in Fig. 5. Proteins experimentally characterized in this work are marked by a red star. Distribution of NAD biosynthetic enzymes in bacterial genomes was taken from the “NAD and NADP cofactor biosynthesis global” subsystem in the SEED genomic database. Functional roles of these enzymes are described in Fig. 1.
The genomic context analysis of pncC genes in bacterial genomes revealed several conserved gene clusters encoding various essential enzymes; however, they were never found in genomic clusters containing other NAD metabolism genes. In most γ-proteobacteria and Firmicutes, pncC precedes the recombinase gene recA. However, these genes are not cotranscribed as an operon because recA has its own promoter controlled by LexA repressor. In most α-proteobacteria, pncC is located in a candidate operon with the isoprenoid biosynthesis gene ispD. In β-proteobacteria, pncC is located in the potential operon with the thiamine monophosphate kinase thiL and phosphatidylglycerophosphatase pgpA. Colocalization of pncC with the lipid metabolism gene pgpA was also found in all δ-proteobacteria. Another lipid metabolism gene, pgsA, encoding phosphatidylglycerophosphate synthase was found in a conserved genomic cluster with pncC in Firmicutes, Actinobacteria, and Chloroflexi. In the Thermus/Deinococcis group, pncC belongs to the same operon with the RNA ligase ligT and recA.
DISCUSSION
The NMN deamidase-encoding gene has been identified through purification of the native enzyme from S. oneidensis cells, followed by partial protein sequencing. Biochemical studies, as well as growth phenotype analysis of S. oneidensis knock-out mutants, proved that in this bacterium the newly assigned enzyme is the only deamidase acting on NMN and confirmed its involvement in the Nm salvage/recycling route. We therefore named it PncC, the acronym first proposed by Foster et al. (19) to describe this enzyme in the PNC. Surprisingly, the results of this work assigned PncC activity to a protein currently annotated as “competence/damage-inducible protein,” CinA. The cinA gene was first described in Streptococcus pneumoniae as a component of the recA operon, a cin (competence induced) operon involved in the bacterium genetic transformation (21, 22). Although many studies reported that the gene was markedly induced during competence, contrasting evidence on the possible CinA-mediated targeting of RecA to the membrane favoring its early interaction with incoming ssDNA did not enable ultimate elucidation of CinA induction significance (39–41). Moreover, very recently, it was found that in Bacillus subtilis, a cinA deletion mutant, showed a reduction in the transformation efficiency, although less pronounced than in S. pneumoniae (42). However, no induction of the protein expression was observed during competence, and CinA was always found localized within the nucleoid, thus not affecting RecA targeting to the membrane. This led the authors to conclude that B. subtilis CinA might play only a minor role in competence (42).
Our results on the assignment of the NMN deamidase function to CinA might reconcile the phenotype of cinA deletion mutants with the enzyme's proposed role in preventing the inhibition of NAD-dependent DNA ligase by NMN (1). Indeed, the burst of DNA ligase activity during recombination events is likely to result in NMN level rise. By contributing to NMN scavenging, NMN deamidase would prevent the ligase inhibition and, given its involvement in NAD recycling, would also ensure continued NAD supply to the ligase reaction. Therefore, the observed reduction of transformation efficiency in cinA-deleted strains might be explained by the loss of NMN deamidase activity. The enzyme localization to the bacterial nucleoid (42) is in keeping with its proposed role in the recombination machinery. The phylogenetic analysis also confirms that the enzyme's role extends beyond that of a merely salvaging enzyme; in fact it is present in the majority of bacterial species, including those able to salvage/recycle Nm via alternative routes. On the other hand, the finding that PncC is absent in some species, including those lacking NadD and thus unable to utilize NaMN as an NAD precursor, clearly indicates that cells must rely on additional enzymes for NMN scavenging. This is in keeping with the observation that, even though S. oneidensis pncC deletion mutant shows NaMN levels significantly lower with respect to the wild type, NMN levels are comparable.4
In this work, we have characterized two representative members of the PncC family: the S. oneidensis enzyme, where the domain responsible for the deamidating activity (PncC, formerly CinA) is fused to a domain similar to the molybdopterin binding domain of enzymes involved in MoCo biosynthesis, and the E. coli enzyme, comprising only the PncC domain. The functional role of the additional domain in the S. oneidensis protein remains to be determined, as well as its possible involvement in the allosteric behavior exhibited by the S. oneidensis enzyme, in contrast to E. coli PncC. Nonetheless, the different domain organization does not seem to affect the catalytic efficiency; in fact, both the E. coli and the S. oneidensis enzymes display similar Km values toward NMN and share the same kcat. The Km in the low micromolar range exhibited by both enzymes points to an efficient deamidating reaction even at very low NMN concentrations and is consistent with the proposed enzyme's role in contributing to NMN level regulation. The PncC family also comprises a few members with a nonfunctional PncC domain, bearing multiple mutations and deletions. The phylogenetic analysis suggests that the distribution of PncC domains is likely the result of multiple evolutionary events, including an ancient domain fusion and multiple posterior domain fissions (e.g. PncC in the common ancestor of Proteobacteria), gene losses/horizontal transfers (e.g. MocF/PncC in S. oneidensis and Mycobacterium tuberculosis), gene duplications, and decay (e.g. YdeJ in Enterobacteria).
The newly identified NMN deamidase is both phylogenetically and structurally distinct from enzymes catalyzing the hydrolysis of amide bonds belonging to known superfamilies. Indeed, in the SCOP database, the crystal structure of A. tumefaciens PncC is the only representative of the novel CinA-like superfamily. The inhibition data obtained in this work argue against PncC being either a thiol- or a metal-dependent amidohydrolase. Although the analysis of conserved residues located in the predicted active site of the A. tumefaciens PncC points to a possible Ser/Thr-dependent mechanism, full classification of NMN deamidase remains an open question and the subject of ongoing studies.
Supplementary Material
This work was partly supported by the Italian Minister of Foreign Affairs, “Direzione Generale per la Promozione del Sistema Paese.” The research at the Pacific Northwest National Laboratory and Sanford Burnham Institute was supported by the U.S. Department of Energy, Office of Biological and Environmental Research, as part of the Genomic Science Program. Their contribution originates from the Genomic Science Program Foundational Scientific Focus Area at the Pacific Northwest National Laboratory.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Tables S1 and S2 and Figs. S1–S5.
N. Raffaelli, unpublished observations.
- PNC
- pyridine nucleotide cycle
- NMN
- nicotinamide mononucleotide
- NaMN
- nicotinic acid mononucleotide
- Na
- nicotinic acid
- Nm
- nicotinamide
- NmR
- nicotinamide riboside
- Qa
- quinolinic acid.
REFERENCES
- 1. Park U. E., Olivera B. M., Hughes K. T., Roth J. R., Hillyard D. R. (1989) J. Bacteriol. 171, 2173–2180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sorci L, K., Rodionov DA, Osterman AL. (2010) in Comprehensive Natural Products: II. Chemistry and Biology (Mander L., Lui H.-W. eds.) pp. 213–251, Elsevier, Oxford [Google Scholar]
- 3. Gazzaniga F., Stebbins R., Chang S. Z., McPeek M. A., Brenner C. (2009) Microbiol. Mol. Biol. Rev. 73, 529–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Preiss J., Handler P. (1958) J. Biol. Chem. 233, 488–492 [PubMed] [Google Scholar]
- 5. Preiss J., Handler P. (1958) J. Biol. Chem. 233, 493–500 [PubMed] [Google Scholar]
- 6. Foster J. W., Baskowsky-Foster A. M. (1980) J. Bacteriol. 142, 1032–1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Grose J. H., Bergthorsson U., Xu Y., Sterneckert J., Khodaverdian B., Roth J. R. (2005) J. Bacteriol. 187, 4521–4530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hanson A. D., Pribat A., Waller J. C., de Crécy-Lagard V. (2010) Biochem. J. 425, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen L., Vitkup D. (2007) Trends Biotechnol. 25, 343–348 [DOI] [PubMed] [Google Scholar]
- 10. Imai T. (1973) J. Biochem. 73, 139–153 [PubMed] [Google Scholar]
- 11. Friedmann H. C., Garstki C. (1973) Biochem. Biophys. Res. Commun. 50, 54–58 [DOI] [PubMed] [Google Scholar]
- 12. Kinney D. M., Foster J. W., Moat A. G. (1979) J. Bacteriol. 140, 607–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Foster J. W., Brestel C. (1982) J. Bacteriol. 149, 368–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Manlapaz-Fernandez P., Olivera B. M. (1973) J. Biol. Chem. 248, 5150–5155 [PubMed] [Google Scholar]
- 15. Zimmerman S. B., Little J. W., Oshinsky C. K., Gellert M. (1967) Proc. Natl. Acad. Sci. U.S.A. 57, 1841–1848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Olivera B. M., Hall Z. W., Anraku Y., Chien J. R., Lehman I. R. (1968) Cold Spring Harb Symp. Quant. Biol. 33, 27–34 [DOI] [PubMed] [Google Scholar]
- 17. Grose J. H., Bergthorsson U., Roth J. R. (2005) J. Bacteriol. 187, 2774–2782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Maggio-Hall L. A., Escalante-Semerena J. C. (2003) Microbiology 149, 983–990 [DOI] [PubMed] [Google Scholar]
- 19. Foster J. W., Kinney D. M., Moat A. G. (1979) J. Bacteriol. 138, 957–961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cheng W., Roth J. (1995) J. Bacteriol. 177, 6711–6717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Martin B., García P., Castanié M. P., Claverys J. P. (1995) Mol. Microbiol. 15, 367–379 [DOI] [PubMed] [Google Scholar]
- 22. Pearce B. J., Naughton A. M., Campbell E. A., Masure H. R. (1995) J. Bacteriol. 177, 86–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bradford M. M. (1976) Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 24. Kitagawa M., Ara T., Arifuzzaman M., Ioka-Nakamichi T., Inamoto E., Toyonaga H., Mori H. (2005) DNA Res. 12, 291–299 [DOI] [PubMed] [Google Scholar]
- 25. Rodionov D. A., Yang C., Li X., Rodionova I. A., Wang Y., Obraztsova A. Y., Zagnitko O. P., Overbeek R., Romine M. F., Reed S., Fredrickson J. K., Nealson K. H., Osterman A. L. (2010) BMC Genomics 11, 494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Overbeek R., Begley T., Butler R. M., Choudhuri J. V., Chuang H. Y., Cohoon M., de Crécy-Lagard V., Diaz N., Disz T., Edwards R., Fonstein M., Frank E. D., Gerdes S., Glass E. M., Goesmann A., Hanson A., Iwata-Reuyl D., Jensen R., Jamshidi N., Krause L., Kubal M., Larsen N., Linke B., McHardy A. C., Meyer F., Neuweger H., Olsen G., Olson R., Osterman A., Portnoy V., Pusch G. D., Rodionov D. A., Rückert C., Steiner J., Stevens R., Thiele I., Vassieva O., Ye Y., Zagnitko O., Vonstein V. (2005) Nucleic Acids Res. 33, 5691–5702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Edgar R. C. (2004) Nucleic Acids Res. 32, 1792–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Felsenstein J. (1989) Cladistics 5, 164–166 [Google Scholar]
- 29. Finn R. D., Mistry J., Tate J., Coggill P., Heger A., Pollington J. E., Gavin O. L., Gunasekaran P., Ceric G., Forslund K., Holm L., Sonnhammer E. L., Eddy S. R., Bateman A. (2010) Nucleic Acids Res. 38, D211–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Roy A., Kucukural A., Zhang Y. (2010) Nat. Protoc 5, 725–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Berendsen H. J., Spoel V. D., Drunen R. V. (1995) Comput. Phys. Commun. 95, 43–56 [Google Scholar]
- 32. Morris G. M., Huey R., Olson A. J. (2008) Current Protocols in Bioinformatics, Chapter 8, Unit 8.14, Wiley Online Library; [DOI] [PubMed] [Google Scholar]
- 33. van Gunsteren W. F., Billeter S. R., Eising A. A., Hünenberger P. H., Krüger P., Mark A. E., Scott W. R., Tironi I. G. (1996) Biomolecular Simulation: The GROMOS96 Manual and User Guide, Zürich, Groningen [Google Scholar]
- 34. Dundas J., Ouyang Z., Tseng J., Binkowski A., Turpaz Y., Liang J. (2006) Nucleic Acids Res. 34, W116–W118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. (2004) J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 36. Rodionov D. A., De Ingeniis J., Mancini C., Cimadamore F., Zhang H., Osterman A. L., Raffaelli N. (2008) Nucleic Acids Res. 36, 2047–2059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schwarz G., Mendel R. R., Ribbe M. W. (2009) Nature 460, 839–847 [DOI] [PubMed] [Google Scholar]
- 38. Arnez J. G., Harris D. C., Mitschler A., Rees B., Francklyn C. S., Moras D. (1995) EMBO J. 14, 4143–4155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Masure H. R., Pearce B. J., Shio H., Spellerberg B. (1998) Mol. Microbiol. 27, 845–852 [DOI] [PubMed] [Google Scholar]
- 40. Mortier-Barrière I., de Saizieu A., Claverys J. P., Martin B. (1998) Mol. Microbiol. 27, 159–170 [DOI] [PubMed] [Google Scholar]
- 41. Bergé M., García P., Iannelli F., Prère M. F., Granadel C., Polissi A., Claverys J. P. (2001) Mol. Microbiol. 39, 1651–1660 [DOI] [PubMed] [Google Scholar]
- 42. Kaimer C., Graumann P. L. (2010) Arch. Microbiol. 192, 549–557 [DOI] [PubMed] [Google Scholar]
- 43. Wu M., Eisen J. A. (2008) Genome Biol. 9, R151. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.