

Abstract
Fetal alcohol spectrum disorder is estimated to affect 1% of live births. The similarities between children with fetal alcohol syndrome and those with mutations in the gene encoding L1 cell adhesion molecule (L1) implicates L1 as a target of ethanol developmental neurotoxicity. Ethanol specifically inhibits the neurite outgrowth promoting function of L1 at pharmacologic concentrations. Emerging evidence shows that localized disruption of the lipid rafts reduces L1-mediated neurite outgrowth. We hypothesize that ethanol impairment of the association of L1 with lipid rafts is a mechanism underlying ethanol's inhibition of L1-mediated neurite outgrowth. In this study, we examine the effects of ethanol on the association of L1 and lipid rafts. We show that, in vitro, L1 but not N-cadherin shifts into lipid rafts following treatment with 25 mM ethanol. The ethanol concentrations causing this effect are similar to those inhibiting L1-mediated neurite outgrowth. Increasing chain length of the alcohol demonstrates the same cutoff as that previously shown for inhibition of L1-L1 binding. In addition, in cerebellar granule neurons (CGN) in which lipid rafts are disrupted with methyl-beta-cyclodextrin (MBCD), the rate of L1-mediated neurite outgrowth on L1-Fc is reduced to background rate and that this background rate is not ethanol sensitive. These data indicate that ethanol may inhibit L1-mediated neurite outgrowth by retarding L1 trafficking through a lipid raft compartment.
The use of ethanol during pregnancy can be detrimental to the offspring, the most severe result being the constellation of developmental defects and abnormalities commonly referred to as fetal alcohol syndrome (FAS). In the central nervous system, FAS is hallmarked by a wide range of defects in brain morphology, including hypoplasia of the corpus callosum and optic nerves, the formation of neuronal heterotopias, microcephaly, disorders in cortical lamination, and hypoplasia of the cerebellar vermis. These deficits can result in disorders such as motor dysfunction, hyperactivity, increased susceptibility to seizures, and deficits in learning and memory (Stratton, Howe, and Battaglia, 1996). The resemblance of FAS to the spectrum of disorders caused by mutations in L1 cell adhesion molecule (L1) has led investigators to explore the possible role of L1 in FAS (Charness, Safran, and Perides, 1994). L1 is a member of the immunoglobulin (Ig) superfamily implicated in a variety of processes in neurohistogenesis, including neurite elongation, axon fasciculation, and migration of neuronal precursors. It is a member of a subgroup of cell adhesion molecules which are related by structure and sequence. Each consists of six Ig-like domains, five fibronectin type III (FNIII)-like domains, and a highly conserved cytoplasmic tail (Kamiguchi, 2003; Winckler and Yap, 2011). L1 mediates cell-cell adhesion via a homophilic binding mechanism. In vitro L1 presented as a culture substrate stimulates neurite outgrowth by binding homophilically to L1 expressed on neurons. The neurite outgrowth of neurons plated on L1 is inhibited by ethanol (Bearer, Swick, O'Riordan, and Cheng, 1999; Watanabe, Yamazaki, Miyazaki, Arikawa, Itoh, Sasaki, Maehama, Frohman, and Kanaho, 2004). We have previously shown that L1 does not signal through the fibroblast growth factor receptor but activates both c-Src (pp60src) and ERK1/2 (Tang, He, O'Riordan, Farkas, Buck, Lemmon, and Bearer, 2006). In addition, ethanol inhibits downstream signaling of L1 including activation of c-Src and ERK1/2, phosphorylation of L1 tyrosines, and the dephosphorylation of tyrosine 1176 in the cytoplasmic domain of L1 (Tang, He, O'Riordan, Farkas, Buck, Lemmon, and Bearer, 2006; Yeaney, He, Tang, Malouf, O'Riordan, Lemmon, and Bearer, 2009). Inhibition of c-Src alone inhibits all of these downstream events, suggesting that ethanol acts by preventing L1 activation of c-Src (Yeaney, He, Tang, Malouf, O'Riordan, Lemmon, and Bearer, 2009).
Recently, L1 and a similar family member, neurofascin, are identified in lipid rafts in the peripheral (P) domain of the growth cone (Nakai and Kamiguchi, 2002; Ren and Bennett, 1998). Lipid rafts are microdomains of the plasma membrane, composed of sphingolipids and cholesterol in the outer exoplasmic leaflet, connected to phospholipids and cholesterol in the inner cytoplasmic leaflet of the lipid bilayer. These microdomains are fluidic but more ordered and tightly packed than the surrounding bilayer (Simons and Ehehalt, 2002; Simons and Gerl, 2010). Current studies suggest that formation of lipid rafts is dynamic (Owen, Gaus, Magee, and Cebecauer, 2010). L1 can be differentially associated with lipid rafts in different stages of development: L1 association with lipid rafts in cerebellum is only detectable between P8 and P28, but not detectable at P56 (Nakai and Kamiguchi, 2002). The molecular basis for the regulation of L1 association with lipid rafts is currently unknown. Targeted disruption of the lipid raft reduces neurite outgrowth on both L1 and N-cadherin, but not laminin (Nakai and Kamiguchi, 2002). Many proteins known to have substantial ethanol toxicity, such as the GABAA receptor (Mihic, 1999), the NMDA receptor (Mihic, 1999), the alpha-amino-3-hydroxy-5-methyl-4 isoxazole proprionic acid receptor (AMPA-R) (Mameli, Zamudio, Carta, and Valenzuela, 2005), the receptor for transforming growth factor beta (Luo and Miller, 1999) and heterotrimeric G proteins (Zhao, Marrero, Song, Oliver, Chin, Simon, Schurr, Zhang, Thoppil, Lee, Nelson, and Kolls, 2003) have all recently been shown to be associated with lipid rafts. We hypothesize that a major mechanism of ethanol toxicity is through disruption of protein-lipid raft interactions.
In the present study, we investigate the redistribution of L1, N-cadherin, beta-tubulin, caveolin-1, c-Src as well as GABAA receptors in lipid rafts of cerebellar granule neurons (CGN) following ethanol exposure in vitro, and ethanol effects on L1-mediated neurite outgrowth in CGN in which lipid rafts have been disrupted. We present evidence that ethanol at pharmacological concentrations may retard L1 trafficking through lipid rafts, identifying L1-lipid raft interactions as a novel target for ethanol inhibition of neurite outgrowth.
Materials and Methods
Antibodies
HRP-conjugated cholera toxin B subunits (CTxB), mouse monoclonal anti-beta III tubulin are obtained from Sigma. Mouse monoclonal anti-transferrin receptor antibody is from Zymed laboratories. Goat polyclonal anti-NCAM L1, rabbit polyclonal anti-caveolin-1 and mouse monoclonal anti-c-Src are from Santa Cruz Biotechnology. Mouse monoclonal anti-N-cadherin antibody is from BD Transduction Laboratories. Mouse monoclonal anti-beta III tubulin is from Invitrogen, and mouse monoclonal anti-GABAA receptor beta 2 subunit antibody is a gift from Dr. Ruth Siegel, Department of Pharmacology, Case Western Reserve University. Alexa Fluor 488 conjugated CTxB and Alexa Fluor 633 conjugated donkey anti-goat IgG are from Molecular Probes. All secondary antibodies for western blot analysis are from Jackson ImmunoResearch.
Preparation of CGN cells
All of the procedures involving animals that are used in the current studies are approved by the University of Maryland School of Medicine Institutional Animal Care and Use Committee and conform to NIH guidelines. Rat CGN are prepared from Sprague-Dawley rat pups (Zivic-Miller) on postnatal day 6 as previously described (Bearer, Swick, O'Riordan, and Cheng, 1999). Viability of these dissociated cells is assessed with trypan blue and is >90%. CGN are plated on prepared dishes, or coverslips in DMEM with 10% FBS. For neurite outgrowth experiments, the media is changed to a serum free defined media at the time of addition of ethanol. Serum free defined media consists of Neurobasal Media (Gibco) with the following additions: 2% B27 supplement (Gibco), 20 mM L-glutamine, 6 g/l glucose, 20 mM HEPES, pH 7.2, penicillin/streptomycin. All the cultures are maintained in a humid atmosphere of 90% air, 10% CO2 at 37°C.
Lipid raft isolation
CGN are cultured in a 150 mm dish grown for 40 h. Three hours prior to harvest, the cells are serum starved. For CGN treated with ethanol, the ethanol is added 1 h before harvest. Prior to harvest, all equipment is cooled to 4°C. Cultures are taken to the cold room where media is removed, and cells are washed with cold Hank's balanced salt solution. The cells are scraped, and lysed for 30 min on ice in 4 ml Tris-buffered saline containing 0.5% Triton X100, 10 mM Na vanadate, 2 μM aprotinin, 100 μM PMSF, 1 μM leupeptin, 10 μg/mL turkey trypsin inhibitor, 1μg/mL pepstatin A, 100 pM cypermethein, phosphatase inhibitor cocktail I and phosphatase inhibitor cocktail II. The lysates are centrifuged at 13,000 × g for 30 min at 4°C. After centrifugation, 4 ml of the supernatant is overlaid on a sucrose density gradient with 40%, 32% and 5% layers. The gradient is centrifuged at 180,000 × g, for 24 h at 4°C. Sequential 1 ml fractions are drawn off the top of the gradient. An aliquot from each fraction is analyzed by western blot analysis for both transferrin receptor and GM1 ganglioside. All GM1 ganglioside containing fractions are combined (lipid raft fractions). All remaining fractions are combined (non-lipid raft fractions). Proteins from each pool are precipitated using chloroform/methanol and resuspended in sample buffer for western blot.
SDS-PAGE and immunoblotting analysis
Cell lysates from each sample are resolved by SDS-PAGE (4 to 15% Tris gradient gel) then transferred to a PVDF membrane. The membrane is incubated with washing buffer (20 mM Tris-buffered saline and 0.1% Tween 20) containing 5% nonfat dry milk (Bio-Rad) for 1 h to block nonspecific protein binding. Primary antibodies are diluted in washing buffer containing 5% nonfat dry milk and are applied to the PVDF membrane at 4°C overnight. After being washed, the blots are incubated with the appropriate horseradish peroxidase-conjugated secondary antibodies (diluted 1:1,000 or 1:2,000 in washing buffer with 5% milk) for 1 h at room temperature. Immunoreactive bands are visualized with the enhanced chemiluminescence system (Amersham Biosciences). Where indicated, blots are stripped to be reprobed with additional primary antibodies. Densitometric quantitation is carried out with the software Kodak ID.
Immunofluorescence and confocal microscopy
CGN plated on poly L-lysine (PLL) coated coverslips are rinsed in phosphate buffered saline (PBS) three times. The cells are fixed in 4% paraformaldehyde for 30 min at room temperature, followed by 3 more washes of PBS. Permeabilization/blocking solutions (3% BSA/0.2% TX-100/PBS) are added to each coverslip. Coverslips are incubated for 1 h at 37°C or overnight at 4°C. Primary antibody is added, and the cells are incubated for 2 h at 37°C and washed. Secondary antibody is added and incubated for 1 h at 37°C. Cells are washed extensively. Confocal microscopy is performed with a Leica TCS SP2 confocal microscope with 63× objective lens using laser excitation at 488 and 633 nm. The colocalizations between two emissions channels are measured using the software Image J. Pixel intensity values greater than threshold level are defined as intense signals. A pixel with intense signals in both wavelength channels is regarded as indicating colocalization.
Preparation of L1-Fc
L1-Fc containing plasmid is prepared as previously described (Tang, He, O'Riordan, Farkas, Buck, Lemmon, and Bearer, 2006). Briefly, the plasmid is stably expressed in NIH 3T3 cells using lipofectamine. Cells are co-transfected with the Neomycin resistant plasmid, pMAMneo (ratio 1:9) (a gift from Susanne Brady-Kalnay, Case Western Reserve University). Transfected cells are selected with G4 18 (Gibco) at 0.9 mg/ml. Clones are selected and subcloned by limited dilution. Supernatants are assayed for L1-Fc. An L1-Fc expressing stably transfected 3T3 clone is propagated using the CELLMAX system (Cellco) using manufacturer's instructions. Serum in the media in the CELLMAX system is gradually removed. L1-Fc is purified from the harvested serum free media using affinity chromatography using protein A-AffiGel (Bio-Rad).
Neurite outgrowth
Control coverslips are coverslips coated with PLL. To prepare an L1 substratum, control coverslips are coated with L1-Fc. At 1 h post plating of CGN, 4 mM methyl-beta-cyclodextrin (MBCD) is added to some cultures. At 2 h post plating, ethanol is added to some cultures. Cells are fixed in 4% paraformaldehyde for 30 min at 12 h following plating and then labeled fluorescently for beta III tubulin. A blinded investigator identifies eligible neurites for measurement using an a priori design. Eligible neurites are the longest neurite of a CGN that are longer than the length of the cell soma, from single isolated neurons and that do not touch other neurites. A Nikon Optiphot-2 fluorescence microscope and the SPOT camera software are used for measurements of neurite outgrowth.
Statistics
Means, standard deviations and p values are calculated using Microsoft Excel software.
Results
Ethanol alters L1 distribution in lipid rafts, but not N-cadherin distribution
To investigate the possibility that L1-lipid raft interactions are disrupted by ethanol, CGN from rat pups on postnatal day 6 are cultured for 40 h and serum starved for 1 h before treated with 25 mM ethanol for 1 h. No L1 is detectable in the pellet from the initial low speed centrifugation (data not shown). Lipid rafts are separated from non-lipid rafts in CGN lysate by Triton X-100 extraction and sucrose density gradient centrifugation. As shown in Fig. 1A, ethanol pretreatment does not cause a significant change in the separation of the marker for the lipid raft compartment, GM1 ganglioside, and the marker for the non-lipid raft compartment, transferrin receptor. The sucrose density gradient fractions containing GM1 ganglioside are pooled as the lipid raft fraction, and the remaining sucrose gradient fractions are pooled into a non-lipid raft fraction. Thus, all L1 is either in the lipid raft or non-lipid raft pools. The proteins are precipitated from these fractions, reconstituted in equal volumes of sample buffer, and immunoblotted with antibody to the cytoplasmic domain of L1. As can be seen in Fig. 1B,C, the amount of L1 in lipid rafts from cells pretreated with 25 mM ethanol for 1 h increases to 43% of total L1, compared to 10% from control cells.
Figure 1.

(A) Ethanol does not alter the separation of lipid rafts from cell lysates. Immunoblots of two sucrose density gradients, control and EtOH treated. Top panel (Control) is of neurons not exposed to ethanol. Bottom panel (EtOH) is of neurons exposed to 25 mM ethanol for 1 hour prior to lysis. Fractions of the sucrose density gradients were blotted for both cholera toxin subunit B (CTxB) as a marker of lipid raft fractions, and for transferrin receptor (TfR) as a marker of non-lipid raft fractions. (B) Ethanol shifts L1 into lipid rafts while not affecting N-cadherin distribution. CGN incubated with or without 25 mM ethanol (EtOH) for 1 h are separated into lipid raft (LR) and non-lipid raft (N) pools. Proteins from each pool are precipitated, and reconstituted into equal volumes of sample buffer. Equal volumes from each pool are loaded onto 2 lanes, LR and N, and immunoblotted for L1, then stripped and blotted for N-cadherin and GM1 ganglioside using cholera toxin subunit B (CTxB) as a marker for lipid rafts. (C) Densitometric analysis of blots (n = 4). The total pixels in the L1 or N-cadherin bands in both the LR and N lanes is determined for both control and ethanol exposed cultures. The % of cell adhesion molecules (CAM) in the LR is calculated by taking the pixels in the LR band and dividing by the sum of the pixels in the N and LR bands. The mean +/− SD is shown. Statistically significant difference is indicated (*Paired t test, P<0.04).
To confirm these results, CGN cells are fluorescently labeled for L1 (red) and GM1 ganglioside (green) and viewed by confocal microscopy. The colocalization of L1 and GM1 ganglioside are determined. The percentage of overlap of the red and green channel is measured in at least 20 cells in 3 separate experiments. As shown in Fig. 2, 40.2 +/− 1.4% of labeled L1 colocalizes with GM 1 ganglioside in ethanol treated cells, significantly more than in control cells (27.6 +/− 1.7%, p<0.02).
Figure 2.

Confocal images confirm ethanol's effect on colocalization of L1 and GM1 ganglioside. CGN grown on PLL are pretreated with or without 25 mM ethanol for 1 h, then fixed and labeled for L1 (red) and GM1 ganglioside (green). Cells are visualized using a Leica TCS SP2 confocal microscope. Images are merged to visualize overlapping of L1 and lipid rafts (yellow).
A) CGN not exposed to ethanol. No yellow signal is seen. B) CGN treated with ethanol. Extensive overlap is seen, as shown by the yellow signal. C) The % of overlap of the red and green channels is measured in at least 20 cells in 3 separate experiments. The mean overlap is calculated for each condition in each experiment. The mean +/− SD of the individual means is shown. Statistically significant difference is indicated (*Paired t test, P<0.02).
Our previous study shows that N-cadherin-mediated neurite outgrowth is not inhibited by pharmacologic concentrations of ethanol (Tang, He, O'Riordan, Farkas, Buck, Lemmon, and Bearer, 2006). Here we test whether ethanol also changes the distribution of N-cadherin in lipid rafts. As shown in Fig. 1B,C, 38.1 +/− 10.2% of N-cadherin is in lipid rafts in control cells and no shift occurs after ethanol treatment.
Ethanol decreases the association of beta-tubulin with lipid rafts and has no effect on the lipid raft association of caveolin-1
L1 endocytosed in the C domain of the growth cone is recycled to the leading edge in vesicles carried by microtubules (Kamiguchi and Lemmon, 2000; Dequidt, Danglot, Alberts, Galli, Choquet, and Thoumine, 2007). Microtubules regulate the lipid raft localization of some proteins (Head, Patel, Roth, Murray, Swaney, Niesman, Farquhar and Insel, 2006). If ethanol traps L1 in the lipid rafts, it may increase the association of L1 with beta III tubulin, thus increasing the beta III tubulin associated with lipid rafts. As shown in Figure 3, the amount of beta III tubulin decreased in lipid rafts exposed to ethanol (51% +/− 5% vs. 40% +/− 8%), opposite to the effect on L1. Chronic alcohol abuse has been shown to increase levels of caveolin-1 in liver, and increased its binding to eNOS (Wang and Abdel-Rahman, 2005). We examined whether our brief, 1 h exposure to ethanol changed caveolin-1 redistribution in lipid rafts potentially trapping L1, and found no effect (Fig. 4).
Figure 3.
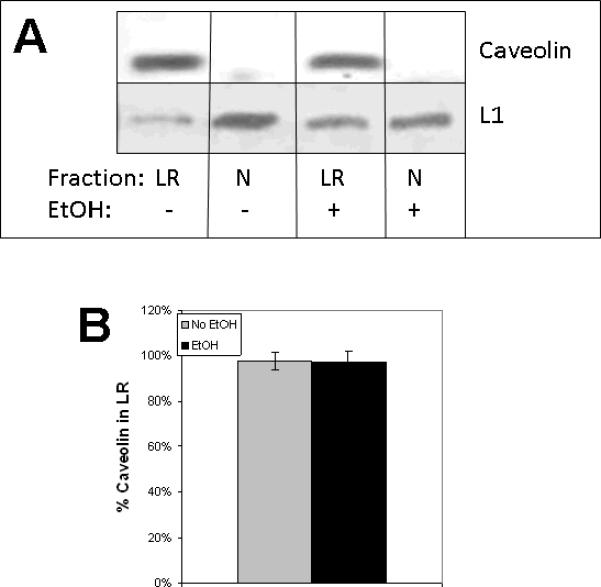
Ethanol has no effect on caveolin-1 association with lipid rafts. A) CGN are treated as described, either no addition (Control) or 25 mM ethanol for 1 h (EtOH), prior to preparation of lipid rafts. Lipid rafts (LR) were isolated from non-lipid rafts (N) by sucrose density gradient, pooled, protein precipitated and immunoblotted for caveolin-1. Blots were stripped and reblotted for L1 and CTxB. B) Blots (n=3) were quantified by densitometry. Mean values +/− SD are shown.
Figure 4.

Ethanol shifts beta III tubulin out of lipid rafts, opposite to the effect on L1. A) CGN are treated as described in Figure 3. Lipid rafts (LR) were isolated from non-lipid rafts (N) by sucrose density gradient, pooled and western blotted for beta III tubulin. B) Blots were stripped and reblotted for L1. C) Blots from 4 separate experiments quantified by densitometry. Shown are mean values +/− SD. *p<0.05, paired t test.
Butanol redistributes L1 in lipid rafts but pentanol does not, in parallel with effects seen on L1-L1 homophilic binding
Ethanol inhibits L1 adhesive properties in some cells (Charness, Safran, and Perides, 1994; Ramanathan, Wilkemeyer, Mittal, Perides, and Charness, 1996) and not in others (Bearer, Swick, O'Riordan, and Cheng, 1999; Vallejo, Hortsch, Dubreuil, 1997). In three separate L1-expressing cell lines, ethanol inhibits 55% of cell aggregation with a half-maximal inhibition at 5 to 10 mM ethanol (Wilkemeyer and Charness, 1998). Short chain alcohols (C<5) mimic the inhibitory effect, but longer chain alcohols (C>5) do not (Charness, Safran, and Perides, 1994). To determine if the effect on L1-lipid raft interactions also parallels the observed alcohol chain length dependence of L1-L1 homophilic binding, we tested the ability of butanol and pentanol at equipotent concentrations to alter L1 distribution in lipid rafts. If the L1-lipid raft disruption causes the reduction of L1 adhesivity, then the same cut off between butanol and pentanol should exist for the effect on lipid rafts (Charness, Safran, and Perides, 1994). As shown in Fig. 5, the redistribution of L1 also occurs with an equipotent concentration of butanol, but not pentanol.
Figure 5.

Butanol redistributes L1 in lipid rafts, but pentanol does not. (A) CGN pretreated with equipotent concentrations of ethanol (EtOH), butanol (BtOH) and pentanol (PtOH) are separated into lipid rafts and non-lipid rafts as described, and immunoblotted for L1. N – Non-lipid raft pool, LR-lipid raft pool. (B) Blots (n=3) are quantified as described in Fig. 1. Mean +/− SD are shown. Statistically significant differences are indicated (*p<0.02, **p<0.003, paired t test).
Other known protein targets of ethanol change their lipid raft distribution in the presence of ethanol
In this study, the effects of ethanol on the lipid raft association of other proteins known to be targets of ethanol toxicity, such as the tyrosine kinase c-Src (Suvarna, Borgland, Wang, Phamluong, Auberson, Bonci, and Ron, 2005) and GABAA receptor (Mihic, 1999) are investigated. As can be seen in Fig. 6, ethanol decreases the distribution of c-Src in lipid rafts in contrast to its effect on L1. Similarly, experiments with GABAA receptor beta 2 subunit show the same effect as c-Src, a decrease of GABAA receptor beta 2 subunit in the lipid raft in the presence of ethanol (Fig. 7). This subunit is particularly important for receptor function as it is required for sensitivity to GABA. The alteration in L1, c-Src and GABAA receptor beta 2 subunit distributions in lipid rafts, known targets for ethanol action, suggests that disruption of protein-lipid raft interactions may be an underlying mechanism of toxicity.
Figure 6.

Ethanol shifts c-Src out of lipid rafts, opposite to the effect on L1. A) CGN are treated as described, either no addition (Control) or 25 mM ethanol for 1 h (EtOH), prior to preparation of lipid rafts. Lipid rafts (LR) were isolated from non-lipid rafts (N) by sucrose density gradient, pooled, protein precipitated and immunoblotted for Src. Blots were stripped and reblotted for L1. B) Blots (n=3) were quantified by densitometry. Mean values +/− SD. *p<0.05, paired t test.
Figure 7.

Ethanol shifts GABAA receptor out of lipid rafts, opposite to the effect on L1. A) CGN are treated as described in Figure 5. Lipid rafts (LR) were isolated from non-lipid rafts (N) by sucrose density gradient and western blotted for GABAA receptor beta 2 subunit. B) Blots from 3 separate experiments quantified by densitometry. Shown are mean values +/− SD. *p<0.05, paired t test.
The redistribution of L1 into lipid rafts occurs at low concentrations of ethanol
Ethanol inhibition of L1-mediated neurite outgrowth has an IC50 of 3 – 5 mM (Bearer, Swick, O'Riordan, and Cheng, 1999). If the inhibition of L1-mediated neurite outgrowth is a result of altered trafficking through lipid rafts, then the concentrations of ethanol that induce redistribution of L1 into lipid rafts should be similar to that which inhibits neurite outgrowth. As shown in Fig. 8, the redistribution of L1 occurs even at the lowest concentration tested, 5 mM ethanol, indicating a similar sensitivity to ethanol of L1 mediated neurite outgrowth.
Figure 8.

L1 redistribution to lipid rafts is dependent on ethanol concentration. A) CGN are prepared as described and cultured overnight. Ethanol is added at the indicated concentrations for 1 h, then cells are harvested and lipid rafts isolated. Both the lipid raft pool (LR) and non-lipid raft pool (N) are immunoblotted for L1 (L1) and cholera toxin B (CTxB). B) The percent of L1 in lipid rafts (% L1 in LR), calculated as described in the text, are plotted for each concentration of ethanol. Shown are means +/− SD (n=3).
Removal of L1 from the lipid raft compartment abolishes ethanol inhibition of neurite outgrowth
Neurite outgrowth on L1 is inhibited by pharmacological treatments that deplete cellular cholesterol or sphingolipids, essential components for lipid rafts (Nakai and Kamiguchi, 2002). Remaining neurite outgrowth on L1 is independent of lipid rafts. If the effects of ethanol are on the lipid rafts, then this remaining neurite outgrowth will be insensitive to ethanol. To directly address this hypothesis, lipid rafts are disrupted by cholesterol depletion using MBCD. The survival of CGN grown in the presence of 4 mM MBCD is first determined. Results from 4 different experiments are shown in Table 1. Cells remain viable up to 12 h of exposure to MBCD with and without 25 mM ethanol with no significant difference to control cell survival at 12 h.
Table 1.
CGN survival in MBCD and/or 25 mM ethanol
| Condition | Mean +/− SE, % survival | P value vs Control, 0h | P value vs Control, 12h |
|---|---|---|---|
| Control, 0 h | 90.75 +/− 1.38 | n/a | n/a |
| Control, 12 h | 80.50 +/− 2.63 | 0.007 | n/a |
| MBCD, 12 h | 76.75 +/− 2.17 | 0.007 | 0.22 |
| MBCD + EtOH, 12 h | 74.75 +/− 3.33 | 0.015 | 0.19 |
To examine whether L1 is still associated with lipid rafts after cholesterol depletion, CGN are grown overnight, then exposed to MBCD for 1 h. Cells are harvested and lipid rafts isolated by sucrose density gradients. Fractions containing non-lipid raft proteins are identified by transferrin receptor (TfR) immunoreactivity. Each fraction is immunoblotted for L1. As shown in Fig. 9A, L1 was strictly located in fractions containing TfR. This result means that all L1 in the MBCD treated cells are located in non-lipid raft compartments.
Figure 9.


L1 mediated neurite outgrowth is dependent on lipid rafts. A) Immunoblot of L1 in sucrose density gradient fractions of CGN treated with or without MBCD. All detectable L1 colocalizes with the transferrin receptor (TfR), a marker for the non-lipid raft fraction. B) Neurite length of CGN in the presence of various reagents. Cells are plated on either PLL alone (Control) or PLL and L1-Fc (L1-Fc). Ethanol (25 mM) and/or MBCD (4mM) are added 2 h after plating. Cells are fixed 12 h after plating, and neurite length is measured. Shown is mean neurite length +/− SD. Paired t test: a, p<0.0002 from control; b, p< 0.0002 from L1 -Fc; c, p<0.02 from Control; d, p not significant from Control; e, p not significant from L1-Fc+MBCD nor Control.
To determine the effect on neurite outgrowth, CGN are cultured on PLL (control), or PLL with L1-Fc. L1-Fc is a chimeric protein containing the extracellular domain of L1 (Bearer, Swick, O'Riordan, and Cheng, 1999; Tang, He, O'Riordan, Farkas, Buck, Lemmon, and Bearer, 2006). One hour after plating, 4 mM MBCD and/or 25 mM ethanol are added to the plates. Cells are fixed 10 h after addition of MBCD and/or ethanol and neurite length is measured. Results are shown in Fig. 9B. As can be seen, cells grown on L1-Fc have longer neurites. Ethanol reduces neurite lengths of CGN grown on L1-Fc by 70% (L1-Fc versus L1-Fc+EtOH). MBCD reduced neurite length of CGN grown on L1-Fc to that of PLL alone (L1-Fc+MBCD versus Control). Addition of ethanol to CGN grown on L1-Fc and in the presence MBCD has no further effect on neurite length (L1-Fc+MBCD versus L1- Fc+MBCD+EtOH).
Discussion
There is growing interest in identifying lipid rafts as targets for ethanol action. Ethanol reportedly blocks the LPS redistribution of CD14 to a lower density fraction of the lipid raft (Dai, Zhang, and Pruett, 2005). Ethanol inhibits the LPS-mediated redistribution of toll-like receptor 4 to the lipid rafts, but has no effect on the peptidoglycan-mediated redistribution of toll like receptor 2 (Dolganiuc, Bakis, Kodys, Mandrekar, and Szabo, 2006). Another study has shown that insulin treatment of adipocytes from rats recruits cCbl, a multifunctional molecule with ubiquitin ligase activity and a protein adaptor function, and TC10, a Rho family small GTPase, to lipid rafts. However, in adipocytes from rats chronically fed ethanol, insulin treatment does not translocate cCbl and TC10 to the lipid raft (Sebastian and Nagy, 2005). The underlying mechanism for the effects of ethanol on the redistribution of proteins is yet to be investigated. L1 mediated neurite outgrowth is dependent on lipid rafts as shown by targeted disruption of lipid rafts in the P but not C domain of growth cones (Nakai and Kamiguchi, 2002) and our results reported here (Fig 9A, B).
The classic lipid raft hypothesis postulates that lipid rafts containing a given set of proteins can change their size and composition in response to intra- or extracellular stimuli. This ability favors specific protein-protein interactions, resulting in the activation of signaling cascades (Simons and Toomre, 2000). Proteins with lipid anchors or transmembrane domains have been shown to dynamically partition into and out of lipid rafts such that only a fraction of proteins are clustered in the lipid rafts (Simons and Vaz, 2004). In the axonal growth cone, L1-mediated adhesive interactions may be dynamically regulated to create a front-versus-rear asymmetry in growth cone-substrate adhesion which is believed to be necessary for growth cone migration (Kamiguchi, 2003; Dequidt, Danglot, Alberts, Galli, Choquet, and Thoumine, 2007). Therefore, the L1-lipid raft interactions should be considered as dynamic, requiring both L1 trafficking to the rafts, and L1 trafficking out of the rafts to the C domain of the growth cone where it is endocytosed (Kamiguchi and Lemmon, 2000; Schaefer, Kamei, Kamiguchi, Wong, Rapoport, Kirchausen, Beach, Landreth, Lemmon and Lemmon, 2002). Disruption of this trafficking leading to accumulation of L1 in the lipid raft would reduce L1-mediated neurite outgrowth. While ethanol induced increase of L1 association with lipid rafts may increase L1 mediated cell adhesion, there are several reports of ethanol decreasing L1-L1 cell adhesion (Charness, Safran and Perides, 1994; Ramanathan,Wilkemeyer, Mittal, Perides and Charness, 1996; Wilkemeyer and Charness, 1998). However, this effect seems both cell type and cell clone dependent (Bearer, Swick, O'Riordan, and Cheng, 1999; Vallejo, Hortsch, Dubreuil, 1997; Wilkemeyer and Charness, 1998). The changes in adhesive strength of lipid raft associated L1 will need to be investigated.
An increase in the amount of L1 in the lipid raft could be caused either by an increase in L1 trafficking to the lipid raft, or a decrease in L1 trafficking out of the lipid raft. Proteins which are temporally associated with rafts do so through the dynamic regulation of the following processes: 1) S-palmitoylation, 2) initial partial immobilization by binding to the cytoskeleton, 3) phosphorylation and dephosphorylation, 4) increased affinity of the protein for raft associated proteins or 5) length of the transmembrane domain (Lucero and Robbins, 2004). A previous study suggested that targeting of L1 family members to lipid rafts is in part dependent on palmitoylation (Ren and Bennett, 1998). Ethanol may increase L1 in lipid rafts by increasing palmitoylation of L1 (Carpenter-Hyland and Chandler, 2006). In addition we have previously shown that ethanol inhibits the tyrosine phosphorylation of L1 (Yeaney, He, Tang, Malouf, O'Riordan, Lemmon, and Bearer, 2009). Phosphorylation of a highly conserved tyrosine in the cytoplasmic domain of neurofascin abolishes ankyrin binding (Garver, Ren, Tuvia and Bennett, 1997; Tuvia, Garver and Bennett, 1997). Thus, ethanol may increase L1 binding to the cytoskeleton and therefore increase its lipid raft association. The changes in the phosphorylation state of L1 alone may increase its association with lipid rafts. L1 may have increased affinity for a lipid raft associated protein, such as c-Src (Lu, Ouyang, Seong, Zhang, Chien and Wang, 2008). While we have shown that L1 activation of c-Src occurs prior to changes in L1 tyrosine phosphorylation (Yeaney, He, Tang, Malouf, O'Riordan, Lemmon and Bearer, 2009), a direct interaction of L1 with c-Src has not been reported. It is unclear whether a direct interaction of L1 with c-Src within the lipid raft has been investigated.
Finally, it must be considered that ethanol has a direct effect on the lipid raft. Ethanol is a solvent and may have therefore altered the characteristics of the lipid raft bilayer membrane. Experiments to isolate effects of ethanol on proteins or on the lipid raft are needed. It is intriguing to speculate that ethanol effects on protein-lipid raft interactions may be a common mechanism of ethanol's toxicity. Here, we show that both c-Src and GABAA receptor, known targets for ethanol toxicity, shift out of lipid rafts in the presence of ethanol. More work is needed to define the effect of ethanol on the lipid raft proteome.
Numerous proteins require trafficking through a lipid raft compartment to initiate signal transduction. Data is accumulating on the importance of lipid rafts in basic cellular biology, implying that impairment of protein-lipid raft interactions plays a role in toxicology/clinical disease (Simons and Gerl, 2010). The involvement of lipid raft dysfunction in human disease is yet relatively unexplored but may represent a novel target for therapeutic action (Escriba, Gonzalez-Ros, Goni, Kinnunen, Vigh, Sanchez-Magraner, Fernandez, Busquets, Horvath, and Barcelo-Coblijn, 2008). FAS may be one example of a clinical disease caused at least in part by a perturbation of protein-lipid raft interactions. Further investigations will have important diagnostic and therapeutic implications for FAS, as well as other clinical diseases caused by protein-lipid raft disruptions.
Acknowledgements
This work was supported by National Institutes of Health Grant AA-0 16398 (to C.F. B.). C.F. Bearer holds the Mary Gray Cobey Professorship in Neonatology. The authors have no conflicts of interest to disclose.
The abbreviations used are
- L1
L1 cell adhesion molecule
- CGN
cerebellar granule neurons
- MBCD
methyl-beta-cyclodextrin
- FAS
fetal alcohol syndrome
- NMDA
N-methyl-D-aspartic acid
- CTxB
cholera toxin subunit B
- SDS-PAGE
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- PLL
poly L-lysine
References
- Bearer CF, Swick AR, O'Riordan MA, Cheng G. Ethanol inhibits L1-mediated neurite outgrowth in postnatal rat cerebellar granule cells. J. Biol. Chem. 1999;274(19):13264–13270. doi: 10.1074/jbc.274.19.13264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter-Hyland EP, Chandler LJ. Homeostatic plasticity during alcohol exposure promotes enlargement of dendritic spines. Eur. J. Neurosci. 2006;24(12):3496–506. doi: 10.1111/j.1460-9568.2006.05247.x. [DOI] [PubMed] [Google Scholar]
- Charness M, Safran R, Perides G. Ethanol inhibits neural cell-cell adhesion. J. Biol. Chem. 1994;269:9304–9309. [PubMed] [Google Scholar]
- Dai Q, Zhang J, Pruett SB. Ethanol alters cellular activation and CD14 partitioning in lipid rafts. Biochem. Biophys. Res. Commun. 2005;332(1):37–42. doi: 10.1016/j.bbrc.2005.04.088. [DOI] [PubMed] [Google Scholar]
- Dequidt C, Danglot L, Alberts P, Galli T, Choquet D, Thoumine O. Fast turnover of L1 adhesions in neuronal growth cones involving both surface diffusion and exo/endocytosis of L1 molecules. Mol. Biol. Cell. 2007;18(8):3131–43. doi: 10.1091/mbc.E06-12-1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolganiuc A, Bakis G, Kodys K, Mandrekar P, Szabo G. Acute ethanol treatment modulates Toll-like receptor-4 association with lipid rafts. Alcohol. Clin. Exp. Res. 2006;30(1):76–85. doi: 10.1111/j.1530-0277.2006.00003.x. [DOI] [PubMed] [Google Scholar]
- Escriba PV, Gonzalez-Ros JM, Goni FM, Kinnunen PK, Vigh L, Sanchez-Magraner L, Fernandez AM, Busquets X, Horvath I, Barcelo-Coblijn G. Membranes: a meeting point for lipids, proteins and therapies. J. Cell. Mol. Med. 2008;12(3):829–875. doi: 10.1111/j.1582-4934.2008.00281.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garver TD, Ren Q, Tuvia S, Bennett V. Tyrosine phosphorylation at a site highly conserved in the L1 family of cell adhesion molecules abolishes ankyrin binding and increases lateral mobility of neurofascin. J. Cell Biol. 1997;137(3):703–14. doi: 10.1083/jcb.137.3.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Head BP, Patel HH, Roth DM, Murray F, Swaney JS, Niesman IR, Farquhar MG, Insel PA. Microtubules and actin microfilaments regulate lipid raft/caveolar localization of adenylyl cyclase signaling components. J. Biol. Chem. 2006;281(36):26391–9. doi: 10.1074/jbc.M602577200. [DOI] [PubMed] [Google Scholar]
- Kamiguchi H. The mechanism of axon growth. Mol. Neurobiol. 2003;28:219–227. doi: 10.1385/MN:28:3:219. [DOI] [PubMed] [Google Scholar]
- Kamiguchi H, Lemmon V. Recycling of the cell adhesion molecule L1 in axonal growth cones. J. Neurosci. 2000;20(10):3676–86. doi: 10.1523/JNEUROSCI.20-10-03676.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S, Ouyang M, Seong J, Zhang J, Chien S, Wang Y. The spatiotemporal pattern of Src activation at lipid rafts revealed by diffusion-corrected FRET imaging. PLoS Comput. Biol. 2008;4(7):e1000127. doi: 10.1371/journal.pcbi.1000127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucero HA, Robbins PW. Lipid rafts-protein association and the regulation of protein activity. Arch. Biochem. Biophys. 2004;426(2):208–224. doi: 10.1016/j.abb.2004.03.020. [DOI] [PubMed] [Google Scholar]
- Luo J, Miller MW. Transforming Growth Factor B1-Regulated cell proliferation and expression of neural cell adhesion molecule in B104 neuroblastoma cells: Differential effects of ethanol. J. Neurochem. 1999;72:2286–2293. doi: 10.1046/j.1471-4159.1999.0722286.x. [DOI] [PubMed] [Google Scholar]
- Mameli M, Zamudio PA, Carta M, Valenzuela CF. Developmentally regulated actions of alcohol on hippocampal glutamatergic transmission. J. Neurosci. 2005;25(35):8027–8036. doi: 10.1523/JNEUROSCI.2434-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihic SJ. Acute effects of ethanol on GABAA and glycine receptor function. Neurochem. Int. 1999;35(2):115–123. doi: 10.1016/s0197-0186(99)00053-4. [DOI] [PubMed] [Google Scholar]
- Nakai Y, Kamiguchi H. Migration of nerve growth cones requires detergent-resistant membranes in a spatially defined and substrate-dependent manner. J..Cell Biol. 2002;159(6):1097–1108. doi: 10.1083/jcb.200209077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen DM, Gaus K, Magee AI, Cebecauer M. Dynamic organization of lymphocyte plasma membrane: lessons from advanced imaging methods. Immunology. 2010;131(1):1–8. doi: 10.1111/j.1365-2567.2010.03319.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanathan R, Wilkemeyer M, Mittal B, Perides G, Charness M. Alcohol inhibits cell-cell adhesion mediated by human L1. J. Cell Biol. 1996;133:381–390. doi: 10.1083/jcb.133.2.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Q, Bennett V. Palmitoylation of neurofascin at a site in the membrane-spanning domain highly conserved among the L1 family of cell adhesion molecules. J. Neurochem. 1998;70(5):1839–49. doi: 10.1046/j.1471-4159.1998.70051839.x. [DOI] [PubMed] [Google Scholar]
- Schaefer AW, Kamei Y, Kamiguchi H, Wong EV, Rapoport I, Kirchhausen T, Beach CM, Landreth G, Lemmon SK, Lemmon V. L1 endocytosis is controlled by a phosphorylation-dephosphorylation cycle stimulated by outside-in signaling by L1. J. Cell Biol. 2002;157(7):1223–1232. doi: 10.1083/jcb.200203024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebastian BM, Nagy LE. Decreased insulin-dependent glucose transport by chronic ethanol feeding is associated with dysregulation of the Cbl/TC10 pathway in rat adipocytes. Am. J. Physiol. Endocrinol. Metab. 2005;289(6):E1077–84. doi: 10.1152/ajpendo.00296.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons K, Ehehalt R. Cholesterol, lipid rafts, and disease. J. Clin. Invest. 2002;110(5):597–603. doi: 10.1172/JCI16390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons K, Gerl MJ. Revitalizing membrane rafts: new tools and insights. Nat. Rev. Mol. Cell Biol. 2010;11(10):688–699. doi: 10.1038/nrm2977. [DOI] [PubMed] [Google Scholar]
- Simons K, Toomre D. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 2000;1(1):31–39. doi: 10.1038/35036052. [DOI] [PubMed] [Google Scholar]
- Simons K, Vaz WL. Model systems, lipid rafts, and cell membranes. Annu. Rev. Biophys. Biomol. Struct. 2004;33:269–295. doi: 10.1146/annurev.biophys.32.110601.141803. [DOI] [PubMed] [Google Scholar]
- Stratton K, Howe C, Battaglia F, editors. Summary. Fetal Alcohol Syndrome: Diagnosis, Epidemiology, Prevention, and Treatment. National Academy Press; Washington, D.C.: 1996. [Google Scholar]
- Suvarna N, Borgland SL, Wang J, Phamluong K, Auberson YP, Bonci A, Ron D. Ethanol alters trafficking and functional N-methyl-D-aspartate receptor NR2 subunit ration via H-Ras. J. Biol. Chem. 2005;36:31450–31459. doi: 10.1074/jbc.M504120200. [DOI] [PubMed] [Google Scholar]
- Tang N, He M, O'Riordan MA, Farkas C, Buck K, Lemmon L, Bearer CF. Ethanol inhibits L1 cell adhesion molecule activation of mitogen-activated protein kinases. J. Neurochem. 2006;96:1480–1490. doi: 10.1111/j.1471-4159.2006.03649.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuvia S, Garver TD, Bennett V. The phosphorylation state of the FIGQY tyrosine of neurofascin determines ankyrin-binding activity and patterns of cell segregation. Proc. Natl. Acad. Sci. U.S.A. 1997;94(24):12957–62. doi: 10.1073/pnas.94.24.12957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallejo Y, Hortsch M, Dubreuil RR. Ethanol does not inhibit the adhesive activity of Drosophila neuroglian or human L1 in Drosophila S2 tissue culture cells. J. Biol. Chem. 1997;272:12244–7. doi: 10.1074/jbc.272.18.12244. [DOI] [PubMed] [Google Scholar]
- Wang X, Abdel-Rahman AA. Effect of chronic ethanol administration on hepatic eNOS activity and its association with caveolin-1 and calmodulin in female rats. Am J Physiol Gastrointest Liver Physiol. 2005;289(3):G579–85. doi: 10.1152/ajpgi.00282.2004. [DOI] [PubMed] [Google Scholar]
- Watanabe H, Yamazaki M, Miyazaki H, Arikawa C, Itoh K, Sasaki T, Maehama T, Frohman MA, Kanaho Y. Phospholipase D2 functions as a downstream signaling molecule of MAP kinase pathway in L1-stimulated neurite outgrowth of cerebellar granule neurons. J. Neurochem. 2004;89:142–151. doi: 10.1111/j.1471-4159.2004.02308.x. [DOI] [PubMed] [Google Scholar]
- Wilkemeyer MF, Charness ME. Characterization of ethanol-sensitive and insensitive fibroblast cell lines expressing human L1. J. Neurochem. 1998;71:2382–2391. doi: 10.1046/j.1471-4159.1998.71062382.x. [DOI] [PubMed] [Google Scholar]
- Winckler B, Yap CC. Endocytosis and endosomes at the crossroads of regulating trafficking of axon outgrowth-modifying receptors. Traffic. 2011 doi: 10.1111/j.1600-0854.2011.01213.x. doi:10.1111/j.1600-0854.2011.01213.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeaney NK, He M, Tang N, Malouf AT, O'Riordan MA, Lemmon V, Bearer CF. Ethanol inhibits L1 cell adhesion molecule tyrosine phosphorylation and dephosphorylation and activation of pp60(src) J. Neurochem. 2009;110(3):779–790. doi: 10.1111/j.1471-4159.2009.06143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao XJ, Marrero L, Song K, et al. Acute alcohol inhibits TNF-alpha processing in human monocytes by inhibiting TNF/TNF-alpha-converting enzyme interactions in the cell membrane. J. Immunol. 2003;170:2923–31. doi: 10.4049/jimmunol.170.6.2923. [DOI] [PubMed] [Google Scholar]