

Abstract
Accumulating evidence supports the theory that breast cancer arises from a subpopulation of mammary stem/progenitor cell which posses the ability to self-renew. However, the involvement of estrogen signaling in regulation of breast cancer stem/progenitor cells has not been fully established, mainly because expression and function of ER-α in breast cancer stem cells remains controversial. Previously, our laboratory cloned a variant of ER-α, ER-α36, and found that ER-α36-mediated non-genomic estrogen signaling plays an important role in malignant growth of triple-negative breast cancer cells. In this study, we found that ER-α36 was highly expressed in ER-negative breast cancer SK-BR-3 cells and mediated non-genomic estrogen signaling such as activation of the MAPK/ERK signaling in these cells. Knock-down of ER-α36 expression in these cells using the shRNA method diminished their responsiveness to estrogen and significantly down-regulated HER2 expression. HER2 signaling activated ER-α36 transcription through an AP1 site in the ER-α36 promoter and ER-α36 physically interacted with HER2. We also found that ER-α36 is highly expressed in a subset of SK-BR-3 cells that was positive for ALDH1, a breast cancer stem cell marker, and knock-down of ER-α36 expression reduced the population of ALDH1 positive cells. Our results thus demonstrated that ER-α36 positively regulates HER2 expression and the population of ALDH1 positive breast cancer cells, and suggested that non-genomic estrogen signaling mediated by ER-α36 is involved in maintenance and regulation of breast cancer stem cells.
Keywords: HER2, ER-α36, ALDH1, Breast cancer stem cells
1 Introduction
Tumor-initiating or -stem cells are a subpopulation of tumor cells capable of initiating and driving tumor growth. Accumulating experimental and clinical evidence supports the hypothesis that breast cancer arises from a subpopulation of mammary stem/progenitor cell which posses the ability to self-renew (reviewed by [1–4]). Al-Hajj et al, were the first to enrich a CD44+/CD24−/low cell population from human breast cancer that displayed cancer stem cell properties and was capable of forming tumors in immuno-compromised mice with higher efficiency than cells with alternate phenotypes [5]. Later, aldehyde dehydrogenase (ALDH) 1 expression and/or its activity were identified to be a functional marker for breast cancer stem/progenitor cells; fewer ALDH1 positive tumor cells than CD44+/CD24−/low tumor cells are required to generate tumors in vivo [6]. The breast cancers with ALDH1high cancer stem cells are associated with more aggressive phenotypes such as estrogen receptor (ER) negativity, high histological grade, HER2 positivity, as well as poor prognosis [6, 7].
Many signaling pathway important for cell growth and survival are involved in maintenance of breast cancer stem/progenitor cells. Recent studies demonstrated that members of human epidermal growth factor receptor (EGFR) such as HER2 plays a pivotal role in regulation of human breast cancer stem/progenitor cells; the EGFR/HER2 dual inhibitor, lapatinib, and the HER2 specific monoclonal antibody, trastuzumab, dramatically decrease populations of CD44+/CD24−/low/ALDH1High cells and tumorsphere-forming efficiency. In addition, the population of ALDH1High cells was increased by up-regulation of “stemness” genes through HER2 over-expression in breast cancer cells [8–10].
However, the involvement of estrogen signaling, a major signaling pathway in breast cancer development, in regulation of breast cancer stem/progenitor cells has not been fully established. A functional and molecular characterization of mouse mammary “side population” (SP) cells showed that 40% of these cells expressed ER-α [11]. In addition, Clarke et. al., reported that ER-α is expressed in a subset of putative normal breast stem/progenitor cells enriched by the SP method and proposed that these ER-positive stem/progenitor cells are directly stimulated by circulating estrogens [12]. However, Sleeman et al. [13] demonstrated that the ER-expressing luminal epithelial subpopulation contains little in vivo stem cell activity; ER expressing cells are distinct from the mammary stem cell population and the effects of estrogen signaling on mammary stem cells are likely to be mediated indirectly [13]. Despite the controversy of receptor expression, mouse mammary stem cells are highly responsive to steroid hormone signaling; ovariectomy markedly diminished mammary stem cell number and outgrowth potential in vivo whereas mammary stem cell activity increased in mice treated with estrogen plus progesterone [14]. Estrogen was also found to expand breast cancer stem cells through paracrine FGF/Tbx3 pathway, indicating the indirect effects of estrogen on stem cell activity [15]. However, Simoes et al., recently reported that estrogen treatment reduced the population of stem cells in the normal human mammary gland and in breast cancer cells [16]; overexpression of embryonic stem cell genes such as NANOG, OCT4 and SOX2 reduced ER-α expression and increased the population of breast cancer stem cells as well as properties associated with malignancy, which argues a negative role of estrogen signaling mediated by ER-α in activities of breast cancer stem cells.
Previously, we identified and cloned a 36 kDa variant of ER-α, ER-α36, that is mainly expressed on the plasma membrane and in the cytoplasm, and mediates non-genomic estrogen signaling [17, 18]. ER-α36 lacks both transcription activation function domains AF-1 and AF-2 of the full-length 66 kDa ER-α (ER-α66), consistent with the fact that ER-α36 has no intrinsic transcriptional activity [18]. ER-α36 is generated from a promoter located in the first intron of the ER-α66 gene [19], indicating that ER-α36 expression is regulated differently from ER-α66, consistent with the findings that ER-α36 is expressed in specimens from ER-negative breast cancer patients and established ER-negative breast cancer cells that lack ER-α66 expression [18, 20, 21]. ER-α36 was found to be over-expressed in triple-negative breast carcinomas [22], and promotes malignant growth of triple-negative breast cancer MDA-MB-231 and MDA-MB-436 cells [23]. Thus, ER-α36-mediated signaling plays an important role in development and progression of ER-negative breast cancer. However, the molecular mechanisms underlying ER-α36 action in ER-negative breast cancer still remains largely unknown.
In the present study, we investigated the role of ER-α36 in ER-negative breast cancer SK-BR-3 cells that express high levels of both ER-α36 and HER2 and revealed a positive feedback loop between ER-α36 and HER2 expression. This positive cross-regulation is involved in regulation of ALDH1 positive population of SK-BR-3 cells.
2 Materials and methods
2.1 Reagents
Polyethylenimine (PEI) and 17β-estradiol (E2) were purchased from Sigma-Aldrich (St. Louis, MO). The dual luciferase assay system was purchased from Promega Corporation (Madison, WI).
We developed an affinity-purified rabbit polyclonal anti-ER-α36 antibody as a custom service from Alpha Diagnostic, Inc. The antibody was raised against a synthetic peptide antigen corresponding to the unique C-terminal 20 amino acids of ER-α36. The antibody was tested and characterized as described before [18].
Anti-ALDH1 antibody was from BD biosciences (Sparks, MD). Anti-HA mouse monoclonal antibody (mAb), anti-phospho-p44/42 ERK (Thr202/Tyr204) (197G2) mouse mAb, anti-p44/42 ERK (137F5) rabbit mAb, anti-phospho-Akt (Ser473), and anti-Akt antibodies were purchased from Cell Signaling Technology (Boston, MA). The anti-HER2, anti-β-actin and the different secondary antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The goat anti-mouse Alexa Fluor 555 and goat anti-rabbit Alexa Fluor 488 antibodies were purchased from Invitrogen (Carlsbad, CA). HER2 inhibitors Lapatinib, AG825, sodium 4-phenylbutyrate (PB) and retinoic acid (RA) were from CalBiochem (San Diego, CA), and the PI3K inhibitor LY294002 was purchased from Tocris Bioscience (Ellisville, MO). The ECL Western Blotting Detection Reagents were from GEHealthcare (Little Chalfont, Buckinghamshire, UK). The “Concert” cytoplasmic RNA purification reagent was purchased from Invitrogen (Carlsbad, CA), and the ProtoScript II RT-PCR kit was obtained from New England BioLabs (Ipswich, MA). Protein A/G plus agarose was obtained from Santa Cruz Biotechnology (Santa Cruz, CA). The ALDEFLUOR assay kit was purchased from Stemcell Technologies (Durham, NC).
2.2 Cell culture
Breast epithelial cell line MCF10A and breast cancer cell lines MCF7, ZR-75-1, T-47D, H3396, SK-BR-3, MDA-MB-231, MDA-MB-436, and MDA-MB-468 were obtained from American Type Culture Collection (ATCC, Manassas, VA). SK-BR-3, MDA-MB-231, MDA-MB-436, and MDA-MB-468 were maintained in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS) and 1% antibiotic-antimycotic. MCF7, ZR-75-1, T-47D, and H3396 were maintained in Improved Minimal Essential Medium (IMEM) from Invitrogen (Carlsbad, CA) supplemented with 10% FBS, 1% non-essential amino-acids, 1% HEPES buffer, 1% antibiotic-antimycotic from Invitrogen (Carlsbad, CA) and 2μg/ml bovine insulin (Sigma, St. Louis, MO). SK-BR-3 cells transfected with the empty expression vector, a control vector expressing shRNA for luciferase or the pER36Sh-1 expression vector were established as described before [23] and were named as SK-BR-3/V, SK-BR-3/L and SK-BR-3/36S, respectively. All cell lines were maintained at 37°C and 5% CO2 in a humidified incubator. For E2 treatment, cells were maintained in phenol red-free media with 2.5% charcoal-stripped fetal calf serum (Hyclone, Logan, UT) for three days, and then in serum-free medium for 24 hours before experimentation. For ERK activation assays, cells were treated with vehicle (ethanol) and indicated concentrations of E2 for different periods of time.
2.3 Cell growth assays
Cells were seeded in 35mm dishes at a density of 2×104 cells/dish in DMEM supplemented with 10% FBS and were then counted with the ADAM automatic cell counter (Digital Bio., Korea) after different time periods. Three dishes were used for each time point and the experiments were repeated three times.
To test the effects of the anti-ER-α36 antibody on growth of ALDHL and ALDHH SK-BR-3 cells, cells sorted after ALDEFLUOR staining were seeded in 35mm dishes at a density of 3×104 cells/dish in medium containing 2.5% charcoal-dextran stripped FBS and 5 or 10 μg/ml of the anti-ER-α36 antibody for six days. Cells were then counted with the ADAM automatic cell counter (Digital Bio., Korea). Three dishes were used for each time point and the experiments were repeated three times.
2.4 ALDEFLUOR assay
ALDH activity was detected using the ALDEFLUOR assay kit (StemCell Technologies) as recommended by the manufacturer. Briefly, SK-BR-3 cells were suspended in ALDEFLUOR assay buffer containing an ALDH substrate, bodipy-aminoacetaldehyde (BAAA), and incubated for 1 hr at 37°C. A specific inhibitor of ALDH, diethylaminobenzaldehyde (DEAB) was used as a negative control. Flowcytometry cell sorting was performed to separate ALDHL and ALDHH SK-BR-3 cells and data was analyzed by FlowJo software (TreeStar, Ashland, OR).
2.5 Immunoblot and immunoprecipitation analysis
For imunoprecipitation assays, cells were washed twice with ice-cold PBS and lysed with the lysis buffer (150 mM NaCl, 20 mM TrisHCl, pH 7.4, 0.1% NP-40) supplemented with protease and phosphatase inhibitors (Sigma, St. Louis, MO) for 30 minutes on ice. Cell lysates were then incubated with indicated primary antibodies, or pre-immune serum as a negative control for 1 hour at 4°C. Protein A/G plus agarose was then added and incubated for another 1 hour at 4°C. Precipitates were then extensively washed with the lysis buffer, re-suspended in loading buffer and separated on SDS-PAGE. Western blot analysis was performed as described before [23].
2.6 DNA transfection and luciferase assay
HEK293 cells were transfected using PEI transfection reagent with the pER36-736-Luc, pER36-584-Luc, or pER36-513-Luc reporter plasmids as described before [19] and an empty expression vector or the expression vector for HER2 (a kind gift from Dr. Laura Hansen at Creighton University). Cells were co-transfected with a cytomegalovirus-0driven Renilla luciferase plasmid, pRL-CMV (Promega, Madison, WI) to establish transfection efficiency. Twenty-four hours after transfection, cells were treated with DMSO (vehicle), 10 μM of Lapatinib, or LY294002 for twenty-four hours. Forty-eight hours after transfection, cell extracts were prepared and luciferase activities were determined and normalized using the Dual-Luciferase Assay System (Promega, Madison, WI) and a TD 20/20 Luminometer (Turner BioSystems, Inc., Sunnyvale, CA) as instructed by the manufacturer.
2.7 RNA purification and RT-PCR
RNA purification and RT-PCR procedures for ER-α36 and actin were performed as described before [23]. HER2 primers were as follows: forward primer: 5′-AGGGAGTATGTGAATGCC-3′; reverse primer: 5′-GGCCACTGGAATTTTCAC-3′. The procedure of PCR for HER2 was carried out as following: first a denaturing at 94°C for 3 minute, then the remaining PCR was performed at 94 °C for 30 seconds, 50 °C for 30 seconds, and 72 °C for 30 seconds (35 cycles). At last, there was a final elongation at 72 °C for 7 minutes.
2.8 DNA mutagenesis
The DNA mutagenesis process was performed as described in [23].
2.9 Indirect immunofluorescent staining
Cells were fixed in cold methanol for 5 minutes, blocked in 10% normal rabbit serum for 20 minutes, and then incubated with anti-ER-α36 (1:100) or anti-ALDH1 (1:200) antibodies at 4°C overnight. Secondary antibodies, anti-rabbit Alexa Fluor 488 and anti-mouse Alexa Fluor 555 (1:200) were then added and incubated for 30 minutes. Sections were then mounted with the mounting medium containing DAPI before inspection under the fluorescent microscope (Nikon, Eclipss E600).
2.11 Statistical analysis
Data were summarized as the mean ± standard error (SE) using GraphPad InStat software program. Tukey-Kramer Multiple Comparisons Test was used, and the significance was accepted for P values of less than 0.05.
3 Results
3.1 ER-α36 mediates non-genomic estrogen signaling in ER-negative breast cancer SK-BR-3 cells
ER-α36 is highly expressed in ~ 40% of ER-negative breast cancer and its expression is significantly correlated with HER2 expression [21]. Recently, we reported a positive feedback loop between EGFR and ER-α36 expression in triple-negative breast cancer MDA-MB-231 and MDA-MB-436 cells; EGFR signaling activates the promoter activity of ER-α36 and ER-α36 stabilizes the steady state levels of EGFR protein [23]. To determine if there exists a similar relationship between HER2 and ER-α36, we first examined HER2 and ER-α36 expression status in eight breast cancer cell lines with normal mammary epithelial MCF10A cells as a control. ER-α36 expression was detected in all of the breast cancer cell lines but not in MCF10A cells (Figure 1A). Among breast cancer cell lines examined, two cell lines ZR-75-1 and SK-BR-3, also expressed HER2. We used ER-negative breast cancer cell line SK-BR-3 that co-expresses high levels of ER-α36 and HER2 for further study.
Fig. 1. Cross-regulation of ER-α36 and HER2 expression in ER-negative breast cancer SK-BR-3 cells.

(A). Western blot analysis of ER-α36 and HER2 expression in different breast cancer cell lines and mammary epithelial MCF10A cells. (B). Knock-down of ER-α36 expression down-regulated HER2 expression. RT-PCR and Western blot analysis of parental SK-BR-3 cells (SK-BR-3/P), SK-BR-3 cells transfected with an empty vector (SK-BR-3/V), with a control vector expressing shRNA for luciferase (SK-BR-3/L) and with the ER-α36 shRNA (SK-BR-3/36S). (C). SK-BR-3/V and SK-BR-3/36S cells treated with E2 (1nM) for different time points were assessed with Western blot analysis using phosphorylation specific or non specific anti-ERK1/2 antibodies. SK-BR-3/36S cells treated with EGF (20ng/ml) were included as a positive control. (D). SK-BR-3/P, SK-BR-3/V and SK-BR-3/36S cells were counted every other days for six days. Three dishes were used for each time points, the experiments were repeated three times and the data represent the mean ± s.e.
To examine ER-α36 function in SK-BR-3 cells, we knocked down ER-α36 expression using the small hairpin RNA (shRNA) method. SK-BR-3 cells were stably transfected with an shRNA expression vector targeting the 3′ UTR of ER-α36 and established a cell line by pooling more than twenty transfectants. SK-BR-3 cells transfected with an empty expression vector or an expression vector for shRNA against re y luciferase were used as controls. Both western blot analysis and reverse transcriptase PCR indicated that ER-α36 expression was knocked-down more than 80% in the shRNA-expression vector-transfected cells compared with control cells (Figure 1B). Intriguingly, HER2 expression was also dramatically down-regulated in cells with knocked-down levels of ER-α36 at both mRNA and protein levels (Figure 1B), indicating that ER-α36 mediated signaling is involved in positive regulation of HER2 expression.
We then examine whether 17β-estradiol (E2β) induced phosphorylation of the MAPK/ERK1/2, a typical non-genomic estrogen-signaling event, in control cells (SK-BR-3/V) that were transfected by an empty expression vector and cells transfected with a shRNA expression vector specific for ER-α36 that express knocked-down levels of ER-α36 (SK-BR-3/36S). Cells were treated with 1 nM of E2β for different time periods and cell lysates were analyzed with western blot using a phospho-specific ERK1/2 antibody. Figure 1C shows that E2β elicited ERK phosphorylation in control SK-BR-3 cells transfected with an empty expression vector but not in cells with ER-α36 expression knocked-down. However, EGF was still able to induce ERK activation in SK-BR-3 cells with knocked-down level of ER-α36 expression (Figure 1C), indicating there was no defect of the MAPK/ERK signaling in the cells with ER-α36 expression knocked-down. Time course analysis revealed that ERK phosphorylation occurred within 5 min after E2β application, peaked at 15 min, declined at 30 min but failed to return to the basal level at 120 min (Figure 1C). These results strongly suggested that ER-α36 mediates non-genomic estrogen signaling in ER-negative breast cancer SK-BR-3 cells.
We then examined the growth rate of these cells by counting cell numbers every other days for six days. As shown in Figure 1D, the growth rate of SK-BR-3/36S was dramatically decreased compared to control and parental cells. Our data thus indicated that signaling pathways mediated by ER-α36 and HER2 are important for proliferation of ER-negative breast cancer SK-BR-3 cells.
3.2 HER2 signaling positively regulates ER-α36 expression
To determine whether HER2 signaling influences ER-α36 expression, we treated SK-BR-3 cells with the HER2 inhibitors Lapatinib, AG825, sodium 4-phenylbutyrate (PB), retinoic acid (RA), and PB together with RA. The levels of ER-α36 expression in these treated cells were analyzed with western blot. Figure 2A shows that treatment with the HER2 inhibitors potently down-regulated ER-α36 expression. To confirm this, human embryonic kidney (HEK) 293 cells that express un-detectable levels of endogenous HER2 and very low levels of ER-α36 were transiently transfected with a HER2 expression vector. Western blot analysis demonstrated that endogenous ER-α36 expression was up-regulated in HEK293 cells transfected with the HER2-expression vector but not in cells transfected with an empty expression vector (Figure 2B). These data thus indicated that ER-α36 protein concentration is subjected to positive regulation of HER2 signaling.
Fig. 2. HER2 signaling positively regulates ER-α36 expression.

(A). HER2 inhibitors down-regulate ER-α36 expression. SK-BR-3 cells were treated with 10 μM of Lapatinib, AG825, sodium 4-phenylbutyrate (PB), retinoic acid (RA) or PB+RA for 48 hours. ER-α36 expression was examined with Western blot analysis. (B). HER2 expression induces endogenous ER-α36 expression. HEK293 cells were transiently transfected with an empty vector (Vector) or a HER2 expression vector (HER2), and 48 hours after transfection, cells were examined for ER-α36 expression with Western blot analysis. The experiments were repeated three times, and the representative results were shown.
3.3 HER2 signaling activates the ER-α36 promoter activity via an Ap1 site
Recently, we reported that EGFR signaling induces the promoter activity of ER-α36 gene via an Ap-1 binding site located in the 5′ flanking sequence of ER-α36 gene [19]. To examine if HER2 signaling also activates ER-α36 promoter activity, HEK293 cells were transiently co-transfected with a HER2 expression vector and a luciferase reporter plasmid driven by the ER-α36 promoter we cloned and characterized before [19]. HER2 co-transfection resulted in a 2–3-fold induction of ER-α36 promoter activity, which was blocked by pre-treatment of the HER2 inhibitor Lapatinib, but not by the PI3K inhibitor LY294002 (Figure 3C), suggesting the PI3K/AKT signaling is not involved in the activation of ER-α36 promoter activity. When a series of 5′ truncated promoter of ER-α36 was used, we found that HER2 expression failed to activate the promoter activity of the pER36-513 reporter plasmid (Figure 3B). Close examination of DNA sequence in the deleted region revealed an AP-1-binding site located at −556 to −537 residues (relative to the transcription initiation site) of the ER-α36 promoter region (Figure 3A). Mutation of this Ap-1 site abrogated induction of ER-α36 promoter activity by HER2 (Figure 3B), indicating that HER2 signaling activates the ER-α36 promoter activity through an AP-1 dependent signaling pathway.
Fig. 3. HER2 signaling activates ER-α36 promoter activity.

(A). Schematic structures of luciferase reporter plasmid driven by different 5′ truncated promoters of ER-α36. The -736, -584, and -513 indicate residues upstream of the transcription initiation site, respectively. An AP1 binding site is also indicated that was mutated in the pER36-mAP1 plasmid. (B). The luciferase activities in HEK293 cells transfected with different reporter plasmids together with an empty expression vector or an expression vector for HER2. Columns: means of four independent experiments; bars, SE. *, p<0.05, for cells transfected with the HER2 expression vector vs an empty expression vector. (C). HEK293 cells were transfected with the pER36-736 reporter plasmid with the empty expression vector or the HER2 expression vector, and then treated with DMSO vehicle, 10 μM of LY294002 or Lapatinib for 24 hours. The luciferase activities were then normalized and analyzed. Results shown in graph are means from four experiments; bars, SE. *, p<0.05 for cells treated with vehicle vs different inhibitors.
3.4 ER-α36 physically interacts with HER2
To elucidate the molecular mechanism by which ER-α36 functions to mediate non-genomic estrogen signaling in SK-BR-3 cells, we examined whether ER-α36 interacts with HER2. SK-BR-3 cells were transiently transfected with an expression vector for HA-tagged ER-α36 and co-immunoprecipitation/western blot assays were performed with cell lysates from transfected cells. Figure 4 shows that HER2 and ER-α36 co-existed in the immunoprecipitates of the anti-HER2 and anti-HA antibodies. Thus, the presence of HER2 and ER-α36 in the same protein complex suggests an interaction between them and also suggests that ER-α36 may function through HER2.
Fig. 4. ER-α36 physically interacts with HER2.
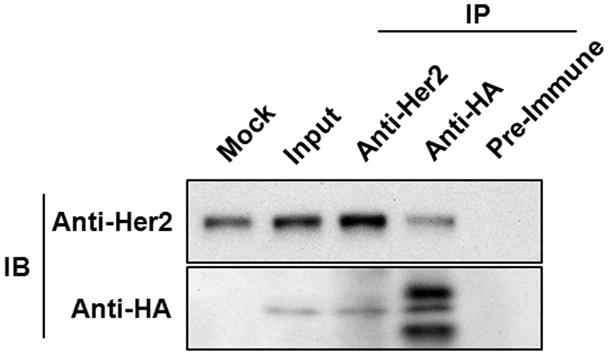
Co-immunoprecipitation and Western blot analysis of ER-α36 and HER2 in SK-BR-3 cells. Cells transiently transfected with an expression vector of HA-tagged ER-α36 were lysed and the cell lysates were immunoprecipitated with anti-HER2 or anti-HA antibodies, or with pre-immune rabbit serum as a negative control. The immunoprecipitates were then separated by SDS-PAGE and probed with anti-HER2 and anti-HA antibodies.
3.5 ER-α36 positively regulates ALDH1 expressing breast cancer cells
Previously, it was reported that the HER2 signaling pathway is involved in the positive regulation of ALDH1 positive breast cancer cells [8–10]. We decided to examine whether ER-α36 is also involved in regulation of ALDH positive breast cancer cells. First, we enriched ALDH1 positive cells from SK-BR-3 cells with flowcytometry cell sorting after stained with the ALDEFLOUR kit. Western blot analysis revealed that the ALDH1 positive/high (ALDH1high) SK-BR-3 cells expressed higher levels of ER-α36 and exhibited higher levels of phosphorylated AKT compared to ALDH1 negative/low (ALDH1low) cells, while no significant changes of HER2 expression were observed (Figure 5A). We also performed the immunofluorescence staining analysis with anti-ALDH1 and ER-α36 antibodies in SK-BR-3/V and SK-BR-3/36S cells. Figure 5C shows that ER-α36 was highly expressed in SK-BR-3/V cells that were also ALDH1 positive. The numbers of ALDH1High cells were significantly decreased in SK-BR-3/36S cells compared to SK-BR-3/V cells (Figure 5B), suggesting that ER-α36 is involved in maintenance of ALDH1High breast cancer cells.
Fig. 5. ER-α36 positively regulates the population of ALDHHigh SK-BR-3 cells.

(A). Western blot analysis of cell lysates of SK-BR-3 cells with ALDH1 low/negative expression (ALDHL) and ALDH1 high expression (ALDHH). (B). Decrease of ALDH1 positive cells in SK-BR-3 cells with ER-α36 expression knocked-down. The numbers of ALDH1 positive cell in 100 counted cells were scored. The results shown are means of four independent experiments; bars, SE. *, p<0.05 for SK-BR-3/V vs SK-BR-3/36S cells. (C). Immunofluorescence staining of SK-BR-3/V and SK-BR-3/36S cells. Cells were fixed and stained by anti-ALDH1 (Red) and anti-ER-α36 (green) antibodies and photographed. The representative photos were shown with X 10 amplification. (D). ALDHL and ALDHH SK-BR-3 cells were treated with 0, 5 or 10 μg/ml of anti-ER-α36 antibody for 6 days. The survived cell numbers were then counted. Results shown in graph are means of three independent experiments; bars, SE. *, p<0.01 for cells treated with control vs cells treated with the antibody.
To further confirm the role of ER-α36-mediated non-genomic estrogen signaling in ALDH1High breast cancer cells, we tested the effects of a specific anti-ER-α36 antibody on ALDH1High cell population. Recently, we demonstrated that the anti-ER-α36 antibody blocked ER-α36-mediated non-genomic estrogen signaling such as activation of the MAPK/ERK signaling [24]. We first enriched ALDH1high cells from SK-BR-3 cells using the ALDEFLOUR kit and flowcytometry, and treated the ALDH1high and ALDH1low cells with different concentrations of the ER-α36 antibody for six days. We found that treatment of these cells with anti ER-α36 antibody significantly inhibited the growth of ALDH1high cells (Figure 5D), indicating that ER-α36-mediated non-genomic estrogen signaling plays an important role in maintenance and proliferation of ALDH1High breast cancer cells.
4 Discussion
Previously, we observed a significant correlation between ER-α36 and HER2 expression in breast cancer patients [21]. Recently, we reported that co-expression of ER-α36 and HER2 were detected 10 out of 19 cases of ER-negative apocrine breast cancer [22]. Here, we used an ER-negative breast cancer cell line SK-BR-3 as a model to study the underlying mechanisms of the correlation between ER-α36 and HER2 expression.
Approximately 20–25 % of breast cancers have an amplification of the HER2 gene or overexpression of its protein product [25]. Overexpression of this receptor in breast cancer is associated with increased disease recurrence and worse prognosis [26, 27]. However, the molecular mechanisms by which breast cancer cells gain HER2 overexpression are largely unknown. Previous studies have demonstrated an interaction between the ER-α66 and HER2 signal transduction [28, 29]. For example, 17β-estradiol is able to reduce HER2 expression in an ER-α66-dependent manner [30], which may provide an explanation to the infrequent co-expression of these two receptors in breast cancer [31–33]. In contrast, a significant positive correlation between HER2 overexpression and ER-β expression has been reported [34, 35]. It was reported that forced expression of ER-β1 in ER-positive breast cancer MCF7 cells induced HER2 expression [36]. Here, we revealed another mechanism for HER2 overexpression in breast cancer cells. We found that HER2 and ER-α36 positively regulate each other’s expression inSK-BR-3 cells; HER2 signaling activates the promoter activity of ER-α36 and ER-α36 activates HER2 transcription. This positive cross-regulation may provide a molecular explanation to the observations that ER-α36 and HER2 expression is significantly correlated in primary breast cancer [21, 22]. These findings are similar to our recent report of a positive feedback loop between ER-α36 and EGFR expression that promotes malignant growth of triple-negative breast cancer cells [23]. Thus, the interplay between growth factor receptors and ER-α36 may play an important role in development and progression of subsets of breast cancer that highly express ER-α36.
In the present study, we also demonstrated ER-α36 physically interacted with HER2 in co-immunoprecipitation assay, which provided a molecular mechanism by which ER-α36 mediates non-genomic estrogen signaling in ER-negative breast cancer SK-BR-3 cells. Consistent with this, we recently found that ER-α36 mediates non-genomic estrogen signaling pathway in triple-negative breast cancer MDA-MB-231 and MDA-MB-436 cells via interaction with the EGFR/Shc/Src complex [23]. Our results thus suggested that growth factor receptors play integral roles in ER-α36-mediated non-genomic estrogen signaling.
The involvement of estrogen signaling in regulation of breast cancer stem/progenitor cells has not been fully established, mainly because expression and function of ER-α66 in breast cancer stem/progenitor cells remains controversial. Thus, our findings demonstrated that ER-α36 is highly expressed in ALDH1high SK-BR-3 cells and knock-down of ER-α36 expression reduced ALDH1high cell population are noteworthy. Previously, the importance of HER2 signaling in maintenance of ALDH1high breast cancer stem cells has been reported [8–10]. Currently, it is not clear whether ER-α36-mediated estrogen signaling is directly involved in positive regulation of ALDH1high breast cancer stem cells or indirectly through activation of HER2 expression. However, the finding that the ER-α36 specific antibody significantly reduced the population of ALDH1high cells in SK-BR-3 cell line suggested that the non-genomic estrogen signaling mediated by ER-α36 is involved in positive regulation of breast cancer stem cells. It is also likely that ER-α36-mediated non-genomic estrogen pathway is involved in the estrogen effects observed in normal mammary stem cells before [14].
In summary, we have shown that ER-α36 positively regulates HER2 expression and the population of ALDH1high breast cancer cells, suggesting that non-genomic estrogen signaling mediated by ER-α36 contributes to development and progression of ER-negative breast cancer that express ER-α36. Thus, ER-α36 is a novel player in non-genomic estrogen signaling that may play important roles in maintenance and regulation of normal mammary stem cells as well as breast cancer stem cells.
Highlights.
ER-α36 mediates non-genomic estrogen signaling in ER-negative breast cancer SK-BR-3 cells.
ER-α36 knock-down diminishes estrogen signaling and downregulates HER2 expression.
HER2 signaling induces ER-α36 expression and ER-α36 physically interacts with HER2.
ER-α36 is highly expressed in ALDH1-positive SK-BR-3 cells, and ER-α36 knock-down reduces their population.
Acknowledgments
This work was supported by NIH grant DK070016 and Nebraska Tobacco Settlement Biomedical Research Program Award (LB-595) to Z.Y. Wang.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dontu G, El-Ashry D, Wicha MS. Breast cancer, stem/progenitor cells and the estrogen receptor. Trends Endocrinol Metab. 2004;15(5):193–197. doi: 10.1016/j.tem.2004.05.011. [DOI] [PubMed] [Google Scholar]
- 2.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414(6859):105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 3.Oliveira LR, Jeffrey SS, Ribeiro-Silva A. Stem cells in human breast cancer. Histol Histopathol. 2010;25(3):371–385. doi: 10.14670/HH-25.371. [DOI] [PubMed] [Google Scholar]
- 4.Dean M. Cancer stem cells: redefining the paradigm of cancer treatment strategies. Mol Interv. 2006;6(3):140–148. doi: 10.1124/mi.6.3.5. [DOI] [PubMed] [Google Scholar]
- 5.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100(7):3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer CG, Liu S, Schott A, Hayes D, Birnbaum D, Wicha MS, Dontu G. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1(5):555–567. doi: 10.1016/j.stem.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morimoto K, Kim SJ, Tanei T, Shimazu K, Tanji Y, Taguchi T, Tamaki Y, Terada N, Noguchi S. Stem cell marker aldehyde dehydrogenase 1-positive breast cancers are characterized by negative estrogen receptor, positive human epidermal growth factor receptor type 2, and high Ki67 expression. Cancer Sci. 2009;100(6):1062–1068. doi: 10.1111/j.1349-7006.2009.01151.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Korkaya H, Paulson A, Iovino F, Wicha MS. HER2 regulates the mammary stem/progenitor cell population driving tumorigenesis and invasion. Oncogene. 2008;27(47):6120–6130. doi: 10.1038/onc.2008.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li X, Lewis MT, Huang J, Gutierrez C, Osborne CK, Wu MF, Hilsenbeck SG, Pavlick A, Zhang X, Chamness GC, Wong H, Rosen J, Chang JC. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Natl Cancer Inst. 2008;100(9):672–679. doi: 10.1093/jnci/djn123. [DOI] [PubMed] [Google Scholar]
- 10.Magnifico A, Albano L, Campaner S, Delia D, Castiglioni F, Gasparini P, Sozzi G, Fontanella E, Menard S, Tagliabue E. Tumor-initiating cells of HER2-positive carcinoma cell lines express the highest oncoprotein levels and are sensitive to trastuzumab. Clin Cancer Res. 2009;15(6):2010–2021. doi: 10.1158/1078-0432.CCR-08-1327. [DOI] [PubMed] [Google Scholar]
- 11.Alvi AJ, Clayton H, Joshi C, Enver T, Ashworth A, Vivanco MM, Dale TC, Smalley MJ. Functional and molecular characterisation of mammary side population cells. Breast Cancer Res. 2003;5(1):R1–8. doi: 10.1186/bcr547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clarke RB, Spence K, Anderson E, Howell A, Okano H, Potten CS. A putative human breast stem cell population is enriched for steroid receptor-positive cells. Dev Biol. 2005;277(2):443–456. doi: 10.1016/j.ydbio.2004.07.044. [DOI] [PubMed] [Google Scholar]
- 13.Sleeman KE, Kendrick H, Robertson D, Isacke CM, Ashworth A, Smalley MJ. Dissociation of estrogen receptor expression and in vivo stem cell activity in the mammary gland. J Cell Biol. 2007;176(1):19–26. doi: 10.1083/jcb.200604065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Asselin-Labat ML, Vaillant F, Sheridan JM, Pal B, Wu D, Simpson ER, Yasuda H, Smyth GK, Martin TJ, Lindeman GJ, Visvader JE. Control of mammary stem cell function by steroid hormone signalling. Nature. 2010;465(7299):798–802. doi: 10.1038/nature09027. [DOI] [PubMed] [Google Scholar]
- 15.Fillmore CM, Gupta PB, Rudnick JA, Caballero S, Keller PJ, Lander ES, Kuperwasser C. Estrogen expands breast cancer stem-like cells through paracrine FGF/Tbx3 signaling. Proc Natl Acad Sci U S A. 2010;107(50):21737–21742. doi: 10.1073/pnas.1007863107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Simoes BM, Piva M, Iriondo O, Comaills V, Lopez-Ruiz JA, Zabalza I, Mieza JA, Acinas O, Vivanco MD. Effects of estrogen on the proportion of stem cells in the breast. Breast Cancer Res Treat. 2010 doi: 10.1007/s10549-010-1169-4. [DOI] [PubMed] [Google Scholar]
- 17.Wang Z, Zhang X, Shen P, Loggie BW, Chang Y, Deuel TF. Identification, cloning, and expression of human estrogen receptor-alpha36, a novel variant of human estrogen receptor-alpha66. Biochem Biophys Res Commun. 2005;336(4):1023–1027. doi: 10.1016/j.bbrc.2005.08.226. [DOI] [PubMed] [Google Scholar]
- 18.Wang Z, Zhang X, Shen P, Loggie BW, Chang Y, Deuel TF. A variant of estrogen receptor-{alpha}, hER-{alpha}36: transduction of estrogen- and antiestrogen-dependent membrane-initiated mitogenic signaling. Proc Natl Acad Sci U S A. 2006;103(24):9063–9068. doi: 10.1073/pnas.0603339103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zou Y, Ding L, Coleman M, Wang Z. Estrogen receptor-alpha (ER-alpha) suppresses expression of its variant ER-alpha 36. FEBS Lett. 2009;583(8):1368–1374. doi: 10.1016/j.febslet.2009.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee LM, Cao J, Deng H, Chen P, Gatalica Z, Wang ZY. ER-alpha36, a novel variant of ER-alpha, is expressed in ER-positive and -negative human breast carcinomas. Anticancer Res. 2008;28(1B):479–483. [PMC free article] [PubMed] [Google Scholar]
- 21.Shi L, Dong B, Li Z, Lu Y, Ouyang T, Li J, Wang T, Fan Z, Fan T, Lin B, Wang Z, Xie Y. Expression of ER-{alpha}36, a novel variant of estrogen receptor {alpha}, and resistance to tamoxifen treatment in breast cancer. J Clin Oncol. 2009;27(21):3423–3429. doi: 10.1200/JCO.2008.17.2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vranic S, Gatalica Z, Deng H, Frkovic-Grazio S, Lee LM, Gurjeva O, Wang ZY. ER-alpha36, a novel isoform of ER-alpha66, is commonly over-expressed in apocrine and adenoid cystic carcinomas of the breast. J Clin Pathol. 2011;64(1):54–57. doi: 10.1136/jcp.2010.082776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang XT, Kang LG, Ding L, Vranic S, Gatalica Z, Wang ZY. A positive feedback loop of ER-alpha36/EGFR promotes malignant growth of ER-negative breast cancer cells. Oncogene. 2011;30(7):770–780. doi: 10.1038/onc.2010.458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kang L, Zhang X, Xie Y, Tu Y, Wang D, Liu Z, Wang ZY. Involvement of estrogen receptor variant ER-alpha36, not GPR30, in nongenomic estrogen signaling. Mol Endocrinol. 2010;24(4):709–721. doi: 10.1210/me.2009-0317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Slamon DJ, Godolphin W, Jones LA, Holt JA, Wong SG, Keith DE, Levin WJ, Stuart SG, Udove J, Ullrich A, et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science. 1989;244(4905):707–712. doi: 10.1126/science.2470152. [DOI] [PubMed] [Google Scholar]
- 26.Goldhirsch A, Glick JH, Gelber RD, Coates AS, Thurlimann B, Senn HJ. Meeting highlights: international expert consensus on the primary therapy of early breast cancer 2005. Ann Oncol. 2005;16(10):1569–1583. doi: 10.1093/annonc/mdi326. [DOI] [PubMed] [Google Scholar]
- 27.Jukkola A, Bloigu R, Soini Y, Savolainen ER, Holli K, Blanco G. c-erbB-2 positivity is a factor for poor prognosis in breast cancer and poor response to hormonal or chemotherapy treatment in advanced disease. Eur J Cancer. 2001;37(3):347–354. doi: 10.1016/s0959-8049(00)00395-6. [DOI] [PubMed] [Google Scholar]
- 28.Ariazi EA, Kraus RJ, Farrell ML, Jordan VC, Mertz JE. Estrogen-related receptor alpha1 transcriptional activities are regulated in part via the ErbB2/HER2 signaling pathway. Mol Cancer Res. 2007;5(1):71–85. doi: 10.1158/1541-7786.MCR-06-0227. [DOI] [PubMed] [Google Scholar]
- 29.Stoica GE, Franke TF, Moroni M, Mueller S, Morgan E, Iann MC, Winder AD, Reiter R, Wellstein A, Martin MB, Stoica A. Effect of estradiol on estrogen receptor-alpha gene expression and activity can be modulated by the ErbB2/PI 3-K/Akt pathway. Oncogene. 2003;22(39):7998–8011. doi: 10.1038/sj.onc.1206769. [DOI] [PubMed] [Google Scholar]
- 30.Read LD, Keith D, Jr, Slamon DJ, Katzenellenbogen BS. Hormonal modulation of HER-2/neu protooncogene messenger ribonucleic acid and p185 protein expression in human breast cancer cell lines. Cancer Res. 1990;50(13):3947–3951. [PubMed] [Google Scholar]
- 31.Ciocca DR, Gago FE, Fanelli MA, Calderwood SK. Co-expression of steroid receptors (estrogen receptor alpha and/or progesterone receptors) and Her-2/neu: Clinical implications. J Steroid Biochem Mol Biol. 2006;102(1–5):32–40. doi: 10.1016/j.jsbmb.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 32.Dowsett M. Overexpression of HER-2 as a resistance mechanism to hormonal therapy for breast cancer. Endocr Relat Cancer. 2001;8(3):191–195. doi: 10.1677/erc.0.0080191. [DOI] [PubMed] [Google Scholar]
- 33.Zeillinger R, Kury F, Czerwenka K, Kubista E, Sliutz G, Knogler W, Huber J, Zielinski C, Reiner G, Jakesz R, et al. HER-2 amplification, steroid receptors and epidermal growth factor receptor in primary breast cancer. Oncogene. 1989;4(1):109–114. [PubMed] [Google Scholar]
- 34.Umekita Y, Souda M, Ohi Y, Sagara Y, Rai Y, Takahama T, Yoshida H. Expression of wild-type estrogen receptor beta protein in human breast cancer: specific correlation with HER2/neu overexpression. Pathol Int. 2006;56(8):423–427. doi: 10.1111/j.1440-1827.2006.01983.x. [DOI] [PubMed] [Google Scholar]
- 35.Choi Y, Pinto M. Estrogen receptor beta in breast cancer: associations between ERbeta, hormonal receptors, and other prognostic biomarkers. Appl Immunohistochem Mol Morphol. 2005;13(1):19–24. doi: 10.1097/00129039-200503000-00004. [DOI] [PubMed] [Google Scholar]
- 36.Lattrich C, Juhasz-Boess I, Ortmann O, Treeck O. Detection of an elevated HER2 expression in MCF-7 breast cancer cells overexpressing estrogen receptor beta1. Oncol Rep. 2008;19(3):811–817. [PubMed] [Google Scholar]