

Abstract
In analysis of bioinformatics data, a unique challenge arises from the high dimensionality of measurements. Without loss of generality, we use genomic study with gene expression measurements as a representative example but note that analysis techniques discussed in this article are also applicable to other types of bioinformatics studies. Principal component analysis (PCA) is a classic dimension reduction approach. It constructs linear combinations of gene expressions, called principal components (PCs). The PCs are orthogonal to each other, can effectively explain variation of gene expressions, and may have a much lower dimensionality. PCA is computationally simple and can be realized using many existing software packages. This article consists of the following parts. First, we review the standard PCA technique and their applications in bioinformatics data analysis. Second, we describe recent ‘non-standard’ applications of PCA, including accommodating interactions among genes, pathways and network modules and conducting PCA with estimating equations as opposed to gene expressions. Third, we introduce several recently proposed PCA-based techniques, including the supervised PCA, sparse PCA and functional PCA. The supervised PCA and sparse PCA have been shown to have better empirical performance than the standard PCA. The functional PCA can analyze time-course gene expression data. Last, we raise the awareness of several critical but unsolved problems related to PCA. The goal of this article is to make bioinformatics researchers aware of the PCA technique and more importantly its most recent development, so that this simple yet effective dimension reduction technique can be better employed in bioinformatics data analysis.
Keywords: principal component analysis, dimension reduction, bioinformatics methodologies, gene expression
INTRODUCTION
In bioinformatics studies, high-throughput profiling techniques have been extensively adopted, leading to high-dimensional measurements. For example, with a typical Affymetrix chip, expressions of ∼40 000 probes can be profiled. In a genome-wide association (GWA) study, a million or more single nucleotide polymorphism (SNPs) can be profiled [1]. As the main focus of this article is on analysis methodologies, to avoid confusion of terminologies, we use microarray gene expression study as a representative example of high-throughput bioinformatics studies. We note that the methodologies discussed are also applicable to other (for example GWA, epigenetic and proteomic) studies. Gene expression data have ‘large d, small n’ characteristic, with the sample size much smaller than the number of genes. Many statistical techniques, for example regression analysis, are not directly applicable. The dimensionality of gene expressions needs to be reduced prior to regression and many other types of analyses. In addition, the nature of bioinformatics studies inevitably leads to data with excessive noises. In a traditional biomedical study, quite often researchers manually select covariates to be measured. Most or all of those covariates are expected to be associated with the response variables. In contrast, in gene profiling studies, only a small number of genes profiled are expected to be associated with the response variables and the majority of the genes are ‘noises’. It is desirable to remove noises in data analysis.
Available approaches that can reduce dimensionality can be classified as variable selection, dimension reduction and hybrid approaches [2]. Among them, hybrid approaches are relatively new and have not been extensively used in bioinformatics studies. Variable selection approaches search for a subset of genes to represent the effects of all genes. In contrast, dimension reduction approaches search for a small number of ‘metagenes’, which are often linear combinations of all genes. As discussed in Ref. [2] and others, performance of variable selection and dimension reduction approaches is data dependent, with no one dominating another. In this article, our goal is to provide in depth review of principal component analysis (PCA), which is a dimension reduction approach. We refer to other publications for generic discussions of variable selection and dimension reduction techniques.
PCA is one of the oldest dimension reduction approaches [3, 4]. It searches for linear combinations of the original measurements called principal components (PCs) that can effectively represent effects of the original measurements. PCs are orthogonal to each other and may have dimensionality much lower than that of the original measurements. Because of its computational simplicity and satisfactory statistical properties, PCA has been extensively used in multiple statistical areas. Most recently, it has been used in bioinformatics studies, particularly gene expression studies, to reduce the dimensionality of high-throughput measurements [5–7].
DEFINITIONS AND APPLICATIONS
Denote 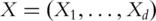 as the expressions of
as the expressions of 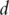 genes. Assume that the gene expressions have been properly normalized and
genes. Assume that the gene expressions have been properly normalized and 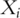 s have been centered to mean zero. To make genes more comparable, sometimes
s have been centered to mean zero. To make genes more comparable, sometimes 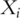 s are also scaled to have variance one. Details of the PCA techniques and its statistical framework have been described in [3, 4]. Denote
s are also scaled to have variance one. Details of the PCA techniques and its statistical framework have been described in [3, 4]. Denote 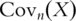 as the
as the  sample variance–covariance matrix computed based on
sample variance–covariance matrix computed based on 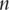 iid observations. In PCA, eigenvalues and eigenvectors of
iid observations. In PCA, eigenvalues and eigenvectors of 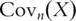 are computed. This can be achieved using standard singular value decomposition (SVD) techniques [8]. PCs are defined as the eigenvectors with non-zero eigenvalues and sorted by the magnitudes of corresponding eigenvalues, with the first PC having the largest eigenvalue. Denote
are computed. This can be achieved using standard singular value decomposition (SVD) techniques [8]. PCs are defined as the eigenvectors with non-zero eigenvalues and sorted by the magnitudes of corresponding eigenvalues, with the first PC having the largest eigenvalue. Denote 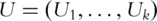 as the
as the 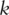 PCs, where
PCs, where 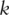 is the rank of
is the rank of 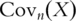 .
.
As PCA is performed on matrices of correlation coefficients, data should satisfy certain assumptions. We refer to chapter 6 of Ref. [9] for details. Particularly for theoretical validity, it is assumed that data is normally distributed. This assumption is intuitive considering that when the mean is not of interest, the normal distribution is fully specified by the variance structure. Gene expression data may or may not satisfy the normality assumption. In theory it is possible to transform gene expressions to achieve normality, although this is rarely done in practice. We note that PCA has been conducted with data obviously not having a normal distribution and shown to have satisfactory performance, although there is a lack of theoretical justification for such observation.
The PCs have the following main statistical properties: (i) 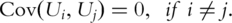 That is, different PCs are orthogonal to each other. In regression analysis, PCs can effectively solve the collinearity problem encountered by gene expressions; (ii)
That is, different PCs are orthogonal to each other. In regression analysis, PCs can effectively solve the collinearity problem encountered by gene expressions; (ii) 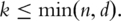 In bioinformatics data analysis, quite often
In bioinformatics data analysis, quite often 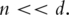 With this property, the dimensionality of PCs can be much lower than that of gene expressions. Thus, the PCs may not have the high-dimensionality problem encountered by gene expressions and have much lower computational cost; (iii) variation explained by PCs decreases, with the first PC explaining the most variation. Often the first few (say three to five) PCs can explain the majority of variation. Thus if the problem of interest is directly related to variation, it suffices to consider only the first few PCs; and (iv) any linear function of
With this property, the dimensionality of PCs can be much lower than that of gene expressions. Thus, the PCs may not have the high-dimensionality problem encountered by gene expressions and have much lower computational cost; (iii) variation explained by PCs decreases, with the first PC explaining the most variation. Often the first few (say three to five) PCs can explain the majority of variation. Thus if the problem of interest is directly related to variation, it suffices to consider only the first few PCs; and (iv) any linear function of  s can be written in terms of
s can be written in terms of 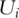 s. That is,
s. That is, 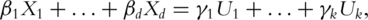 where
where  and
and  are coefficients. When focusing on the linear effects of gene expressions, using PCs are equivalent to using original gene expressions.
are coefficients. When focusing on the linear effects of gene expressions, using PCs are equivalent to using original gene expressions.
In gene expression analysis, PCs have been referred to as ‘metagenes’, ‘super genes’, ‘latent genes’ and others. Applications of PCA in gene expression analysis may include but are not limited to the following areas. (i) Exploratory analysis and data visualization [10]. With the extremely high dimensionality of gene expressions, it is impossible to graphically examine data. With PCA, we are able to project the  (which is usually very large) dimensional gene expressions onto a small number of (say two or three) PCs. We are then able to visualize gene expressions in a projected 2- or 3D space. We refer to Ref. [6] for data examples; (ii) clustering analysis. The first few PCs can usually capture most of the variation in gene expressions. In contrast, the rest of the PCs are often assumed to capture only the residual noises. As described in Ref. [11], we can first project gene expressions onto a small number of PCs and then use the PCs (as opposed to original gene expressions) for clustering genes or samples; (iii) regression analysis. In pharmacogenomic studies, quite often an important goal is to construct predictive models for disease outcomes such as prognosis or response to treatment. As the dimensionality of gene expressions is much larger than the sample size, straightforward regression analysis will result in saturated models and unreasonable estimates. As shown in Ref. [12] and references therein, it is possible to first conduct PCA and then use the first few PCs as covariates in regression analysis. With the low dimensionality of PCs, standard regression analysis techniques are directly applicable. Beyond the aforementioned areas, PCA has also been used in image processing and compression, immunology, molecular dynamics, small angle scattering and information retrieval [13].
(which is usually very large) dimensional gene expressions onto a small number of (say two or three) PCs. We are then able to visualize gene expressions in a projected 2- or 3D space. We refer to Ref. [6] for data examples; (ii) clustering analysis. The first few PCs can usually capture most of the variation in gene expressions. In contrast, the rest of the PCs are often assumed to capture only the residual noises. As described in Ref. [11], we can first project gene expressions onto a small number of PCs and then use the PCs (as opposed to original gene expressions) for clustering genes or samples; (iii) regression analysis. In pharmacogenomic studies, quite often an important goal is to construct predictive models for disease outcomes such as prognosis or response to treatment. As the dimensionality of gene expressions is much larger than the sample size, straightforward regression analysis will result in saturated models and unreasonable estimates. As shown in Ref. [12] and references therein, it is possible to first conduct PCA and then use the first few PCs as covariates in regression analysis. With the low dimensionality of PCs, standard regression analysis techniques are directly applicable. Beyond the aforementioned areas, PCA has also been used in image processing and compression, immunology, molecular dynamics, small angle scattering and information retrieval [13].
In studies such as [11], PCA is conducted with all genes measured. In addition, it is also possible to incorporate the hierarchical structure of genes in PCA-based analysis. For example, in Ref. [12] and others, the pathway structure is accounted for and PCA is conducted on genes within the same pathways. Here the PCs are used to represent the effects of pathways. In Ref. [14] and others, the network structure is accounted for and PCA is conducted on genes within the same network modules. Here PCs are used to represent the effects of modules of tightly connected genes. Following a similar strategy, PCA can be conducted for any pre-defined clusters of genes.
Many existing software packages can be used to conduct PCA. In fact, any software that can conduct SVD can be used for PCA. Examples of available PCA packages include: (i) R: the prcomp function, (ii) SAS: procedures PRINCOMP and FACTOR, (iii) SPSS: factor function (data reduction), (iv) MATLAB: princomp, (v) NIA array analysis tool (http://lgsun.grc.nia.nih.gov/ANOVA/) and others.
NON-STANDARD APPLICATIONS
The applications of PCA discussed in the above section are ‘standard’ in the sense that PCA is conducted straightforwardly with gene expression measurements. In this section, we review two recent studies of ‘non-standard’ applications of PCA. These studies do not change the way how PCA is conducted. However, in these studies, the PCA is no longer applied to the original gene expression measurements.
Accommodating interactions
In gene-based whole-genome analysis, because of the extremely high dimensionality, it is usually difficult to investigate interactions (particularly among all pairs of genes). In recent studies [12; manuscript under review], PCA has been used to study interactions.
For simplicity of notation, assume that there are only two pathways containing 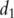 and
and 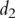 genes, respectively. Denote their gene expressions as
genes, respectively. Denote their gene expressions as  and
and 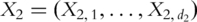 , respectively. Without loss of generality, consider regression analysis where the goal is to use functions of
, respectively. Without loss of generality, consider regression analysis where the goal is to use functions of 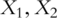 as covariates and predict response variables. In PCA-based regression analysis, we first conduct PCA on genes within the two pathways separately. Denote
as covariates and predict response variables. In PCA-based regression analysis, we first conduct PCA on genes within the two pathways separately. Denote 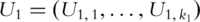 and
and 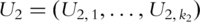 as the
as the 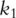 and
and 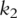 PCs for pathways 1 and 2, respectively.
PCs for pathways 1 and 2, respectively. 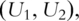 which have dimensionality much lower than that of
which have dimensionality much lower than that of 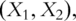 are used as covariates in downstream analysis. This approach has been adopted in studies such as Ref. [14].
are used as covariates in downstream analysis. This approach has been adopted in studies such as Ref. [14].
In recent studies, Ma and Kosorok [12], Ma et al. [manuscript under review] and others propose the following alternative ways of constructing PCA-based covariates:
(A1) Conduct PCA on genes within the two pathways separately. Denote
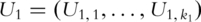 and
and 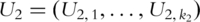 as the PCs for pathways 1 and 2, respectively.
as the PCs for pathways 1 and 2, respectively.  are used as covariates in downstream analysis. Here
are used as covariates in downstream analysis. Here 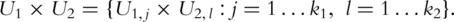
(A2) For
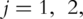 conduct PCA with
conduct PCA with 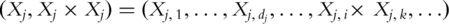 , which is the set composed of original gene expressions and their second-order interactions. With slight abuse of notation, still use
, which is the set composed of original gene expressions and their second-order interactions. With slight abuse of notation, still use 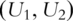 to denote the PCs.
to denote the PCs. 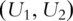 are used as covariates in downstream analysis.
are used as covariates in downstream analysis.
Using PCs (as opposed to original gene expressions) in regression analysis has been adopted in many studies. Using (A1) and (A2) as opposed to the simple PCA has only been proposed in most recent studies. With (A1), interactions between pathways are accommodated via the interactions among PCs from different pathways. This approach is feasible as the dimensionality of PCs is much smaller than that of original gene expressions. For example, in cancer prognosis studies [12], less than 10 PCs can be sufficient to represent pathways composed of hundreds of genes. With (A2), interactions among genes within the same pathways are accommodated. As the number of genes per pathway can be much smaller than the total number of genes, it is feasible to consider the set composed of original gene expressions and their interactions and conduct PCA.
Analysis of cancer microarray studies in [12; manuscript under review] suggest that it is computationally feasible to accommodate interactions using (A1), (A2) and their extensions. Incorporating interactions among genes has led to the identification of important pathways missed by using linear terms only. Incorporating interactions among pathways may significantly improve prediction performance measured using log-rank statistic and concordance index in prognosis studies. On the negative side, incorporating interactions using (A1) and (A2) also has drawbacks. Consider for example two pathways each with 10 PCs. When using the PCs as covariates in regression analysis, the total number of covariates is 20. For microarray studies with moderate to large sample sizes (say >100 as in many cancer microarray studies), standard regression analysis is directly applicable. However, with (A1) when the interactions are accounted for, the total number of covariates is 120 (20 first-order terms and 100 interaction terms). This may significantly increase computational cost. More importantly, additional regularization may have to be introduced in the estimation procedure. For example, in Ma et al. [manuscript under review], the thresholding regularization is used for estimation.
Following a similar strategy, it is possible to extend (A1) and (A2) in multiple ways. For example, it is possible to consider third- or higher order interactions. It is also possible to combine (A1) and (A2), first conduct PCA with genes (within the same pathways) and their interactions and then accommodate interactions among pathways. Such extensions have not been applied in practical data analysis, with concerns on high computational cost and lack of interpretability. We note that, although the above discussions have been in the context of gene pathways, they are applicable to whole-genome analysis and gene network-based analysis with only minor modifications.
Conducting PCA with estimating equations
When using PCA in regression analysis, the approaches described above and most published studies conduct PCA with gene expressions and/or their functions. There are also studies that take advantage of the special forms of estimating equations under certain data and model setup and conduct PCA with estimating equations [15, 16]. In what follows, we present an example of analyzing prognosis data under the additive risk model [16].
Denote 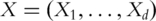 as the expressions of
as the expressions of 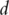 genes. Denote
genes. Denote 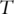 as the survival time, which can be progression-free, overall or other types of survival. Denote
as the survival time, which can be progression-free, overall or other types of survival. Denote 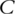 as the censoring time. Under right censoring, one observation consists of
as the censoring time. Under right censoring, one observation consists of 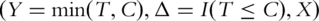 where
where 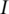 is the indicator function. Under the additive risk model, the hazard function is
is the indicator function. Under the additive risk model, the hazard function is 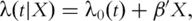 where
where 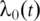 is the unknown baseline hazard function,
is the unknown baseline hazard function,  is the regression coefficient and
is the regression coefficient and 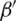 is the transpose of
is the transpose of 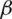 . Assume
. Assume 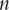 iid observations
iid observations 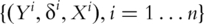 .
.
Define the cumulative baseline hazard function 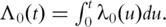 For the i-th subject, denote
For the i-th subject, denote 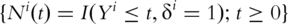 and
and  as the observed event process and at-risk process, respectively.
as the observed event process and at-risk process, respectively. 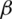 can be estimated by solving.
can be estimated by solving.
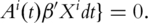 Here
Here 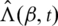 is the estimate of
is the estimate of 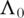 satisfying
satisfying 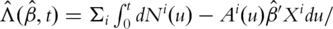
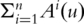 . The resulting estimate of
. The resulting estimate of 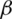 satisfies the estimating equation
satisfies the estimating equation 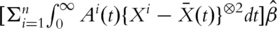
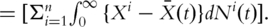 Here
Here 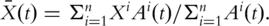 Note that the term
Note that the term 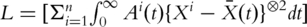 is symmetric, semi-positive definite and mimics a variance–covariance matrix. Following a similar strategy as with PCA and conducting SVD, there exist matrices
is symmetric, semi-positive definite and mimics a variance–covariance matrix. Following a similar strategy as with PCA and conducting SVD, there exist matrices 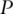 and
and 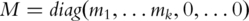 such that
such that  and
and 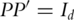 (the
(the  identity matrix). Denote
identity matrix). Denote 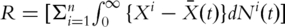 . Define the general inverse of
. Define the general inverse of 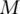 as
as 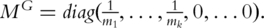 Then from the estimating equation, we have
Then from the estimating equation, we have 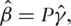 where
where 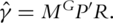 Numerical study suggests that with high dimensional gene expression data, some components of
Numerical study suggests that with high dimensional gene expression data, some components of 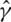 can have estimated variances several orders larger than the other components. With PCA, focusing only on the PCs with large eigenvalues may improve stability of estimates. Motivated by such considerations, PCA-based estimates can be defined by removing PCs with small eigenvalues in the decomposition of L. Specifically, denote
can have estimated variances several orders larger than the other components. With PCA, focusing only on the PCs with large eigenvalues may improve stability of estimates. Motivated by such considerations, PCA-based estimates can be defined by removing PCs with small eigenvalues in the decomposition of L. Specifically, denote 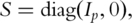 where
where 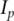 is the p-dimensional identity matrix with
is the p-dimensional identity matrix with  Define the PCA-based estimate as
Define the PCA-based estimate as  and
and 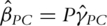 . Analysis of cancer prognosis studies suggests that this estimating equation-based PCA approach uses only a small number of PCs. The estimates are more stable and the prediction performance is significantly better than alternatives particularly including step-wise variable selection approaches.
. Analysis of cancer prognosis studies suggests that this estimating equation-based PCA approach uses only a small number of PCs. The estimates are more stable and the prediction performance is significantly better than alternatives particularly including step-wise variable selection approaches.
This approach and the one in Ref. [15] have been motivated by the special forms of the estimating equations. After computing the estimating equations, they can be realized using standard PCA software. Under other data and model settings, estimating equations with similar properties may or may not exist. Thus, the estimating equation-based PCA needs to be conducted on a case-by-case basis. Another possible drawback of this approach is the lack of interpretability. With standard PCA, for example, the first PC is the linear combination of genes that explains the most variation. However, with the estimating equation-based PCA, the matrix L is not a variance–covariance matrix. Thus, the PCs do not have simple interpretations.
EXTENSIONS OF PCA TECHNIQUES
In recent studies, several PCA-based approaches have been proposed. Among the approaches reviewed below, the supervised PCA and sparse PCA are designed to analyze the same type of data as the standard PCA. However, they have the sparity property not shared by the standard PCA and significantly better numerical performance. The functional PCA has been developed to analyze time-course data, which cannot be analyzed using the standard PCA.
Supervised PCA
A unique feature of whole-genome studies is that most genes profiled are not expected to be associated with the response variables. Statistically speaking, this corresponds to sparse models with only a few genes having non-zero regression coefficients. When using PCs (as opposed to original gene expressions) as covariates in regression analysis, as the PCs are linear combinations of all genes, the models are inevitably dense with all genes having non-zero regression coefficients. Supervised PCA has been proposed to solve this problem [17, 18].
Consider a simple example where the goal is to construct a prediction model for a continuous response variable 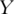 using
using  the expressions of d genes. For simplicity, assume the linear regression model. The supervised PCA consists of two main steps:
the expressions of d genes. For simplicity, assume the linear regression model. The supervised PCA consists of two main steps:
(S1) For
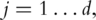 fit the linear regression model
fit the linear regression model 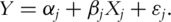 This is the marginal model consisting of the intercept
This is the marginal model consisting of the intercept 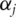 and only one gene. Compute a ranking statistic for gene
and only one gene. Compute a ranking statistic for gene  Examples of the ranking statistic include the absolute magnitude of
Examples of the ranking statistic include the absolute magnitude of 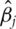 (the estimate of
(the estimate of 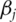 ), its significance level, the value of the likelihood and others. Rank the d genes based on their ranking statistics;
), its significance level, the value of the likelihood and others. Rank the d genes based on their ranking statistics;(S2) Select the top
 genes and conduct downstream PCA using only those
genes and conduct downstream PCA using only those 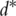 genes.
genes.
We describe the supervised PCA for continuous response variables. Extension to other types of response variables can be easily carried out. With categorical and survival response variables, receiver operating characteristic (ROC)-based statistics have been used for ranking and can be more robust than parametric regression-based statistics [19]. Compared with the standard PCA, the supervised PCA has an extra screening step (S1). As discussed above, with standard PCA, the PCs are constructed in an unsupervised manner. Thus, all genes, including those associated with response variables as well as noises, are included in the PCs. With the screening step, genes not associated with the response variables can be effectively removed and only genes likely to be associated with the response variables are used in downstream PCA-based analysis. A byproduct is the reduced computational cost. The screening step only involves simple models with a single covariate and can be conducted in a parallel manner. With a small number of selected genes, (S2) has lower computational cost than PCA with all genes. Numerical studies in Ref. [17, 20, 21] suggest that the supervised PCA has better prediction performance than quite a few alternatives, particularly including the standard PCA. In addition, with only a small number of genes, the PCs may have better interpretability. Studies such as Ref. [21] provide software for realizing the supervised PCA.
Sparse PCA
The sparse PCA has been described in Ref. [22, 23] and motivated by similar considerations as with the supervised PCA. Consider, for example, the first PC 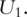 It is the linear combination of all d genes. That is,
It is the linear combination of all d genes. That is, 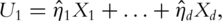 where
where 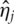 s are non-zero and refereed to as ‘loadings’. With standard PCA, all loadings are non-zero and the PCs and downstream analysis results may be in conflict with the sparse nature of models with gene expression data and lack interpretability.
s are non-zero and refereed to as ‘loadings’. With standard PCA, all loadings are non-zero and the PCs and downstream analysis results may be in conflict with the sparse nature of models with gene expression data and lack interpretability.
It is easy to note that 
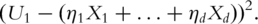 That is, the loadings can be ‘recovered’ from linear regression analysis. The essence of the sparse PCA is to use a small number of genes to approximate the PCs, so that the loadings of PCs are sparse with coefficients corresponding to most genes equal to zero. The sparse PCA consists of the following steps:
That is, the loadings can be ‘recovered’ from linear regression analysis. The essence of the sparse PCA is to use a small number of genes to approximate the PCs, so that the loadings of PCs are sparse with coefficients corresponding to most genes equal to zero. The sparse PCA consists of the following steps:
(S1) Construct the PCs as in standard PCA. Denote
 as the PCs;
as the PCs;(S2) For
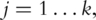 compute the sparse loadings. Particularly, a penalized estimation approach has been proposed, which computes the loadings as
compute the sparse loadings. Particularly, a penalized estimation approach has been proposed, which computes the loadings as 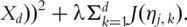 Here
Here 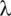 is the data-dependent tuning parameter. It balances sparsity and goodness-of-fit and can be obtained using for example cross validation.
is the data-dependent tuning parameter. It balances sparsity and goodness-of-fit and can be obtained using for example cross validation. 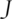 is the penalty function. Using penalization to generate sparse models has been thoroughly discussed in Ref. [2] and reference therein. The simplest penalty is perhaps the Lasso penalty with
is the penalty function. Using penalization to generate sparse models has been thoroughly discussed in Ref. [2] and reference therein. The simplest penalty is perhaps the Lasso penalty with  . The sparse PCs are then defined as
. The sparse PCs are then defined as 
(S3) Use the sparse PCs in downstream analysis.
When the penalty has a Lasso-type formulation, the sparse PCA can be realized using the R package elasticnet (http://cran.r-project.org/web/packages/elasticnet/). With other penalties, to the best of our knowledge, there is no available software yet. Numerical studies in Ref. [22] suggest that compared with standard PCA, the sparse PCA has significantly better interpretability by introducing sparsity. It may also have better prediction performance by removing noises. Compared with the supervised PCA, the sparse PCA may have higher computational cost, as the penalized estimation can be computationally expensive. To the best of our knowledge, there is still no study comprehensively comparing the performance of supervised PCA versus sparse PCA.
Functional PCA
Approaches discussed above are all for the analysis of snapshot gene expression data. That is, gene expressions are only measured at one time point. Another type of studies measure gene expressions consecutively at multiple time points. The data so obtained has been referred to as ‘time-course gene expression data’ [24]. For a specific gene, its expression can no longer be described using a single number 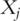 . Rather, a function of time
. Rather, a function of time 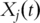 is needed, where
is needed, where  denotes time.
denotes time.
Consider for example the first PC. With snapshot gene expression data, computation of the first PC is equivalent to solving 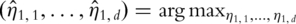
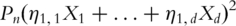 subject to
subject to 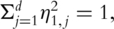 where
where 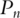 is the empirical measure based on n iid observations. It can be shown that this maximization formulation is equivalent to SVD with the sample variance–covariance matrix. In contrast, with time-course gene expression data, computation of the first PC is equivalent to solving
is the empirical measure based on n iid observations. It can be shown that this maximization formulation is equivalent to SVD with the sample variance–covariance matrix. In contrast, with time-course gene expression data, computation of the first PC is equivalent to solving 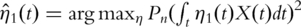 subject to
subject to 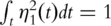 , where
, where 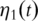 is a function of time. Loosely speaking, all simple summation with the simple PCA needs to be replaced with integration over time. The statistical basis of the functional PCA has been laid in Ref. [25]. In practice,
is a function of time. Loosely speaking, all simple summation with the simple PCA needs to be replaced with integration over time. The statistical basis of the functional PCA has been laid in Ref. [25]. In practice, 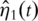 can be estimated using a sieve approach with, for example, spline basis functions [24].
can be estimated using a sieve approach with, for example, spline basis functions [24].
Functional PCA has been used in Ref. [24, 26] for clustering time-course gene expression data. The time-dependent nature of gene expressions inevitably brings along complications to estimation and inference. However, except for the difference in computing the PCA, using functional PCA and simple PCA share the same spirit. Numerical studies in the aforementioned articles show that the performance of functional PCA (with time-course data) is similar to that of simple PCA (with snapshot data).
DISCUSSION
How to interpret PCA results
When there are only a small number of covariates [4], the PCs may have simple interpretations. However, with gene expression data, the PCs are linear combinations of thousands of genes, which make them difficult to interpret. In certain studies [16], researchers examine a few genes with the highest loadings. In our limited numerical study, we find that usually the loadings are ‘continuously distributed’, with no obvious jumps. Thus, it is usually difficult to tease out a few important genes by investing loadings. A few studies seem to suggest that the PCs may correspond to latent causes (e.g. of diseases). However, our limited literature review suggests that there is no study that has satisfactorily interpreted the PCs. PCs from supervised and sparse PCA may be constructed using only a small number of genes. Thus, they may have better interpretability. As discussed in Ref. [2] and references therein, the lack of interpretability is shared by most if not all dimension reduction methods, whereas variable selection and hybrid methods may enjoy better interpretability.
How to choose the number of PCs
The PCs are constructed with the goal to explain variation. Usually, the first few PCs can explain the majority of variation. However, it may take a large number of PCs to explain all the variation. It is not desirable to keep all PCs. In Ref. [3], there are some common rules of thumb for choosing how many PCs to retain. Examples include keeping enough PCs such that the cumulative variance explained by the PCs is >50–70%. An alternative is to keep all PCs with eigenvalues > 1. In many bioinformatics studies [14, 20, 27], it is suggested that the first one or two PCs may be sufficient. However, such a statement is based on empirical observations from analysis of a small number of datasets and lacks solid statistical justification. In clustering analysis, there is in fact a result showing that the first few PCs may not contain cluster information. Chang [28] considers an example with a mixture of two multivariate normal distributions with different means but with an identical within-cluster covariance matrix. It is shown that the first few PCs may contain less cluster structure information than other PCs. Ma and Kosorok [12] and Ma et al. (manuscript under review) conduct numerical studies and show that PCs other than the first one or two may contribute significant predictive power in regression analysis. Such a finding is not surprising, considering that the PCs are constructed in an unsupervised manner. There is no biological or statistical reason why the latent variables (PCs) that explain variation in covariates are predictive for response variables.
Our extensive literature review suggests that choosing the proper number of PCs remains an open problem. We agree with the statement [12] which says ‘in practical data analysis, we suggest that researchers explore different number of PCs and select the proper number based on, for example, biological implications and predictive power… (in regression analysis)’.
Theoretical justifications
For the theoretical validity of PCA, the PCs need to be consistently estimated. In ‘classic’ statistical analysis with fixed 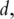 the consistency is usually trivial [4]. In bioinformatics studies, data has the ‘large
the consistency is usually trivial [4]. In bioinformatics studies, data has the ‘large 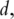 small
small 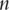 ’ structure. It has been proved that, under mild assumptions, if
’ structure. It has been proved that, under mild assumptions, if 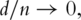 then the variance–covariance matrix and the first fixed number of PCs can be consistently estimated. Such a consistency result, although insightful, is not quite useful for bioinformatics studies. In recent studies, it has been shown that if certain assumptions on the variance matrix hold, then less strict requirements on
then the variance–covariance matrix and the first fixed number of PCs can be consistently estimated. Such a consistency result, although insightful, is not quite useful for bioinformatics studies. In recent studies, it has been shown that if certain assumptions on the variance matrix hold, then less strict requirements on 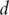 can be assumed. For example [29], the ‘bandable’ assumption is made and it is proved that if
can be assumed. For example [29], the ‘bandable’ assumption is made and it is proved that if 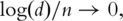 estimation of PCs is consistent. Note that this result allows the number of genes to be much larger than the sample size. The bandable assumption postulates that the expression of a gene is only correlated with those of a small number of genes and not or only weakly correlated with those of most genes. Such an assumption is perhaps reasonable considering that the expression of a gene tends to be correlated only with those of genes with similar biological functions. In practical data analysis, we suggest first computing the variance–covariance matrix and then count how many elements are above a certain cutoff, say 0.3. If there are only a small percentage of the elements are above the cutoff, then the bandable assumption is perhaps reasonable and the estimates of PCs are expected to be consistent even with
estimation of PCs is consistent. Note that this result allows the number of genes to be much larger than the sample size. The bandable assumption postulates that the expression of a gene is only correlated with those of a small number of genes and not or only weakly correlated with those of most genes. Such an assumption is perhaps reasonable considering that the expression of a gene tends to be correlated only with those of genes with similar biological functions. In practical data analysis, we suggest first computing the variance–covariance matrix and then count how many elements are above a certain cutoff, say 0.3. If there are only a small percentage of the elements are above the cutoff, then the bandable assumption is perhaps reasonable and the estimates of PCs are expected to be consistent even with 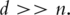
As discussed above, PCA has also been used in pathway and network based analysis. In such analysis, the consistency of estimated PCs within each pathway or network module follows from the argument above. In addition, it is required that the consistency over all pathways/modules is uniform. This requirement may put further constraint on the sample size and number of genes [30].
CONCLUSION
In bioinformatics data analysis, a unique challenge arises from the high dimensionality of measurements. PCA is a classic dimension reduction approach. Because of its computational simplicity, it has been extensively used in bioinformatics studies and shown to have satisfactory performance. In recent studies, there have been modifications and extensions of the PCA. Several examples are reviewed in this article. Those approaches advance from PCA and have been shown to have better prediction performance and interpretability in data analysis. Despite its successes, as pointed out in the above section, PCA also has certain limitations. There are a few open questions, including for example choosing the proper number of PCs, that remain to be solved. Examining published studies suggest that the performance of PCA (and in fact all dimension reduction and variable selection approaches) is data dependent. There is no guarantee that PCA can always outperform alternatives. However, because of its extremely low computational cost, PCA can be a preferred dimension reduction tool in many studies.
In general, PCA is only powerful when the biomedical question is related to the highest variation in data. When such a condition is not satisfied, PCA may need to be replaced by alternatives. A closely related but significantly different approach is ICA (independent component analysis). With ICA, an independence condition is optimized. Different independent components (ICs) represent different non-overlapping information. ICA can give more meaningful components than optimization of only the variance. There are also other alternatives to PCA. As our main goal is an in depth review of PCA, we defer comprehensive discussions of other dimension reduction methods to future studies.
SUPPLEMENTARY DATA
Supplementary data are available online at http://bib.oxfordjournals.org/.
Key Points.
In bioinformatics data analysis, PCA has been extensively used for dimension reduction. It has an intuitive interpretation, low computational cost and satisfactory empirical performance.
In recent studies, PCA has been used to accommodate interactions. In addition, it has been conducted on estimating equations as opposed to the original covariates.
Extensions including the supervised PCA and sparse PCA have better sparity property (and hence more lucid interpretability) and superior numerical performance. The functional PCA can analyze time-course functional data.
There are several open problems related to PCA that need to be carefully addressed in future studies.
FUNDING
This study was supported in part by award LM009754 and CA142774 from National Institute of Health, DMS0904181 from National Science Foundation and Research of Longitudinal Data Analysis Methodology and Its Application (2009JJD910002) from Key Research Institute of Humanities and Social Sciences Program (China).
Acknowledgements
The authors would like to thank the associate editor and three referees for careful review and insightful comments.
Biographies
Shuangge Ma obtained his PhD in Statistics from University of Wisconsin, Madison. He is an Assistant Professor in School of Public Health, Yale University.
Ying Dai is PhD candidate in Department of Planning and Statistics, School of Economics, Xiamen University, P.R. China.
References
- 1.Wong S. The Practical Bioinformatician. World Scientific Publishing Company; 2004. [Google Scholar]
- 2.Ma S, Huang J. Penalized feature selection and classification in bioinformatics. Briefings in Bioinformatics. 2008;9:392–403. doi: 10.1093/bib/bbn027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jolliffe IT. Principal Component Analysis. New York: Springer; 1986. [Google Scholar]
- 4.Johnson RA, Wichern DW. Applied Multivariate Statistical Analysis. Upper Saddle River, NJ: Prentice Hall; 2001. [Google Scholar]
- 5.Knudsen S. Cancer Diagnostics with DNA Microarrays. Hoboken, NJ: John Wiley and Sons; 2006. [Google Scholar]
- 6.McLachlan GJ, Do KA, Ambroise C. Analyzing Microarray Gene Expression Data. Wiley-Interscience; 2004. [Google Scholar]
- 7.Sharov AA, Dudekula DB, Ko MSH. A web-based tool for principal component and significance analysis of microarray data. Bioinformatics. 2005;21:2548–9. doi: 10.1093/bioinformatics/bti343. [DOI] [PubMed] [Google Scholar]
- 8.Golub GH, Van Loan CF. Matrix Computations. Baltimore: John Hopkins University Press; 1996. [Google Scholar]
- 9.Hatcher L, Stepanski EJ. A Step-by-step Approach to Using the SAS System for Univariate and Multivariate Statistics. Books by Users Press; 1994. [Google Scholar]
- 10.Hibbs MA, Dirksen NC, Li K, et al. Visualization methods for statistical analysis of microarray clusters. BMC Bioinformatics. 2005;6:115. doi: 10.1186/1471-2105-6-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yeung KY, Ruzzo WL. Principal component analysis for clustering gene expression data. Bioinformatics. 2001;17:763–74. doi: 10.1093/bioinformatics/17.9.763. [DOI] [PubMed] [Google Scholar]
- 12.Ma S, Kosorok MR. Identification of differential gene pathways with principal component analysis. Bioinformatics. 2009;25:882–9. doi: 10.1093/bioinformatics/btp085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wall ME, Rechtsteiner A, Rocha LM. Singular value decomposition and principal component analysis. In: Berrar DP, Dubitzky W, Granzow M, editors. A Practical Approach to Microarray Data Analysis. Norwell, MA: Kluwer; 2003. [Google Scholar]
- 14.Langfelder P, Horvath S. Eigengene networks for studying the relationships between co-expression modules. BMC Syst Biol. 2007;1:54. doi: 10.1186/1752-0509-1-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ma S. Principal component analysis in linear regression survival model with microarray data. J Data Sci. 2007;5:183–98. [Google Scholar]
- 16.Ma S, Kosorok MR, Fine JP. Additive risk models for survival data with high dimensional covariates. Biometrics. 2006;62:202–10. doi: 10.1111/j.1541-0420.2005.00405.x. [DOI] [PubMed] [Google Scholar]
- 17.Bair E, Tishirani R. Semi-supervised methods to predict patient survival from gene expression data. PLoS Biol. 2004;2:511–22. doi: 10.1371/journal.pbio.0020108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bair E, Hastie T, Dabashis P, et al. Prediction by supervised principal components. J Acous Soc Am. 2006;102:119–37. [Google Scholar]
- 19.Li J, Fine J. Weighted area under the receiver operating characteristic curve and its application to gene selection. JRSSC. 2010;59:673–92. doi: 10.1111/j.1467-9876.2010.00713.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen X, Wang L, Hu B, et al. Pathway-based analysis for genome-wide association studies using supervised principal components. Genetic Epidemiol. 2010;34:716–24. doi: 10.1002/gepi.20532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen X, Wang L, Smith JD, et al. Supervised principal component analysis for gene set enrichment of microarray data with continuous or survival outcomes. Bioinformatics. 2008;24:2474–81. doi: 10.1093/bioinformatics/btn458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zou H, Hastie T, Tibshirani R. Sparse principal component analysis. J Comp Graph Stat. 2006;15:262–86. [Google Scholar]
- 23.D’Aspremont A, Ghaou LEL, Jordan MI, et al. In: Proceedings of the Neural Information Processing Systems (NIPS) Neural Information Processing Systems Foundation; 2004. A direct formulation for sparse PCA using semidefinite programming. [Google Scholar]
- 24.Luan Y, Li H. Clustering of time-course gene expression data using a mixed-effects model with B-splines. Bioinformatics. 2003;19:474–82. doi: 10.1093/bioinformatics/btg014. [DOI] [PubMed] [Google Scholar]
- 25.Ramsay JO, Silverman BW. Functional Data Analysis – Methods and Case Studies. New York: Springer; 2002. [Google Scholar]
- 26.Song JJ, Lee HJ, Morris JS, et al. Clustering of time-course gene expression data using functional data analysis. Comput Biol Chem. 2007;31:265–74. doi: 10.1016/j.compbiolchem.2007.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saris CG, Horvath S, van Vught PW, et al. Weighted gene co-expression network analysis of the peripheral blood from amyotrophic lateral sclerosis patients. BMC Genomics. 2009;10:405. doi: 10.1186/1471-2164-10-405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chang WC. On using principal components before separating a mixture of two multivariate normal distributions. Appl Stat. 1983;32:267–75. [Google Scholar]
- 29.Bickel PJ, Levina E. Covariance regularization by thresholding. Ann Stat. 2008;36:2577–604. [Google Scholar]
- 30.Kosorok MR, Ma S. Marginal asymptotics for “large p, small n” paradigm: with applications to microarray data. Ann Stat. 2007;35:1456–86. [Google Scholar]