

Abstract
The human pathogen Pseudomonas aeruginosa produces a variety of virulence factors including pyoverdine, a non-ribosomally produced peptide siderophore. The maturation pathway of the pyoverdine peptide is complex and provides a unique target for inhibition. Within the pyoverdine biosynthetic cluster is a periplasmic hydrolase, PvdQ, that is required for pyoverdine production. However, the precise role of PvdQ in the maturation pathway has not been biochemically characterized. We demonstrate herein that the initial module of the nonribosomal peptide synthetase PvdL adds a myristate moiety to the pyoverdine precursor. We extracted this acylated precursor, called PVDIq, from a pvdQ mutant strain and show that the PvdQ enzyme removes the fatty acid catalyzing one of the final steps in pyoverdine maturation. Incubation of PVDIq with crystals of PvdQ allowed us to capture the acylated enzyme and confirm through structural studies the chemical composition of the incorporated acyl chain. Finally, because inhibition of siderophore synthesis has been identified as a potential antibiotic strategy, we developed a high throughput screening assay and tested a small chemical library for compounds that inhibit PvdQ activity. Two compounds that block PvdQ have been identified and their binding within the fatty acid binding pocket structurally characterized.
Keywords: Siderophore biosynthesis, NTN Hydrolase, Pyoverdine, High-Throughput Assay Development
Pseudomonas aeruginosa is an opportunistic gram-negative pathogen that causes nosocomial infections and chronic lung infections in cystic fibrosis patients ((1), (2)) These infections are established in the form of a biofilm that is relatively insensitive to immune responses and antibiotics (3). This native resistance and persistent infection in the face of current antibacterial drugs has far reaching consequences for patient morbidity and mortality and also demonstrates a need to identify new strategies and therapies to combat this pathogen. Targeting novel essential bacterial pathways that are responsible for the acquisition of essential nutrients is one possible mechanism for development of new anti-infective agents (4).
Iron is a necessary trace element for nearly all living organisms and plays key catalytic and structural roles in proteins (5). Despite its relative abundance, free iron (Fe3+) acquisition poses a challenge to bacteria due to toxicity and poor solubility. As a result, bacteria have evolved synthetic pathways to produce and secrete high affinity sequestering agents called siderophores that bind to iron and are actively transported back into the cell (6). In many bacteria, specialized peptide siderophores are produced by modular enzymes known as non-ribosomal peptide synthetases (NRPSs). These enzymes are molecular assembly lines, organized with multiple catalytic domains joined in a single protein (7). To produce the siderophore compounds, many NRPS proteins work in concert with other NRPSs or accessory proteins. These supplementary enzymes are involved in the synthesis of building blocks, siderophore maturation and export, import of the Fe3+-siderophore complex, or the removal of Fe3+ from the imported siderophore (8).
Pyoverdine is the primary iron siderophore produced by P. aeruginosa and has been associated with infection in multiple disease models (5). Multiple isoforms of pyoverdine have been identified in P. aeruginosa (Figure 1), all of which are composed of a cyclic peptide chain synthesized by the four large cytoplasmic NRPSs (PvdL, PvdI, PvdJ, PvdD), a chemically modified dihydroxyquinoline-based chromophore that is responsible for iron binding, and an N-terminal side chain bound to the chromophore ((9), (10)). Along with the NRPSs that produce the peptide chain, eleven other proteins have been identified that are critical to pyoverdine production (11). These proteins play a significant role in pyoverdine synthesis, including cyclization, export, and final maturation in the periplasmic space ((5), (12)). Several proteins are well characterized, including the ornithine hydroxylase PvdA (13), the aminotransferase PvdH (14), and the hydroxyornithine transformylase PvdF (15) Although the exact roles of many of the tailoring enzymes are not known, their involvement in this essential siderophore catalytic pathway presents them as attractive targets for new antibiotic development (16).
Figure 1.
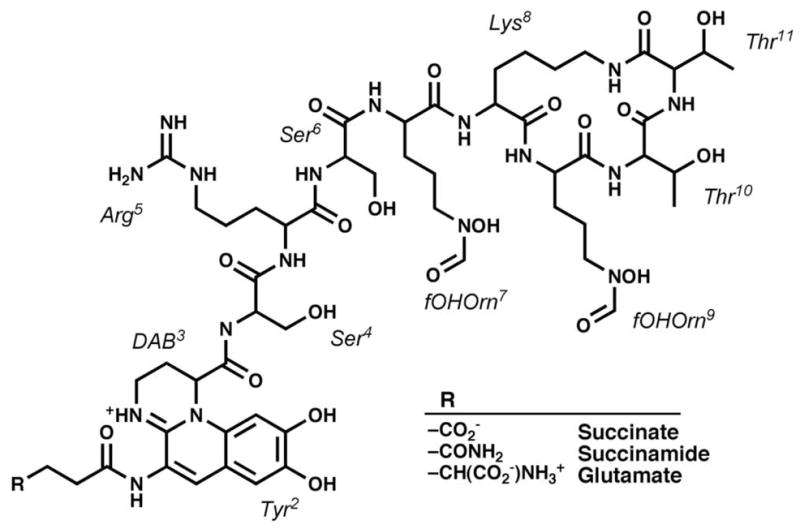
The pyoverdine siderophore produced by the human pathogen Pseudomonas aeruginosa. Several isoforms have been identified that vary at the N-terminal residue.
A potential strategy for identifying new treatments for P. aeruginosa infections is finding small molecules that interfere with maturation and expression of critical siderophores or quorum sensors, both of which have been implicated in biofilm formation ((17), (18)) and bacterial virulence (19). High throughput screening (HTS) methods could be used to identify compounds that disrupt the maturation processes in these metabolic pathyways, and indeed this method has already been proven effective in identifying potential small molecule inhibitors of bacterial signaling molecules (20). In this regard, we have investigated the fatty acylase PvdQ, a critical protein in pyoverdine synthesis.
PvdQ belongs to the NTN hydrolase family (21), whose members are produced as inactive proteins and autoproteolytically cleaved to produce active α/β-heterodimers. The N-terminus of the β–chain is preferentially a serine that is responsible both for autoproteolysis and the catalytic cleavage of acyl esters (22). PvdQ was identified as a periplasmic quorum quenching protein that cleaves acyl homoserine lactones (23). The crystal structure has been reported in this context (21) for both native PvdQ and a covalently acylated enzyme. However, the location of PvdQ in the pyoverdine synthetic operon and recent reports suggest that its primary role is in pyoverdine maturation.
Quax et al (24) have demonstrated that a pvdQ knockout shows multiple reduced virulence factors, including pyoverdine production, growth in iron-limiting media, swarming motility, and biofilm formation. These phenotypes could not be linked with the presence or absence of acylated signaling molecules. Additionally, an acyl-pyoverdine precursor termed PVDIq has recently been isolated from a pvdQ mutant, providing a direct link between PvdQ and maturation of the pyoverdine precursor (25).
Herein, we describe the fatty acid adenylating activity of the initial module of PvdL, demonstrating the chemical nature of the acylated pyoverdine peptide precursor. The acyl pyoverdine is purified and shown through biochemical analysis to serve as a substrate for PvdQ. We additionally describe the development and validation of a high throughput screen for small molecule inhibitors of PvdQ. The assay exploits the promiscuous activity of PvdQ to produce colorimetric or fluorimetric by-products. Results from initial small library screenings are presented and validated by kinetic analyses. Structural studies are also presented showing PvdQ covalently acylated with the acyl moiety substrate derived from the acyl-pyoverdine precursor, as well as bound to two novel small molecule inhibitors identified from the library screen. These biochemical and structural studies demonstrate that the described assay is a viable method for identification of small molecule inhibitors that may prevent pyoverdine maturation and hinder cell growth under iron limiting conditions.
Results and Discussion
Substrate Preference of the PvdL Module 1 Adenylation Domain
NRPS proteins are modular enzymes that catalyze the formation of many biologically important peptides including peptide antibiotics and peptide siderophores (26). Most commonly, each module of an NRPS is responsible for incorporation of a single amino acid into the final peptide product. Each module contains a peptidyl carrier protein (PCP) domain that is covalently modified with a pantetheine cofactor. During peptide synthesis, amino acids and the nascent peptide are bound as thioesters to the PCP pantetheine. Upstream of the PCP domains are adenylation domains that catalyze a two-step reaction to first activate the amino acid through an adenylation reaction and then transfer the amino acid to the pantetheine thiol. These two step adenylation and thioesterification reactions are functionally similar to acyl-CoA synthetases and place the NRPS adenylation domains within the ANL superfamily of adenylating enzymes (27). Finally, each module contains a condensation domain that catalyzes peptide bond formation between activated amino acids on adjacent PCP domains.
Pyoverdine is produced by the activity of four NRPS proteins, termed PvdL, PvdI, PvdJ, and PvdD (5). PvdL (PA2424, NP_251114) is a four module NRPS protein (28). The three C-terminal adenylation domains have all had substrates assigned and activate the first three amino acids in the pyoverdine peptide backbone. The N-terminal module was predicted to add an acyl chain to the pyoverdine peptide (28), but no acyl precursor had been identified until Yeterian et al. characterized a fluorescent pyoverdine precursor from a pvdQ deletion strain with a m/z that was 226 Da higher than mature pyoverdine (25). Given the presence of the known acylase PvdQ (PA2385, NP_251075) in the pyoverdine synthetic operon, and its requirement for mature pyoverdine production, we identified the preferred substrate of the N-terminal PvdL module to correlate with the preferred substrate of PvdQ. Purified PvdL module 1 (PvdLM1) adenylation domain was examined using a pyrophosphate exchange assay and demonstrated clear preference with the C14 medium chain acid as a substrate (Table 1). Activity was also apparent using other medium chain fatty acids (C10, C12). Activity with short chain acids (C4, C6, C8) and long chain acids (C16, C18) were comparable to background. These results identify myristate as the preferred acyl substrate for the PvdLM1 adenylation domain.
Table 1.
Pyrophosphate Exchange Activity of PvdL Module 1
| Fatty Acid Chain Length | CPM (×103)a |
|---|---|
| C6 | 18.4 ± 3.9 |
| C8 | 26.7 ± 0.7 |
| C10 | 87.6 ± 24.4 |
| C12 | 79.4 ± 11.5 |
| C14 | 160.0 ± 14.1 |
| C16 | 26.1 ± 2.8 |
| C18 | 7.3 ± 3.4 |
Counts of 32P-pyrophosphate incorporated into ATP through the reversible adenylation reaction as described in Methods.
Cleavage of the acyl chain from pyoverdine precursors
To confirm that PVDIq has the fatty acid chain incorporated by the PvdLM1 adenylation domain and show that PvdQ is capable of removing this chain we purified the precursor from culture media. A pvdQ mutant (ΔQ) will grow to saturation in a minimal media lacking an iron chelator where trace iron is sufficient to support growth. Pyoverdine isoforms were purified by HPLC and monitored at 410 nm. Multiple isoforms of pyoverdine were evident reflecting heterogeneity at the N-terminal side chain and the chromophore (29).
Wild type pyoverdine elutes much earlier in an acetonitrile gradient (~40 %) than PVDIq (~80 %) due to the fatty acid chain (Figure 2). Incubations with purified PvdQ showed that the PVDIq peak could be shifted to ~43 % on the acetonitrile gradient. PvdQ does not affect the wild type peak (data not shown). Mass spectrometry analyses of these fractions showed major peaks for the wild type as m/z 1,333.61 (corresponding to PVDI + H+) and PVDIq as m/z 1559.84 (PVDIq + H+). The mass of PVDIq shifted to m/z 1349.64 upon incubation with enzyme. Molecular structures for the major observed molecular masses are provided in the Supplemental Material. We note that the amount of PVDIq present in the mutant strain media is drastically reduced compared to the pyoverdine found in wild type cell media. It is not clear through our results if the PVDIq molecule is actively exported or simply is released from lysed cells.
Figure 2.

HPLC profile of extracted culture supernatants and enzymatically cleaved supernatant. Experimentally determined m/z ratios of the major peak and expected chemical structures are shown. Because much less pyoverdine precursor is produced from the mutant cell line compared to mature pyoverdine from the wild-type cells, the HPLC runs of pvdQ mutants used approximate seven times more sample. The chemical nature of the peaks at retention times 37–38 min of both wild-type and mutant samples are unknown.
Assay development for high throughput screening of PvdQ
PvdQ presents an attractive target for inhibition through chemical probe development. The protein is necessary for pyoverdine production (30) and it has been demonstrated that PvdQ or alternate pyoverdine synthetic proteins (9) are necessary for virulence. PvdQ uses a catalytic nucleophile, which has proven amenable to inhibition in other serine hydrolases. With the previous data supporting a role of PvdQ in pyoverdine maturation, we developed a screening assay for PvdQ activity.
To create a high throughput assay, multiple potential substrates were tested, including 4-nitrophenyl (pNP) esters of short (octanoate), medium (laurate, myristate), and long chain (palmitate) fatty acids. The long and short chain substrates did not show any signal above background. Both medium chain substrates showed fairly low catalytic efficiencies. However, this property proved beneficial in the high-throughput screen. Optimization and quality assessment of the assay conditions were tested by varying enzyme and substrate concentrations, incubation times, and reagent stability. The assays were insensitive to DMSO concentrations up to 1.5 %. We also investigated an alternative fluorogenic substrate, 4-methyllumbelliferyl (4MU) laurate, to minimize interference of the colorimetric assay from the library compounds.
For the colorimetric pNP substrate, the myristate ester showed a ~10-fold decrease in KM towards PvdQ and a ~3 fold increase in catalytic efficiency compared to the laurate ester. For the fluorogenic substrate, only a laurate ester was available precluding a comparison for substrates with differing chain lengths (Table 2).
Table 2.
Enzymatic Activity of PvdQ
| Substrate | KM (mM) | kcat (min−1) | kcat/Km (M−1s−1) |
|---|---|---|---|
| pNP-Laurate | 11.4 | 119.3 | 175 |
| pNP-Myristate | 1.4 | 44.4 | 521 |
| 4MU-Laurate | 0.75 | 74.6 | 1660 |
Kinetic values were determined as described in Methods
Utilizing conditions and concentrations from the steady state kinetics, high throughput assay quality (31) was assessed by calculating the Z′ factor, S/N, and S/B for both reporters. After optimization, the colorimetric assay had a Z score of 0.87, a S/N value of 33.97, and a S/B value of 2.15. Respective values for the fluorometric assay were 0.84, 24.15, and 16.96. Statistical values for both reporters show that a high throughput assay would be a viable tool for determining prospective inhibitors of PvdQ, although library compounds may interfere with both assays via similar optical properties. Two potential positive controls were also examined, the serine hydrolase protease inhibitor phenylmethyl sulfonylfluoride (PMSF) and isopropyldodecyl fluorophosphonate (IDFP), a known inhibitor of a mechanistically related fatty acid amide hydrolase (32). PvdQ activity was insensitive to PMSF whereas IDFP gave an IC50 of 345 μM (not shown)
High Throughput Screening and Hit Evaluation of PvdQ
Following optimization, the assay was probed using the LOPAC 1280 library of bioactive compounds. Each compound was screened at 100 μM in duplicate for both the chromogenic and fluorogenic substrate with positive and negative controls on each plate. Assay substrates and the PvdQ enzyme dilutions were prepared fresh each day, with two to four plates being run on individual days. Z′ factors on the individual plates varied from 0.7 to 0.9 and was 0.6 over all plates. Compounds that showed inhibition effects greater than 20% (1.1% of the 1280 tested compounds) for both substrates were screened again. 11 of the 14 compounds were confirmed by rescreening from the LOPAC 1280 source plates in the high throughput assay, and 7 of these were purchased (based on score, availability, and price) for further evaluation.
The positive compounds from the high throughput screen were then more rigorously examined with the same conditions that were used to obtain kinetic constants for pNP myristate. The IC50 values for the prospective inhibitors were calculated by generating a dose response curve based on percent inhibition of PvdQ.
Five of the seven compounds failed in the second round screening. LOPAC compound 150 (bromoenol lactone (BEL)) causes precipitation of PvdQ. LOPAC compound 464 (Dipyridamole) interfered with the pNP absorption spectrum disrupting measurement, and LOPAC compound 844 (ZM 39923 hydrochloride) showed inhibition but no dose response curve over the range tested. Finally, LOPAC compounds 1018 (Bay11-7802) and 1105 (1-(4-Hexyphenyl)-2-propane-1-one) showed no reproducible inhibition compared to the initial screening with the LOPAC 1280 source plates.
However, two remaining hits (Figure 3), LOPAC compounds 922 (NS2028, 4H-8-Bromo-1,2,4-oxadiazolo[3,4-d]benz[b][1,4]oxazin-1-one) and 1019 (SMER28, 6-Bromo-N-2-propenyl-4-quinazolinamine) both displayed dose-dependent inhibition, with IC50 values of 145 μM and 66 μM respectively. These inhibition values were reproducible and more specific than IDFP, providing confidence that the two leads from the high throughput screen were valid and worth pursuing in crystallization trials.
Figure 3.

Two inhibitors of PvdQ identified in the high-throughput screening assay.
Structural Analysis of PvdQ bound to the acyl chain derived from PVDIq
The PvdQ structure has previously been solved and described by Bokhove et al (21). Our lab had independently solved the structure of PvdQ utilizing selenium multiwavelength anomalous dispersion phasing methods. We solved the structure of the native enzyme, which showed a co-purifying octanoate molecule bound non-covalently in the fatty acid binding pocket. We also incubated native crystals with pNP myristate to obtain the acylated enzyme intermediate with a molecule of myristate bound to Serβ217. The structures of native (3L91) and pNP myristate acylated (3L94) PvdQ have been deposited with the Protein Data Bank (Supplemental Data). PvdQ is part of the NTN-hydrolase superfamily and demonstrates a ‘V’-shaped structure formed from two interleaved chains. The vertex of the ‘V’ creates a hydrophobic pocket that presents the catalytic serine (Serβ217) responsible for the acyl hydrolysis mechanism of the enzyme (Figure 4). The majority of the substrate cavity is interior to the structure and thus sequestered from the solvent.
Figure 4.

Three-dimensional representation of PvdQ. (a) Secondary structure representation showing the ‘V’ architecture. Orange depicts the alpha chain, while blue shows the beta-chain that initiates with the catalytic serine at residue 217 (b) Mixed secondary (orange α-chain) and surface area (blue β-chain) representation of PvdQ. The covalently acylated enzyme is shown with the myristate depicted with yellow sticks for carbon atoms and red sticks for oxygen. Panel b is rotated approximately 30° around the Y-axis for visualization of the ligand.
We first used pNP-myristate to form the acylated enzyme (Supplemental Data). Successfully soaking ligand into preformed crystals to obtain the covalently modified enzyme raised the possibility of soaking acyl-pyoverdine isolated from the ΔQ strain into unliganded crystals. We transferred the crystals from a pH of 7.5 to pH of 5 to promote protonation of the catalytic serine and possibly capture the acyl-pyoverdine substrate in the active site. We soaked crystals in the presence of PVDIq for varying times. At 10 min, the active site showed no density for a fatty acid, demonstrating that the copurifying octanoate was removed from the protein. At 2 h, however, clear density was seen for a fatty acid (Figure S1). Despite the lower pH inubation, the reaction still occurred in the crystal, providing a structure of the acylated enzyme where the acyl group is specifically derived from the pyoverdine precursor (Figure 5A). Electron density unambiguously identified a covalent link between Serβ217 and a straight chain fatty acid provided by PVDIq. Refinement of this chain was undertaken with fatty acids of varying length. Refining 12 or 16 carbon fatty acids showed difference density clearly indicating neither fit well to the experimental data. Chains containing 13, 14, or 15 carbons could all be refined reasonably well to the observed data. However, given the prevalence of even numbered straight chain fatty acids and the data provided by mass spectrometry, the final model is refined with a myristate bound to Serβ217. This covalent-acyl intermediate between PvdQ and the PVDIq fatty acid chain (Figure 5A) runs from the catalytic center along the entire length of the hydrophobic pocket. Interaction of this myristate in the pocket is coordinated mostly by Van der Waal’s forces with the exception of the hydrogen bonding of the carboxyl end with Serβ217, Valβ286, and Asnβ485.
Figure 5.

The liganded catalytic active site of PvdQ. (a) Structure of PvdQ bound to the myristate group derived from incubation of the enzyme with PVDIq. (b) Structure of PvdQ bound to NS2028. (c) Structure of PvdQ bound to SMER28. (d) Superposition of myristoylated enzyme with SMER28. Protein residues are shown in grey and yellow for acylated enzyme and in blue for side chains from the SMER28 structure. In panels a-c, the electron density is calculated with coefficients of the form Fo-Fc generated prior to the inclusion of ligands in the active site. The density is contoured at 2.5σ (grey) and at 8σ (red) in panels b and c.
Structural Analysis of PvdQ bound to novel inhibitors
We then examined the structure of PvdQ bound to the two inhibitors, NS2028 and SMER28 (Figures 5B, 5C). Structural alignment of the two inhibitor structures show RMS distance values below < 0.2 Å for all Cα positions. The two inhibitors both show strong electron density in the PvdQ binding pocket, and contain a bromine atom that allows for definitive placement and orientation. The inhibitors display a strong hydrophobic bonding interface at the deepest interior wall of the substrate pocket and are further stabilized with hydrogen bonding to Asnβ273. No interaction is observed with either inhibitor at the catalytic residues of PvdQ, or with the hydrogen bonding residues that appear to coordinate the acyl-pyoverdine amide for the nucleophilic attack by the serine. Instead the inhibitors bind deep within the hydrophobic pocket that accommodates the fatty acid. This occupancy in the recess of the pocket suggests that the inhibitors are preventing the acyl substrate from presenting the carboxylate in the correct orientation to Serβ217, as opposed to interfering with the catalytic mechanism itself.
This observation is further borne out by the position of Pheβ240. As noted by Bokhove et al (21) this phenyalanine provides a “gate” mechanism that separates the PvdQ acyl pocket from the surrounding solvent. For Serβ217 to initiate the nucleophilic attack on the carbonyl carbon of the substrate, Pheβ240 must transition to an open state and allow the terminal methyl end of the fatty acid chain into the recesses of the substrate pocket. Both inhibitor structures show Pheβ240 in a closed position with the inhibitor maintained in the recess by hydrogen bonding to Asnβ273. This asparagine residue adopts different conformations of the carboxamide group depending on the ligand that is present, with the NS2028 and the covalent acyl-intermediate structures sharing one orientation and the SMER28 structure showing an alternate conformation with a rotation around the β carbon allowing the carbamoyl oxygen to present itself to the nitrogen that precedes the aliphatic tail of SMER28. It might be anticipated that the fatty acid binding pocket for the linear myristate chain is too narrow to accommodate the cyclic inhibitors. In contrast, there are no side chain movements other than Asnβ273 and Pheβ240. The acyl binding pocket is therefore large enough to accept the inhibitor molecules.
Insights into the Maturation of Pyoverdine
The PvdL NRPS produces the peptide foundation for the pyoverdine chromophore. Biosynthesis is initiated with a myristate moiety by the Module 1 adenylation domain being passed to Module 2 of PvdL where canonically an L-Glu is incorporated. Subsequently, Tyr and 2,4-diaminobutyrate (28) are activated and condensed to form the chromophore precursor. The PvdQ substrate is fluorescent (25) suggesting that cyclization, or partial cyclization, of the chromophore does occur prior to myristate removal. Hydrolysis of the myristic fatty acid would be expected to leave the amine on the N-terminus of the PVD peptide, consistent with our observations. However, most isoforms of pyoverdine that have been identified are missing this amine. Interestingly, this would mean that PvdQ is not the last step before export but that further modifications would occur. Additionally, if the only substrate for Module 2 were L-Glu, as is generally reported (5), it is difficult to imagine reasonable chemical mechanisms that could use the remaining periplasmic proteins PvdN, PvdO, and PvdP to form the all of the observed pyoverdine isoforms.
An alternate explanation is that PvdL Module 2 does not only activate L-Glu, but is capable of using multiple amino acids to produce the chemical diversity noted among pyoverdine isoforms. Adenylate enzymes have been shown to have promiscuity (33) and informatic predictions have evolved to use not only the ten residue fingerprint from the active site (34) but also determine the properties and dimensions of the surrounding residues. These adenlyate enzyme substrate predictors provide substrate recognition based on “clustering” of amino acid properties. In this scenario PvdL module 2 is predicted to utilize either Asp, Asn, Glu, or Gln (35). Indeed, our mass spectrometry results support an aspartic acid residue as the primary substrate of module 2, in agreement with results of Schalk (25).
This study demonstrates that the PvdL Module 1 adenylation domain is specific to a myristic fatty acid and that this myristate is removed by PvdQ in the periplasm. The myristate chain may direct the precursor to the periplasm or may serve some other unknown function. To complete our understanding of the pyoverdine maturation process, it will be necessary to understand the function and order of operation of the remaining enzymes. The development of specific chemical probes for the proteins involved in pyoverdine biosynthesis will help elucidate these mechanistic details. The assay described herein will allow for the screening of large chemical libraries, which may yield an improved inhibitor for PvdQ. Combined with similar probes or targeted knockout mutants of other pyoverdine synthetic enzymes, it will allow for the further exploration of the pyoverdine pathway and may ultimately provide insights into P. aeruginosa pathogenesis that can be used for therapeutic development.
Methods
Protein Expression and Purification of PvdQ and PvdL Module 1
The pvdQ gene (NP_251075) was amplified from P. aeruginosa genomic DNA using primers to incorporate restriction sites at the 5′ and 3′ ends of the gene. The gene was then subcloned into a modified pET15b vector. To avoid having a 5xHis affinity tag associated with the protein product, the pvdQ gene was cloned into the plasmid NcoI (5′) and NdeI (3′) restriction sites, replacing the sequence that codes for a 5x His and protease site. The resulting plasmid, pED482, encodes native PvdQ with no extraneous residues. The final plasmid was transformed into E. coli BL21(DE3) for protein production. Native PvdQ was purified from the cellular periplasm to allow for autoproteolytic processing to the α/β-heterodimer. Cells were grown in 2xYT + 0.1 % glycerol for 4 hours at 37 °C - 300 RPM and then incubated at 25 °C - 250 RPM for 24 hours. The periplasmic proteins were harvested according to osmotic lysis methods (36). Cells were harvested by centrifugation at 5000×g for 10 min (10 °C) and resuspended in 40 ml room temperature 0.01 M Tris-HCl (pH 8.0) per gram of cell paste. The cells were then centrifuged at 3500×g for 10 min at 10 °C and resuspended in 40 ml room temperature 0.5 M Sucrose, 0.03 M Tris-HCl (pH 8.0), 0.002 M Na2EDTA per g. The swelled cells were then centrifuged at 4000×g for 10 min at 10 °C and resuspended in 25 ml 4 °C 0.001 M Tris-HCl (pH 8.0) per g. Finally, cells lacking the periplasm were removed with a final centrifugation at 5000×g for 10 min at 4 °C. The supernatant was adjusted to 0.05 M Tris-HCl (pH 8.0) with 1.5 M Tris-HCl (pH 8.0) and further clarified by centrifugation. All of the remaining purification steps were performed at 4 °C.
The periplasmic lysate was subjected to 60 % ammonium sulfate precipitation with solid ammonium sulfate. The lysate was equilibrated for 1 hour, and the precipitate was harvested by centrifugation and resuspended in 0.05 M Tris-HCl (pH 8.0). The protein was passed over a 5 ml HiTrap desalting column (GE-Healthcare) in 0.05 M Tris-HCl (pH 8.0). The fractions were pooled based on their OD280, then run over a 5 ml HiTrapQ FF column (GE-Healthcare) using the same buffer as described by Quax et al. (23). The protein, found in the initial flow through by SDS-Page analysis, was pooled, concentrated, and dialyzed overnight against a gel filtration buffer (0.05 M Tris-HCl (pH 8.0), 0.05 M NaCl, and 0.2 mM TCEP). Gel Filtration was performed on a Superdex 200 HiLoad 16/60 column and protein was concentrated to 10 mg/ml using an extinction coefficient at 280 nm of 118100 M−1 cm−1.
PvdL Module 1 (PvdLM1) was cloned using PCR from genomic DNA with primers incorporating a 5′ NdeI restriction site and a 3′ BamHI site. Module 1 of PvdL was defined as residues 2–657 of PvdL (NP_251114) and includes both the adenylation and PCP domains. The PCR product was subcloned into a modified pET15bTEV vector for protein expression and purification (37) and confirmed by DNA sequencing. The plasmid was transformed into E. coli BL21(DE3) and cells were grown in M9 minimal media to an OD600 of ~0.6 at 37 °C. Protein expression was induced by the addition of 0.75 mM IPTG and cells were incubated overnight. Cells were harvested at 4800×g for 20 minutes and lysed using a microfluidizer (M-110EH Microfluidizer, Microfluidics Corporation) in a buffer containing 50 mM Tris (pH 7.5), 150 mM NaCl, 10 mM imidazole, and 0.2 mM TCEP. Membrane fractions, nucleic acid, and unlysed cells were removed by centrifugation at 45,000 rpm for 45 minutes. Protein was purified using a His-trap immobilized metal ion affinity column (GE-Healthcare); lysate was loaded over the column, and washed with 50 mM imidazole. Bound PvdLM1 was eluted with 300 mM imidazole. The His-tag was removed from the protein by dialysis in a cleavage buffer [50 mM Tris (pH 8.0), 150 mM NaCl, 0.2 mM TCEP, 0.5 mM EDTA] with TEV protease added at a molar ratio of ~1:80. Cleavage continued overnight. The protein was clarified and passed over the His-trap column a second time to remove any uncleaved protein, TEV protease, and the cleaved tag. Protein from the flowthrough was analyzed by SDS page for purity, dialyzed against a final storage buffer [20 mM Tris (pH 7.5), 40 mM NaCl, 0.2 mM TCEP], and concentrated to ~10 mg/mL using a 280 nm extinction coefficient of 63680 M−1 cm−1.
Pyrophosphate Exchange Assay for PvdL Module 1
The pyrophosphate-exchange assay was used to measure the activity of the adenylate-forming half-reaction of Module 1 of PA2424 (PvdLM1) ((38), (39)). In this assay, PvdLM1 was allowed to catalyze the reversible incorporation of radiolabel into ATP using various short to long chain fatty acid substrates. Specifically the reaction contained 2 mM ATP, 0.2 mM NaPPi, 50 mM HEPES (pH 8.0), 100 mM NaCl, 10 mM MgCl2, 1.5 μCi/mL of 32PPi, and 10 mM of linear fatty acids ranging from C6 to C18. The reaction was initiated with 10 μM enzyme, incubated at 37 °C for 10 min, and quenched with a solution containing 1.2 % (w/v) activated charcoal, 0.1 M unlabeled PPi, and 0.35 M perchloric acid. The charcoal pellet was washed twice with 1 mL of H2O and resuspended in 0.5 mL of H2O for scintillation counting. Each assay was evaluated using five replicates and results were recorded as counts per minute. During this assay it was evident that PvdL had co-purified with a fatty acid substrate with the ‘no substrate’ reaction demonstrating a noticeably higher background than the reaction lacking enzyme. This co-purification of a fatty acid has been observed with other adenylate forming enzymes and experimental structural data has even shown electron density in native ‘unliganded ‘ proteins ((40),(41)). Attempts to remove the contaminating ligand through gel filtration and extensive dialysis were not successful. Values for the ‘No Enzyme’ and ‘co-purified fatty acid’ controls were 0.119 × 103 and 30.6 × 103 respectively.
Isolation and Characterization of Pyoverdine Isoforms
Wild type Pseudomonas aeruginosa (ATCC 15692), and knockout mutants of PA2385 (Manoil Lab Transposon Mutant Collection, University of Washington) were grown to saturation in SM9 medium and pyoverdine from the culture media harvested using the method of Budzikiewicz (42). Cells were removed from the media through centrifugation, 10–30 ml of media was adjusted to 50mM Tris (pH 8.0) and 50mM NaCl. For analysis of the activity of PvdQ on the isolated pyoverdine or pyoverdine precursor, the sample was incubated with 0.2 mM PvdQ or an equal volume of buffer for negative controls. This solution was then passed through a 1 ml octyl Amprep minicolumn (GE Healthcare, Piscataway, NJ), washed with 2 ml of H2O and 1 ml of 10 % methanol, and eluted with 50 % methanol. The eluate was evaporatively dried and reconstituted in 3:97:0.1 acetonitrile/H2O/formic acid. These samples were then analyzed by HPLC on a C18 reverse phase column (4.62×50 mm), washed with 3 % acetonitrile, and eluted with a 3–100 % gradient. Samples were detected by absorption at 410 nm.
For mass spectrometry the peak eluted from the HPLC was dried by evaporation, resuspended in 80% acetonitrile, spotted on a Maldi plate, and analyzed by Maldi-TOF mass spectrometry. For crystallization trials, samples were dried by evaporation and resuspended in 20 μL of 100 % DMSO. Samples reconstituted in parallel with the crystallization ligand had an optical density of 0.285 at 410 nm.
Assessment of PvdQ Kinetic Activity
The activity of PvdQ was measured against fluorometric (4-Methylumbelliferyl laurate (4MU-laurate), Research Organics) or colorimetric [4-nitrophenyl myristate (pNP-Myristate) or 4-nitrophenyl laurate (pNP-laurate), Fluka] substrates. Kinetic reactions were carried out on an Agilent 8453 spectrophotometer. For pNP-myristate the A410 of liberated p-nitrophenol was measured, while 4MU-Laurate was monitored at A340. Reactions were carried out according to the method described in Lin et al. (43). The required amount of powdered substrate was initially dissolved in 2-hydroxypropane. This was added to a buffer containing 1 % Triton X-100 and 5 mM sodium acetate buffer pH 5.0, and warmed to 37 °C. Final concentrations of all components in the reaction were 5 % 2-hydroxypropane, 0.5 % Triton X-100, 2.5 mM sodium acetate buffer pH 5.0, 27.5 mM Tris pH 8.0, 27.5 mM NaCl, 0.2 μM PvdQ, and substrate concentration varying between 0.2–1.5 mM. Steady state kinetics with the myristate and laurate esters of pNP were performed under conditions where the rate for PvdQ hydrolysis was linear for up to 3 minutes. Initial rates were calculated using 120 seconds of data and used to determine KM and kcat values for prospective substrates. Reactions were performed in triplicate and the values analyzed using Dynafit software (44). Because we observed a copurified octanoate molecule in the crystal structure of the native enzyme, we examined the affect of octanoate on the kinetics. Concentrations of 0.1 – 1 μM had no effect on the reaction at two min (the linear portion of the assay) or at 30 min (data not shown).
Assay Conditions for High Throughput Screening
The LOPAC 1280 chemical library (Sigma Chemicals) was assayed at concentrations of 100 μM in 96-well plates. For the colorimetric assay, Whatman clear polystyrene flat bottom plates were read in the Tecan Genios plate reader at 340 nm. Fluorescence measurements were made using CoStar black polystyrene flat bottom plates in a BioTek Synergy plate reader with an excitation at 351 nm and emission read at 430 nm. Reaction volumes were 100 μL with the final concentrations of 1 mM pNP-myristate or 0.8 mM 4MU-Laurate, 0.5 % Triton X-100, 5 % 2-hydroxypropane, 50 mM Tris pH 8.0, and 50 mM NaCl. The reactions were initiated with the addition of 10 μL of protein, resulting in final protein concentrations of 200 nM for the colorimetric assay and 100 nM for the fluorometric assay.
Equations for Range (R), sum of standard deviations (SSD), signal to noise (S/N), and Z′ were calculated according Zhang. et al (31), initial Z′ scores were calculated from 12 wells for two independent preps with readings taken every ten minutes over a two hour time course. An optimum Z′ factor was obtained for both substrates at the 45 minute mark. Positive controls consisted of substrate and enzyme with 1 % DMSO; negative controls contained boiled enzyme and 1 % DMSO. Z′ factors during the inhibition screen were measured for each plate using 8 control wells. Assay parameters were calculated with the following equations:
where ODmax, ODmin, σmax and σmin are means and standard deviations of positive and negative control reactions. The signal to background (S/B) is defined as the ratio of ODmax to ODmin (45)
Crystallization and Structure Determination
PvdQ crystals were optimized from leads first identified in the 1536 microbatch crystallization screen provided by the Center for High Throughput Structural Biology (46). Crystals of the native protein were grown at 20°C by hanging drop vapor diffusion using a 1:1 mixture of protein and precipitant containing 10–15 % PEG 4000, 50–100 mM RbCl, and 50 mM Hepes (pH 7.5). Native crystals were mounted in nylon loops and cryo-protected by transferring them through five solutions containing increasing amounts of ethylene glycol (8 %, 12 %, 16 %, 20 %, 24 %) for approximately 20 seconds each and stored in liquid nitrogen. The final cryo-protectant solution contained 24 % ethylene glycol, 15 % PEG 4000, 0.1 M RbCl, and 50 mM Hepes (pH 7.5). SeMet crystals were optimized to 17–22 % PEG 4000, 80–100 mM RbCl, and 50 mM Hepes (pH 7.5).
While this work was underway, Bokhove et al (21) reported the crystal structure of the native PvdQ protein and PvdQ bound to several acyl substrates. We had determined the structure independently using heavy atom selenomethionine protein and MAD phasing (Supplemental Material).
To obtain the structure of the acylated protein, unliganded PvdQ crystals were transferred through a series of solutions from pH 7.5 to 5 at 0.5 pH unit intervals for approximately 5 minutes each. Cryoprotectant solutions containing increasing concentrations of ethylene glycol (8 %, 12 %, 16 %, 20 %, 24 %) were also made at pH 5 containing the PVDIq ligand. Crystals were allowed to soak in the 8 % wash for multiple time points (10 min, 60 min, 2 hours, 4 hours, 24 hours) and then transferred rapidly through the remaining cryo solutions that also contained appropriate substrate concentrations. A dataset from the crystal soaked for 10 min displayed no electron density in the active site attributable to the ligand, while soaks longer than two hours reduced crystal quality (Figure S1). Crystals that were soaked for two hours provided a dataset with density for the acylated enzyme.
Inhibitors that were identified from the high throughput screen were co-crystallized with PvdQ in the native conditions. Inhibitor compounds were dissolved in 100 % DMSO to 50 mM and incubated with protein at 1.5x molar excess for 45 minutes on wet ice prior to crystallization setup. Crystals were grown in the same manner as unliganded PvdQ protein and cryo protection was similar with the addition of 1 mM ligand at each of the five steps. During model building of the liganded structures both SMER28 and NS2028 were refined with multiple orientations and occupancies. In all instances there remained some positive and negative density near the bromine atoms of the ligands. It is likely that there is some degree of dehalogenation of the ligands due to radiation damage.
Datasets were acquired either remotely at the Stanford Synchotron Radiation Laboratory (47) or at the Cornell High Energy Synchotron Source (CHESS). All liganded complexes were determined using difference Fourier methods starting from the coordinates of the p-nitrophenyl myristate PvdQ structure. In all cases, substrates and waters were removed from the starting model to avoid bias. Iterative model-building and refinement were continued to completion for all liganded PvdQ structures using COOT (48) and REFMAC (49). Data collection and refinement statistics can be found in Table 3.
Table 3.
Crystallographic Data for PvdQ acylated with myristate derived from the acyl pyoverdine precursor and PvdQ bound to two inhibitors.
| Acylated PvdQ | PvdQ + SMER28 | PvdQ + NS2028 | |
|---|---|---|---|
| Data Collection | |||
| PDB Code | 3SRA | 3SRB | 3SRC |
| Synchrotron Beamline | SSRL 11-1 | SSRL 11-1 | SSRL 9-1 |
| Wavelength (Å) | 0.97918 | 0.97946 | 0.97945 |
| Resolution (Å) | 2.3 | 1.75 | 2.0 |
| Space Group | C2221 | C2221 | C2221 |
| Unit Cell parameters (Å) | |||
| a | 120.2 | 121.0 | 120.6 |
| b | 166.6 | 166.7 | 165.8 |
| c | 93.7 | 93.9 | 94.0 |
| Rmergea(%) | 11.4 (35.2) | 3.9 (18.7) | 7.6 (47.6) |
| Completenessa | 99.4 (99.8) | 99.6 (100.0) | 97.2 (78.2) |
| I/σa | 12.0 (2.9) | 21 (4.0) | 9.9 (1.7) |
| No. observations | 171381 | 508693 | 270625 |
| No. reflections | 41892 | 87481 | 61822 |
| Refinement | |||
| Refinement resolution (Å) | 29.3 – 2.3 | 35.0 – 1.8 | 39.0 – 2.0 |
| R-factora (%) | 18.2 (20.6) | 18.8 (22.5) | 19.8 (27.9) |
| R-freea (%) | 21.7 (24.1) | 20.3 (28.5) | 22.2 (32.6) |
| Wilson B-value(Å2) | 31.85 | 20.249 | 30.8 |
| Average B-factor | |||
| Overall (Å2) | 27.5 | 21.1 | 32.9 |
| Solvent (Å2) | 28.8 | 25.7 | 35.3 |
| Ligand (Å2) | 37.1 | 15.5 | 49.1 |
| RMS deviation from ideal | |||
| Bond lengths (Å) | 0.010 | 0.007 | 0.009 |
| Bond angles (°) | 1.191 | 1.053 | 1.265 |
Values in parentheses represent the highest resolution shell.
Supplementary Material
Acknowledgments
We thank Jimmy Theriault and Jose Perez (Broad Institute) for helpful suggestions. This research was supported in part by grants from the National Institutes of Health (GM-068440, MH-092076), the Cystic Fibrosis Foundation (GULICK0710), and the Max and Victoria Dreyfus Foundation. Diffraction data were collected at the Cornell High Energy Synchrotron Source which is supported by the NSF under award DMR 0225180 and the NIH through NCRR award 5 P41 RR001646-23, and at the Stanford Synchrotron Radiation Lightsource, a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the NIH, NCRR, Biomedical Technology Program, and the National Institute of General Medical Sciences.
Footnotes
Supporting Information Available
Additional crystallographic analysis of PvdQ and analysis of pyoverdine isoforms are available in Supplemental Material. This information is available free of charge via the Internet at http://pubs.acs.org
References
- 1.Fischbach MA, Walsh CT. Antibiotics for emerging pathogens. Science. 2009;325:1089–1093. doi: 10.1126/science.1176667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Davies JC. Pseudomonas aeruginosa in cystic fibrosis: pathogenesis and persistence. Paediatr Respir Rev. 2002;3:128–134. doi: 10.1016/s1526-0550(02)00003-3. [DOI] [PubMed] [Google Scholar]
- 3.Costerton JW, Lewandowski Z, Caldwell DE, Korber DR, Lappin-Scott HM. Microbial biofilms. Annu Rev Microbiol. 1995;49:711–745. doi: 10.1146/annurev.mi.49.100195.003431. [DOI] [PubMed] [Google Scholar]
- 4.Clatworthy AE, Pierson E, Hung DT. Targeting virulence: a new paradigm for antimicrobial therapy. Nat Chem Biol. 2007;3:541–548. doi: 10.1038/nchembio.2007.24. [DOI] [PubMed] [Google Scholar]
- 5.Visca P, Imperi F, Lamont IL. Pyoverdine siderophores: from biogenesis to biosignificance. Trends Microbiol. 2007;15:22–30. doi: 10.1016/j.tim.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 6.Sandy M, Butler A. Microbial iron acquisition: marine and terrestrial siderophores. Chem Rev. 2009;109:4580–4595. doi: 10.1021/cr9002787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koglin A, Walsh CT. Structural insights into nonribosomal peptide enzymatic assembly lines. Nat Prod Rep. 2009;26:987–1000. doi: 10.1039/b904543k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Quadri LE, Sello J, Keating TA, Weinreb PH, Walsh CT. Identification of a Mycobacterium tuberculosis gene cluster encoding the biosynthetic enzymes for assembly of the virulence-conferring siderophore mycobactin. Chem Biol. 1998;5:631–645. doi: 10.1016/s1074-5521(98)90291-5. [DOI] [PubMed] [Google Scholar]
- 9.Meyer JM, Neely A, Stintzi A, Georges C, Holder IA. Pyoverdin is essential for virulence of Pseudomonas aeruginosa. Infect Immun. 1996;64:518–523. doi: 10.1128/iai.64.2.518-523.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fuchs R, Schafer M, Geoffroy V, Meyer JM. Siderotyping--a powerful tool for the characterization of pyoverdines. Curr Top Med Chem. 2001;1:31–57. doi: 10.2174/1568026013395542. [DOI] [PubMed] [Google Scholar]
- 11.Ochsner UA, Wilderman PJ, Vasil AI, Vasil ML. GeneChip expression analysis of the iron starvation response in Pseudomonas aeruginosa: identification of novel pyoverdine biosynthesis genes. Mol Microbiol. 2002;45:1277–1287. doi: 10.1046/j.1365-2958.2002.03084.x. [DOI] [PubMed] [Google Scholar]
- 12.Baysse C, Budzikiewicz H, Uria Fernandez D, Cornelis P. Impaired maturation of the siderophore pyoverdine chromophore in Pseudomonas fluorescens ATCC 17400 deficient for the cytochrome c biogenesis protein CcmC. FEBS Lett. 2002;523:23–28. doi: 10.1016/s0014-5793(02)02915-0. [DOI] [PubMed] [Google Scholar]
- 13.Meneely KM, Barr EW, Bollinger JM, Jr, Lamb AL. Kinetic mechanism of ornithine hydroxylase (PvdA) from Pseudomonas aeruginosa: substrate triggering of O2 addition but not flavin reduction. Biochemistry. 2009;48:4371–4376. doi: 10.1021/bi900442z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vandenende CS, Vlasschaert M, Seah SY. Functional characterization of an aminotransferase required for pyoverdine siderophore biosynthesis in Pseudomonas aeruginosa PAO1. J Bacteriol. 2004;186:5596–5602. doi: 10.1128/JB.186.17.5596-5602.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McMorran BJ, Shanta Kumara HM, Sullivan K, Lamont IL. Involvement of a transformylase enzyme in siderophore synthesis in Pseudomonas aeruginosa. Microbiology. 2001;147:1517–1524. doi: 10.1099/00221287-147-6-1517. [DOI] [PubMed] [Google Scholar]
- 16.Somu RV, Boshoff H, Qiao C, Bennett EM, Barry CE, 3rd, Aldrich CC. Rationally designed nucleoside antibiotics that inhibit siderophore biosynthesis of Mycobacterium tuberculosis. J Med Chem. 2006;49:31–34. doi: 10.1021/jm051060o. [DOI] [PubMed] [Google Scholar]
- 17.Banin E, Vasil ML, Greenberg EP. Iron and Pseudomonas aeruginosa biofilm formation. Proc Natl Acad Sci U S A. 2005;102:11076–11081. doi: 10.1073/pnas.0504266102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang L, Nilsson M, Gjermansen M, Givskov M, Tolker-Nielsen T. Pyoverdine and PQS mediated subpopulation interactions involved in Pseudomonas aeruginosa biofilm formation. Mol Microbiol. 2009;74:1380–1392. doi: 10.1111/j.1365-2958.2009.06934.x. [DOI] [PubMed] [Google Scholar]
- 19.Viducic D, Ono T, Murakami K, Katakami M, Susilowati H, Miyake Y. rpoN gene of Pseudomonas aeruginosa alters its susceptibility to quinolones and carbapenems. Antimicrob Agents Chemother. 2007;51:1455–1462. doi: 10.1128/AAC.00348-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hentzer M, Riedel K, Rasmussen TB, Heydorn A, Andersen JB, Parsek MR, Rice SA, Eberl L, Molin S, Hoiby N, Kjelleberg S, Givskov M. Inhibition of quorum sensing in Pseudomonas aeruginosa biofilm bacteria by a halogenated furanone compound. Microbiology. 2002;148:87–102. doi: 10.1099/00221287-148-1-87. [DOI] [PubMed] [Google Scholar]
- 21.Bokhove M, Nadal Jimenez P, Quax WJ, Dijkstra BW. The quorum-quenching N-acyl homoserine lactone acylase PvdQ is an Ntn-hydrolase with an unusual substrate-binding pocket. Proc Natl Acad Sci U S A. 2010;107:686–691. doi: 10.1073/pnas.0911839107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brannigan JA, Dodson G, Duggleby HJ, Moody PC, Smith JL, Tomchick DR, Murzin AG. A protein catalytic framework with an N-terminal nucleophile is capable of self-activation. Nature. 1995;378:416–419. doi: 10.1038/378416a0. [DOI] [PubMed] [Google Scholar]
- 23.Sio CF, Otten LG, Cool RH, Diggle SP, Braun PG, Bos R, Daykin M, Camara M, Williams P, Quax WJ. Quorum quenching by an N-acyl-homoserine lactone acylase from Pseudomonas aeruginosa PAO1. Infect Immun. 2006;74:1673–1682. doi: 10.1128/IAI.74.3.1673-1682.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nadal Jimenez P, Koch G, Papaioannou E, Wahjudi M, Krzeslak J, Coenye T, Cool RH, Quax WJ. Role of PvdQ in Pseudomonas aeruginosa virulence under iron-limiting conditions. Microbiology. 2010;156:49–59. doi: 10.1099/mic.0.030973-0. [DOI] [PubMed] [Google Scholar]
- 25.Yeterian E, Martin LW, Guillon L, Journet L, Lamont IL, Schalk IJ. Synthesis of the siderophore pyoverdine in Pseudomonas aeruginosa involves a periplasmic maturation. Amino Acids. 2010;38:1447–1459. doi: 10.1007/s00726-009-0358-0. [DOI] [PubMed] [Google Scholar]
- 26.Strieker M, Tanovic A, Marahiel MA. Nonribosomal peptide synthetases: structures and dynamics. Curr Opin Struct Biol. 2010;20:234–240. doi: 10.1016/j.sbi.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 27.Gulick AM. Conformational dynamics in the Acyl-CoA synthetases, adenylation domains of non-ribosomal peptide synthetases, and firefly luciferase. ACS Chem Biol. 2009;4:811–827. doi: 10.1021/cb900156h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mossialos D, Ochsner U, Baysse C, Chablain P, Pirnay JP, Koedam N, Budzikiewicz H, Fernandez DU, Schafer M, Ravel J, Cornelis P. Identification of new, conserved, non-ribosomal peptide synthetases from fluorescent pseudomonads involved in the biosynthesis of the siderophore pyoverdine. Mol Microbiol. 2002;45:1673–1685. doi: 10.1046/j.1365-2958.2002.03120.x. [DOI] [PubMed] [Google Scholar]
- 29.Kilz S, Lenz C, Fuchs R, Budzikiewicz H. A fast screening method for the identification of siderophores from fluorescent Pseudomonas spp. by liquid chromatography/electrospray mass spectrometry. J Mass Spectrom. 1999;34:281–290. doi: 10.1002/(SICI)1096-9888(199904)34:4<281::AID-JMS750>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 30.Lamont IL, Martin LW. Identification and characterization of novel pyoverdine synthesis genes in Pseudomonas aeruginosa. Microbiology. 2003;149:833–842. doi: 10.1099/mic.0.26085-0. [DOI] [PubMed] [Google Scholar]
- 31.Zhang JH, Chung TD, Oldenburg KR. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 32.Nomura DK, Blankman JL, Simon GM, Fujioka K, Issa RS, Ward AM, Cravatt BF, Casida JE. Activation of the endocannabinoid system by organophosphorus nerve agents. Nat Chem Biol. 2008;4:373–378. doi: 10.1038/nchembio.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Villiers BR, Hollfelder F. Mapping the limits of substrate specificity of the adenylation domain of TycA. Chembiochem. 2009;10:671–682. doi: 10.1002/cbic.200800553. [DOI] [PubMed] [Google Scholar]
- 34.Stachelhaus T, Mootz HD, Marahiel MA. The specificity-conferring code of adenylation domains in nonribosomal peptide synthetases. Chem Biol. 1999;6:493–505. doi: 10.1016/S1074-5521(99)80082-9. [DOI] [PubMed] [Google Scholar]
- 35.Rausch C, Weber T, Kohlbacher O, Wohlleben W, Huson DH. Specificity prediction of adenylation domains in nonribosomal peptide synthetases (NRPS) using transductive support vector machines (TSVMs) Nucleic Acids Res. 2005;33:5799–5808. doi: 10.1093/nar/gki885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koshland D, Botstein D. Secretion of beta-lactamase requires the carboxy end of the protein. Cell. 1980;20:749–760. doi: 10.1016/0092-8674(80)90321-9. [DOI] [PubMed] [Google Scholar]
- 37.Kapust RB, Waugh DS. Controlled intracellular processing of fusion proteins by TEV protease. Protein Expr Purif. 2000;19:312–318. doi: 10.1006/prep.2000.1251. [DOI] [PubMed] [Google Scholar]
- 38.Rusnak F, Faraci WS, Walsh CT. Subcloning, expression, and purification of the enterobactin biosynthetic enzyme 2,3-dihydroxybenzoate-AMP ligase: demonstration of enzyme-bound (2,3-dihydroxybenzoyl)adenylate product. Biochemistry. 1989;28:6827–6835. doi: 10.1021/bi00443a008. [DOI] [PubMed] [Google Scholar]
- 39.Babbitt PC, Kenyon GL, Martin BM, Charest H, Slyvestre M, Scholten JD, Chang KH, Liang PH, Dunaway-Mariano D. Ancestry of the 4-chlorobenzoate dehalogenase: analysis of amino acid sequence identities among families of acyl:adenyl ligases, enoyl-CoA hydratases/isomerases, and acyl-CoA thioesterases. Biochemistry. 1992;31:5594–5604. doi: 10.1021/bi00139a024. [DOI] [PubMed] [Google Scholar]
- 40.Hisanaga Y, Ago H, Nakagawa N, Hamada K, Ida K, Yamamoto M, Hori T, Arii Y, Sugahara M, Kuramitsu S, Yokoyama S, Miyano M. Structural basis of the substrate-specific two-step catalysis of long chain fatty acyl-CoA synthetase dimer. J Biol Chem. 2004;279:31717–31726. doi: 10.1074/jbc.M400100200. [DOI] [PubMed] [Google Scholar]
- 41.Kochan G, Pilka ES, von Delft F, Oppermann U, Yue WW. Structural snapshots for the conformation-dependent catalysis by human medium-chain acyl-coenzyme A synthetase ACSM2A. J Mol Biol. 2009;388:997–1008. doi: 10.1016/j.jmb.2009.03.064. [DOI] [PubMed] [Google Scholar]
- 42.Meyer JM, Stintzi A, De Vos D, Cornelis P, Tappe R, Taraz K, Budzikiewicz H. Use of siderophores to type pseudomonads: the three Pseudomonas aeruginosa pyoverdine systems. Microbiology. 1997;143(Pt 1):35–43. doi: 10.1099/00221287-143-1-35. [DOI] [PubMed] [Google Scholar]
- 43.Lin SF, Chiou CM, Yeh CM, Tsai YC. Purification and partial characterization of an alkaline lipase from Pseudomonas pseudoalcaligenes F-111. Appl Environ Microbiol. 1996;62:1093–1095. doi: 10.1128/aem.62.3.1093-1095.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kuzmic P. Program DYNAFIT for the analysis of enzyme kinetic data: application to HIV proteinase. Anal Biochem. 1996;237:260–273. doi: 10.1006/abio.1996.0238. [DOI] [PubMed] [Google Scholar]
- 45.Lazo JS, Brady LS, Dingledine R. Building a pharmacological lexicon: small molecule discovery in academia. Mol Pharmacol. 2007;72:1–7. doi: 10.1124/mol.107.035113. [DOI] [PubMed] [Google Scholar]
- 46.Luft JR, Collins RJ, Fehrman NA, Lauricella AM, Veatch CK, DeTitta GT. A deliberate approach to screening for initial crystallization conditions of biological macromolecules. J Struct Biol. 2003;142:170–179. doi: 10.1016/s1047-8477(03)00048-0. [DOI] [PubMed] [Google Scholar]
- 47.McPhillips TM, McPhillips SE, Chiu HJ, Cohen AE, Deacon AM, Ellis PJ, Garman E, Gonzalez A, Sauter NK, Phizackerley RP, Soltis SM, Kuhn P. Blu-Ice and the Distributed Control System: software for data acquisition and instrument control at macromolecular crystallography beamlines. J Synchrotron Radiat. 2002;9:401–406. doi: 10.1107/s0909049502015170. [DOI] [PubMed] [Google Scholar]
- 48.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 49.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.