

Abstract
BACKGROUND AND PURPOSE
Besides a significant reduction of low-density lipoprotein (LDL) cholesterol, statins moderately increase high-density lipoprotein (HDL) levels. In vitro studies have indicated that this effect may be the result of an increased expression of apolipoprotein (apo)A-I, the main protein component of HDL. The aim of the present study was to investigate in vivo the effect of rosuvastatin on apoA-I expression and secretion in a transgenic mouse model for human apoA-I.
EXPERIMENTAL APPROACH
Human apoA-I transgenic mice were treated for 28 days with 5, 10 or 20 mg·kg−1·day−1 of rosuvastatin, the most effective statin in raising HDL levels. Possible changes of apoA-I expression by treatment were investigated by quantitative real-time RT-PCR on RNA extracted from mouse livers. The human apoA-I secretion rate was determined in primary hepatocytes isolated from transgenic mice from each group after treatment.
KEY RESULTS
Rosuvastatin treatment with 5 and 10 mg·kg−1·day−1 did not affect apoA-I plasma levels, whereas a significant decrease was observed in mice treated with 20 mg·kg−1·day−1 of rosuvastatin (−16%, P < 0.01). Neither relative hepatic mRNA concentrations of apoA-I nor apoA-I secretion rates from primary hepatocytes were influenced by rosuvastatin treatment at each tested dose.
CONCLUSIONS AND IMPLICATIONS
In human apoA-I transgenic mice, rosuvastatin treatment does not increase either apoA-I transcription and hepatic secretion, or apoA-I plasma levels. These results support the hypothesis that other mechanisms may account for the observed HDL increase induced by statin therapy in humans.
Keywords: human apolipoprotein A-I, rosuvastatin, HDL, transgenic mice, cholesterol
Introduction
Epidemiological studies have established that low plasma high-density lipoprotein (HDL) cholesterol levels are a major risk factor for cardiovascular disease (Barter and Rye, 2006). It is well established that HDL-cholesterol (HDL-C) is a strong and independent inverse predictor of coronary artery disease even in patients with normal levels of low-density lipoprotein cholesterol (LDL-C) (Gordon and Rifkind, 1989).
Statin treatment is recommended as a first-line drug therapy because of its well-established efficacy in decreasing cardiovascular events in both primary and secondary prevention (Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults, 2001). The main mechanism of action of statins is the reduction of cholesterol biosynthesis by inhibition of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase activity, resulting in up-regulation of hepatic LDL receptor activity and consequently marked reduction of LDL cholesterolaemia (Brown and Goldstein, 1986). In addition to LDL-cholesterol (LDL-C), plasma triglycerides are lowered by statins, possibly as a consequence of diminished very-low-density lipoprotein (VLDL) production and induction of receptor-mediated remnant clearance (Vega and Grundy, 1998). Furthermore, statin treatment moderately increases plasma HDL-C concentration in a range between 4% and 10% (McTaggart and Jones, 2008). Although modest, this increase may, however, provide important protection from cardiovascular disease, because it has been estimated that cardiovascular risk decreases by 2–3% for every 10 mg·L−1 increase in HDL-C levels (Gordon and Rifkind, 1989).
The mechanisms behind the effect of statins on HDL have not been yet clarified. In hepatic cells, inhibition of HMG-CoA reductase by statins has been shown to increase peroxisome proliferator activated-receptor α (PPARα) activity and, as found with the fibrates, to elevate the hepatic synthesis of apolipoprotein (apo)A-I, the main protein component of HDL, with consequent increase of HDL particle formation (Martin et al., 2001). A second explanation relates to the metabolic interaction between HDL and triglyceride-rich atherogenic lipoproteins mediated by the cholesteryl ester transfer protein (CETP). The VLDL reduction by statins would reduce the CETP-mediated cholesteryl ester transfer from HDL to VLDL, thus increasing HDL-C levels (Guerin et al., 2000; de Haan et al., 2008).
It was thus of interest to evaluate, in vivo, the possible impact on HDL of statin treatment in a naturally CETP-deficient animal model, the mouse, transgenic for human apoA-I expression (Rubin et al., 1991). This transgenic model has previously been shown to respond to PPARα activators, such as fibrates (Berthou et al., 1996). Human apoA-I transgenic mice were treated for 28 days with three different doses of rosuvastatin. Among statins, rosuvastatin appears to be the most effective in increasing HDL plasma concentrations (McTaggart and Jones, 2008) and, unlike other statins, maintains a relatively constant effect across doses (Jones et al., 2003). The results of the present study indicate that, in human apoA-I transgenic mice, rosuvastatin did not affect human apoA-I expression, at each tested dose.
Methods
Animals and treatments
All animal care and experimental procedures were in accordance with institutional guidelines that are in compliance with national (D.L. no. 116, G.U. Suppl. 40, 18 February 1992, Circolare no. 8, G.U. July 1994) and international laws and policies (EEC Council Directive 86/609, OJL 358, 1, 12 December 1987; Guide for the Care and Use of Laboratory Animals, Institute of Laboratory Animal Research, Commission on Life Sciences, National Research Council, 1996). The study was performed on forty male 20- to 25-week-old C57BL/6 transgenic mice expressing human apoA-I in the absence of murine apoA-I (hA-I/A-IKO). This was achieved by multiple crosses between human apoA-I transgenic mice (hA-I) (Rubin et al., 1991), kindly provided by Dr Edward M. Rubin, and mice lacking murine apoA-I (A-IKO) due to gene targeting (Williamson et al., 1992), kindly provided by Dr Nobuyo Maeda. The transgenic model expresses human apoA-I through the microinjection of an 11 Kb genomic fragment of the human apoA-I gene; this fragment contains the PPARα response element known to confer fibrate responsiveness to the human apoA-I gene (Berthou et al., 1996) and potentially implicated in the HDL increase by statin treatment (Martin et al., 2001). Transgenic mice were randomly divided into four groups of ten mice each and fed a standard diet for rodents (control group) for 28 days or the same standard diet containing 25, 50 or 100 mg·kg−1 of rosuvastatin (AstraZeneca, UK).
Lipid/Lipoprotein analyses
Before and at the end of treatment, after an overnight fast, blood was collected, under isofluorane anaesthesia, from the retro-orbital plexus into tubes containing 0.1% (w/v) EDTA and centrifuged in a microcentrifuge for 10 min at 5900×g at 4°C.
Plasma total cholesterol and triglycerides were measured by enzymatic methods (ABX Diagnostics, Montpellier, France). HDL-C levels were measured after precipitation of apoB-containing lipoproteins with PEG 8000 (20% w/v) in 0.2 M glycine, pH 10 (Schultz et al., 1992). Human apoA-I concentrations were determined by immunoturbidimetric assays, using a sheep antiserum specific for human apoA-I (LTA, Milan, Italy).
To analyse plasma lipoprotein distribution, plasma was pooled for each group and applied onto a Superose 6 10/30 column (Pharmacia) for fast protein liquid chromatography (FPLC) analysis (van Gent and van Tol, 1990). On the collected fractions, total cholesterol and triglycerides were measured by enzymatic methods.
To determine HDL particle size distribution, total lipoproteins (d < 1.215 g·mL−1) were isolated by salt gradient ultracentrifugation (Havel et al., 1955). An aliquot of pooled plasmas from each group was adjusted to a density of 1.215 g·mL−1 with solid KBr and centrifuged for 6 h at 4°C at 541 000×g in a Beckman TL100 ultracentrifuge equipped with a Beckman TL100.3 rotor. HDL particle size distribution was determined by non-denaturing polyacrylamide gradient gel electrophoresis (GGE), essentially as described by Nichols et al. (Nichols et al., 1986). Aliquots (20 µL) of the total lipoprotein fraction were loaded onto a non-denaturing 4–30% polyacrylamide gradient gel and separated by electrophoresis for 25 h at 125 V at 4°C. The proteins were stained with Coomassie R-250 and HDL particle size was determined by computer-assisted scanning densitometry, as previously described (Nichols et al., 1986).
Phospholipid transfer protein (PLTP) activity
PLTP activity was measured in mouse plasma before and after treatment, as previously described (Klein et al., 2006), using a commercially available fluorescence activity assay (Roar Biomedical Inc., New York, NY, USA) according to the manufacturer's instructions. This fluorimetric assay measures the transfer (unquenching) of fluorescent phosphatidylcholine from donor to acceptor synthetic liposomes.
Quantitative real-time PCR
At the end of the dietary treatment, five hA-I/A-IKO mice from each group were killed and total RNA was isolated from mouse livers using the NucleoSpin RNA extraction kit (Macherey-Nagel, Duren, Germany) according to the manufacturer's instructions. One µg of DNase-treated total RNA was reverse-transcribed using TaqMan reverse transcription reagents from Applied Biosystems (Foster City, CA, USA). PCR was performed using SYBR Green Supermix (Bio-Rad Laboratories, Hercules, CA, USA) on an iCycler Optical System (Bio-Rad Laboratories, Hercules, CA, USA). The selected primers used for amplification of human apoA-I cDNA were AGCTTGCTGAAGGTGGAGGT (in exon 4) and ATCGAGTGAAGGACCTGGC (in exon 3). The primers amplify a 154-bp product. The housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used for normalization. All of the procedures and calculation of the results were carried out according to the manufacturer's recommendations.
Hepatic human apoA-I secretion rate
The apolipoprotein secretion rate was determined in primary hepatocytes isolated from three hA-I/A-IKO mice of each group at the end of the dietary treatment. Animals were fasted for 5 h and anesthetized with 5% sodium pentobarbital, and the hepatocytes were prepared with slight modification of a previously described method (Hayek et al., 1993). Cell viability was assessed by Trypan blue staining, and 5 × 105 live cells were plated on 35 mm plates. The culture medium (Williams' E medium; Sigma, St. Louis, MO, USA) was changed after 4 h of incubation at 37°C. The next day, to assess the human apolipoprotein secretion rate, cells were washed once with phosphate-buffered saline, pre-incubated for 1 h in leucine-free Dulbecco's modified Eagle's medium without serum, and then incubated for 30, 60, 90, 120 min with 1 mL of the same medium containing 200 µCi·mL−1[3H]leucine (PerkinElmer Life Sciences). The medium was centrifuged at 12 000×g at 4°C for 5 min to remove cell debris. Radiolabelled human apolipoproteins were quantitatively isolated from cell lysates by immunoprecipitation using a rabbit polyclonal anti-human apoA-I antibody (DAKO, Glostrup, Denmark). The immunoprecipitate was further purified by SDS-PAGE under reducing conditions. A band corresponding to human apoA-I was excised from the gel; the label was extracted with Solvable (Packard Instruments Co., Inc., Meriden, CI, USA) and counted (Azrolan et al., 1995). The results were normalized to cellular protein content of each plate determined by the Bradford method (Bradford, 1976). The data presented are the means of triplicate measurements and are representative of three independent experiments.
Cell treatments
Primary hepatocytes were prepared from hA-I/A-IKO mice as described above (see Hepatic human apoA-I secretion rate). Cells (5 × 105 per dish) were plated on 35 mm plates and the culture medium (Williams' E medium; Sigma) was changed after 4 h of incubation at 37°C. The next day, cells were washed once with phosphate-buffered saline and incubated for a total of 48 h with various concentrations of statins in Williams' medium containing 2.5% serum. Medium supplemented with the drugs was renewed after 24 h incubation. At the end of treatment, cells were harvested and total RNA was isolated as described above (see Quantitative real-time PCR). Human apoA-I cDNA was quantified by real-time PCR on an Applied Biosystems 7000 sequence detector as described. The housekeeping gene cyclophilin was used for normalization.
Fenofibrate and simvastatin were purchased from Sigma (St. Louis, MO, USA), cerivastatin from Toronto Research Chemicals Inc (North York, Ontario, Canada), whereas pitavastatin was kindly provided by Kowa Company Ltd. (Tokyo, Japan).
Statistical analysis
Data are expressed as mean ± SD. Group differences were tested for statistical significance by analysis of variance for repeated measurements with one grouping factor or one-way anova, followed by a Bonferroni's post hoc test. A value of P < 0.05 was considered statistically significant.
Results
Treatments
Daily food intake was monitored during the study and no differences were observed among groups (data not shown). Based on food consumption, it was calculated that the three groups of mice fed the rosuvastatin-containing diets took, approximately, 5, 10 or 20 mg·kg−1·day−1 of rosuvastatin.
Lipid/apolipoprotein analyses
To evaluate the influence of rosuvastatin treatment on lipid/apolipoprotein plasma levels, blood samples were taken from each mouse before and after treatment. In controls, as expected, apoA-I and lipid plasma levels remained unchanged over the 28 day period (Figure 1). Treatment with 5 and 10 mg·kg−1·day−1 of rosuvastatin did not affect apoA-I plasma levels, whereas a significant decrease was observed in mice treated with the highest dose of rosuvastatin (Figure 1A). Rosuvastatin treatment did not significantly modify HDL-C levels in mice treated with the two lower doses, whereas the highest rosuvastatin dose caused a significant reduction in HDL-C (Figure 1B). Unlike humans, the main cholesterol-carrying lipoprotein in the mouse is HDL and this feature is even more pronounced in mice overexpressing human apoA-I (Rubin et al., 1991), that is, in the transgenic model used in this study. Variations in total cholesterol with drug treatment reflected therefore the trend observed for HDL-C levels, although no significant variations were observed in each treated group (Figure 1C). Rosuvastatin treatment did not induce any significant change in triglyceride levels at each tested dose (Figure 1D).
Figure 1.
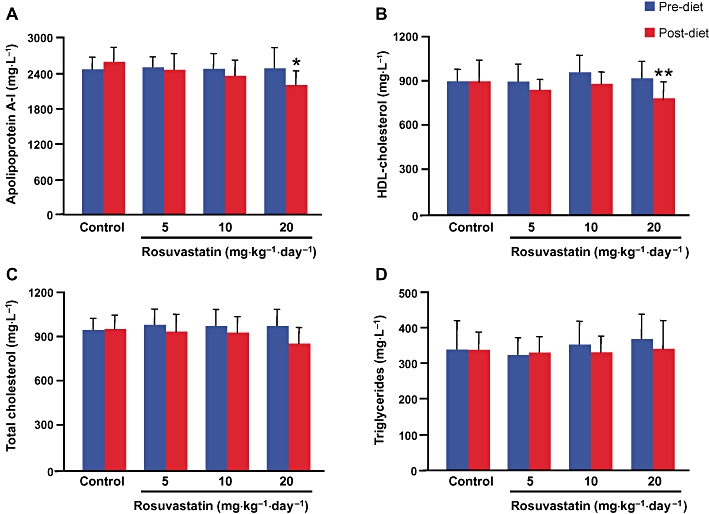
Effect of rosuvastatin treatment on human apolipoprotein A-I (A), HDL-cholesterol (B), total cholesterol (C) and triglyceride (D) plasma levels. Data are expressed as mean ± SD. *P < 0.01, *P < 0.005 significantly different from pretreatment. HDL, high-density lipoprotein.
Plasma collected before and at the end of treatment was analysed by FPLC to assess possible variations in cholesterol and triglyceride distribution among lipoprotein fractions (Figure 2). FPLC confirmed that over 80% of plasma cholesterol was mostly carried in HDL particles. No variations were detected in the control group before and after the 28 days treatment (Figure 2A); the drug treatment caused a slight decrease of the HDL-C content that was more pronounced with the highest dose (Figure 2B–D). Moreover, rosuvastatin appeared to shift the HDL peak towards larger particles. No changes in the other lipoprotein fractions were observed in each treated group. The triglyceride content in the lipoprotein fractions was not modified by the pharmacological treatment, possibly because of the extremely low triglyceride starting concentration and the almost negligible VLDL levels of this mouse model (data not shown).
Figure 2.
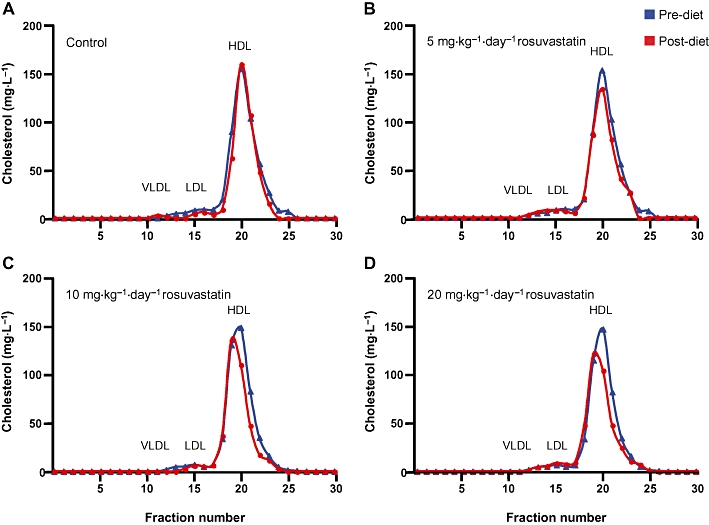
Fast protein liquid chromatography (FPLC) profile of plasma cholesterol before and after rosuvastatin treatment at each tested dose. VLDL, very low-density lipoprotein; LDL, low-density lipoprotein; HDL, high-density lipoprotein.
Changes in the HDL particle size induced by treatment were assessed by non-denaturing GGE of total lipoproteins and subsequent densitometric analysis (Figure 3). As previously reported (Rubin et al., 1991; Chiesa et al., 1998), human apoA-I transgenic mice are characterized by the presence of two major HDL subpopulations with diameters of about 9.8 and 8.5 nm and a shoulder corresponding to larger HDL particles (10.3 nm in diameter). Whereas no variations were detected in the control group (Figure 3A), rosuvastatin treatment caused a redistribution of HDL subpopulations (Figure 3B–D). With increasing doses of rosuvastatin, a relative decrease in the amount of smaller particles and an increase in that of larger particles was observed.
Figure 3.
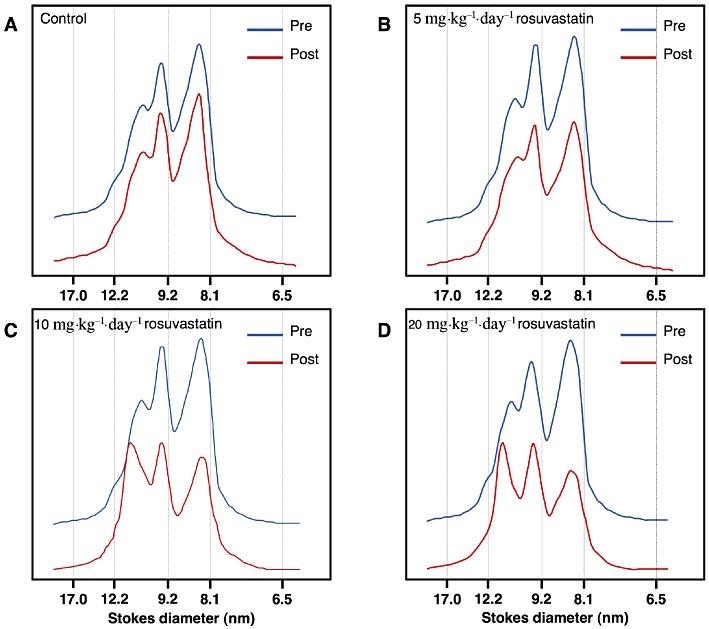
Non-denaturing 4–30% gradient gel electrophoresis of mouse plasma lipoproteins (d < 1.215 g·mL−1) before and after rosuvastatin treatment. Proteins were stained with Coomassie R-250 and HDL particle size was determined by computer-assisted scanning densitometry.
Phospholipid transfer activity
To try to explain the redistribution of HDL particle size towards larger particles by rosuvastatin treatment, PLTP activity was measured in mouse plasma and results are shown in Figure 4. PLTP activity was significantly decreased by rosuvastatin treatment at each tested dose. Moreover, the reduction was proportional to the rosuvastatin dose used.
Figure 4.
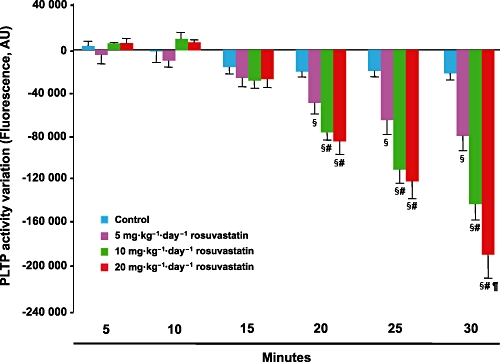
Mean change in phospholipid transfer protein (PLTP) activity measured in mouse plasma after rosuvastatin treatment. Data are expressed as mean ± SD. AU = arbitrary units; §P < 0.05 significantly different from control; #P < 0.05 versus rosuvastatin at 5 mg·kg−1·day−1; ¶P < 0.05 significantly different from rosuvastatin at 10 mg·kg−1·day−1.
Hepatic human apoA-I expression and secretion
To evaluate a possible influence of rosuvastatin on human apoA-I expression, at the end of the pharmacological treatment, mice were killed and hepatic mRNA concentrations of human apoA-I were measured by real-time RT-PCR. As reported in Figure 5, the relative mRNA concentration of apoA-I was not affected by rosuvastatin treatment at any tested dose.
Figure 5.
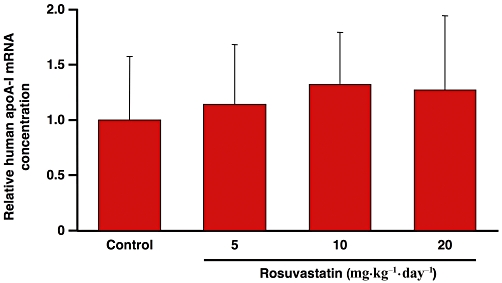
Relative mRNA concentrations of human apolipoprotein A-I in the liver of control mice or mice treated with three different rosuvastatin doses. Values are normalized to the reference gene for GAPDH and are expressed relative to the levels in control mice set as 1. Each bar represents mean values ± SD.
To assess the impact of rosuvastatin treatment on apoA-I secretion, the human apoA-I secretion rate was determined using primary hepatocytes obtained from a subset of transgenic mice from each group. As shown in Figure 6, secretion of human apoA-I was linear for all the groups; no differences were observed in the secretion rates between controls and animals treated with 5 and 10 mg·kg−1·day−1 of rosuvastatin (P > 0.05). Although not significant, apoA-I secretion rate in the group treated with the highest dose of rosuvastatin tended to be lower than that of control animals.
Figure 6.
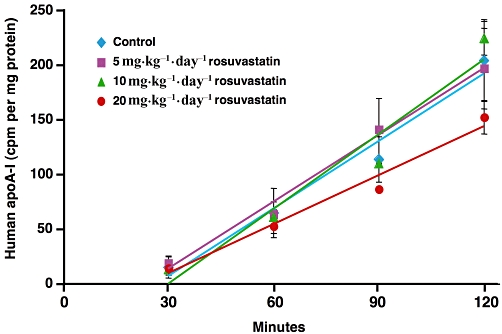
In vitro analysis of human apolipoprotein A-I (apoA-I) secretion from primary hepatocytes obtained from control mice or mice treated with 5, 10, 20 mg·kg−1·day−1 of rosuvastatin. Data are representative of three separate experiments and each point is the mean ± SD of triplicate measurements.
Human apoA-I expression in primary hA-I/A-IKO mouse hepatocytes
To investigate if the results obtained with rosuvastatin in vivo could be translated to other statins, primary hepatocytes from hA-I/A-IKO mice were incubated with rosuvastatin (5, 10, 20 µM), cerivastatin (0.25, 0.5, 1 µM), pitavastatin (2.5, 5, 10 µM) and simvastatin (2.5, 5, 10 µM). These concentration ranges have been previously demonstrated to be effective in increasing apoA-I expression in HepG2 cells (Martin et al., 2001; Maejima et al., 2004; Qin et al., 2008). Whereas fenofibrate increased human apoA-I expression, no variations were observed for each statin, at every concentration tested (Figure 7).
Figure 7.
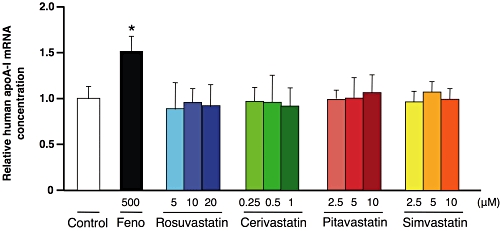
Relative mRNA concentrations of human apolipoprotein A-I in primary hepatocytes isolated from hA-I/A-IKO mice. Hepatocytes were treated for 48 h with fenofibrate (Feno, 500 µM), rosuvastatin (5, 10, 20 µM), cerivastatin (0.25, 0.5, 1 µM), pitavastatin (2.5, 5, 10 µM), and simvastatin (2.5, 5, 10 µM). Values are normalized to the reference gene for cyclophilin and are expressed relative to the levels in control (untreated) set as 1. Data are representative of three separate experiments and each bar represents the mean ± SD of triplicate measurements. *P < 0.05 significantly different from control.
Discussion
The development of statins has been a major advance in cardiovascular medicine, mainly because of the great efficacy of these molecules in lowering plasma LDL-C levels (Jones et al., 2003). However, in view of the strong inverse relationship between HDL levels and cardiovascular risk, it is important to assess the manner and degree of statin effects on these lipoproteins. Placebo-controlled trials have demonstrated that statin treatment moderately, but constantly, increases HDL-C levels, whereas it affects apoA-I levels to a lesser degree and not always significantly (Davidson et al., 1997; 2002a,b; Bays et al., 2004). Consistent with these observations, is the reporting of the formation of larger, cholesterol-rich HDL particles by statins (Asztalos and Schaefer, 2003).
The molecular basis for the effect of statin treatment on HDL levels has received limited attention and it has been investigated mostly in vitro. The present study had the objective of evaluating, in vivo, the effect of rosuvastatin on apoA-I expression and HDL-C levels in a mouse model expressing human apoA-I.
Previous studies on HepG2 cells had indicated that an elevation of apoA-I mRNA levels was involved in the observed HDL increase by statin treatment (Martin et al., 2001). The authors identified a statin response element in the human apoA-I promoter that coincided with a PPARα response element known to confer fibrate responsiveness to the human apoA-I gene. To test this hypothesis in vivo, in the present study a transgenic model was chosen, expressing human apoA-I through the microinjection of an 11 Kb genomic fragment of the human apoA-I gene containing the PPARα response element (Rubin et al., 1991). Indeed, in this same transgenic model, fibrate treatment had been associated with a strong increase in hepatic human apoA-I mRNA and an elevation of human apoA-I and HDL-C plasma concentrations (Berthou et al., 1996). Interestingly, fibrates had an opposite effect on mouse apoA-I expression, which was significantly down-regulated by the pharmacological treatment (Berthou et al., 1996). Thus, to avoid any confounding effect on murine apoA-I, the animal model used in the present study expressed human apoA-I in the absence of mouse apoA-I through crosses between human apoA-I transgenic mice and knock-out animals for murine apoA-I.
In the present study, biochemical evaluations on whole plasma and FPLC fractions did not show any increase of either apoA-I or HDL-C levels with rosuvastatin treatment at each tested dose. Specifically, no variations were observed with 5 and 10 mg·kg−1·day−1 of rosuvastatin, whereas the 20 mg·kg−1·day−1 dose significantly decreased apoA-I and HDL-C. Hepatic apoA-I expression, evaluated by quantitative real-time PCR, was not modified by the pharmacological treatment. Similar results were obtained in vitro, by incubating primary hA-I/A-IKO hepatocytes with different doses of rosuvastatin. Moreover, in vitro assessment of the effect of other statins on apoA-I expression showed no variation of human apoA-I mRNA levels. These results are in contrast with previous investigations, all performed in vitro, in human hepatoma cells (Martin et al., 2001; Maejima et al., 2004; Qin et al., 2008). Although possible limitations of the experimental model used may account for the results obtained, it should be noted that this animal model is indeed responsive to PPARα agonists as also shown by other investigators (Berthou et al., 1996; Rotllan et al., 2011; Srivastava et al., 2011). Altogether, our results suggest that transcriptional activation of apoA-I may not be the major mechanism explaining the HDL increase triggered by statin treatment.
In order to assess whether rosuvastatin treatment could post-transcriptionally influence human hepatic apoA-I production, apoA-I secretion was measured on primary hepatocytes isolated from the different treatment groups. No differences were observed among groups, except for a lower, although not significant, secretion rate in the mice treated with the highest rosuvastatin dose. These results do not support the hypothesis that rosuvastatin treatment may increase HDL levels by directly affecting human apoA-I production. Clinical studies generally show a trend towards increase of apoA-I levels (Brown et al., 2002; Davidson et al., 2002a; Capuzzi et al., 2003; Hunninghake et al., 2004), although there is no real evidence for increased apoA-I production in subjects receiving rosuvastatin treatment. Indeed, investigations on the effect of rosuvastatin on the kinetics of apoA-I in patients affected by metabolic syndrome (Ooi et al., 2008) or type 2 diabetes (Verges et al., 2009) showed no variations or even a reduction in apoA-I production rate versus placebo.
The unexpected result obtained with the highest dose, that is, a reduction of HDL-C and human apoA-I after rosuvastatin treatment, could possibly be a consequence of hepatic cholesterol depletion due to the inhibition of HMG-CoA reductase, that may reduce hepatic HDL production (Brewer et al., 2004). This possibility is partially supported by the slight reduction of apoA-I secretion observed for primary hepatocytes of mice treated with the highest rosuvastatin dose. Alternatively, the increase of LDL receptor activity by statin treatment, by enhancement of the catabolism of apoE-containing lipoproteins, could accelerate the catabolism of apoE-containing HDL particles and thus reduce plasma HDL levels. However, Western blot analysis on plasma from controls and rosuvastatin treated mice showed that apoE levels were not modified by the treatment (data not shown).
Finally, GGE analysis of HDL particles showed, in rosuvastatin treated mice, a relative increase of larger particles and a relative decrease of smaller HDL particles. This variation in HDL size was modest with the lower rosuvastatin dose, but increased progressively at higher doses. The appearance of larger HDL particles after statin treatment has been frequently described in clinical studies (Asztalos and Schaefer, 2003; Ikewaki et al., 2009) and it is generally considered a consequence of a reduced CETP-mediated exchange of triglycerides and cholesteryl esters between HDL and VLDL particles (Guerin et al., 2000). In the present study, although mice lack CETP, a redistribution of HDL particles, towards larger subpopulations, was also observed. These results may imply that other mechanisms could be involved in the interconversion of HDL particles occurring after statin treatment. A recent study reported that statins increased HDL size by lowering PLTP activity, thus decreasing pre-β-HDL formation (Dallinga-Thie et al., 2009). Indeed, in the present study, a reduced PLTP activity by rosuvastatin treatment was observed.
In summary, in human apoA-I transgenic mice lacking CETP, rosuvastatin treatment did not increase apoA-I transcription and hepatic secretion, or apoA-I plasma levels. These results indirectly support the hypothesis that the effect of statins on HDL levels observed in humans can be mostly attributed to reduced CETP activity.
Acknowledgments
This study was supported by AstraZeneca, Italy. We thank Dr Lorenzo Arnaboldi, Professor Maurizio Crestani and coworkers for helpful discussion.
Glossary
Abbreviations
- apo
apolipoprotein
- AU
arbitrary units
- CETP
cholesteryl ester transfer protein
- HDL
high-density lipoprotein
- HDL-C
HDL-cholesterol
- HMG-CoA
3-hydroxy-3-methylglutaryl coenzyme A
- LDL
low-density lipoprotein
- LDL-C
LDL-cholesterol
- PLTP
phospholipid transfer protein
- VLDL
very-low-density lipoprotein
Conflict of interest
The authors state no conflict of interest.
References
- Asztalos BF, Schaefer EJ. High-density lipoprotein subpopulations in pathologic conditions. Am J Cardiol. 2003;91:12E–17E. doi: 10.1016/s0002-9149(02)03383-0. [DOI] [PubMed] [Google Scholar]
- Azrolan N, Odaka H, Breslow JL, Fisher EA. Dietary fat elevates hepatic apoA-I production by increasing the fraction of apolipoprotein A-I mRNA in the translating pool. J Biol Chem. 1995;270:19833–19838. doi: 10.1074/jbc.270.34.19833. [DOI] [PubMed] [Google Scholar]
- Barter PJ, Rye KA. Relationship between the concentration and antiatherogenic activity of high-density lipoproteins. Curr Opin Lipidol. 2006;17:399–403. doi: 10.1097/01.mol.0000236365.40969.af. [DOI] [PubMed] [Google Scholar]
- Bays HE, Ose L, Fraser N, Tribble N, Quinto K, Rejes R, et al. A multicenter, randomized, double-blind, placebo-controlled, factorial design study to evaluate the lipid-altering efficacy and safety profile of the ezetimibe/simvastatin tablet compared with ezetimibe and simvastatin monotherapy in patients with primary hypercholesterolemia. Clin Ther. 2004;26:1758–1773. doi: 10.1016/j.clinthera.2004.11.016. [DOI] [PubMed] [Google Scholar]
- Berthou L, Duverger N, Emmanuel F, Langouet S, Auwerx J, Guillouzo A, et al. Opposite regulation of human versus mouse apolipoprotein A-I by fibrates in human apolipoprotein A-I transgenic mice. J Clin Invest. 1996;97:2408–2416. doi: 10.1172/JCI118687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Brewer HB, Jr, Remaley AT, Neufeld EB, Basso F, Joyce C. Regulation of plasma high-density lipoprotein levels by the ABCA1 transporter and the emerging role of high-density lipoprotein in the treatment of cardiovascular disease. Arterioscler Thromb Vasc Biol. 2004;24:1755–1760. doi: 10.1161/01.ATV.0000142804.27420.5b. [DOI] [PubMed] [Google Scholar]
- Brown WV, Bays HE, Hassman DR, McKenney J, Chitra R, Hutchinson H, et al. Efficacy and safety of rosuvastatin compared with pravastatin and simvastatin in patients with hypercholesterolemia: a randomized, double-blind, 52-week trial. Am Heart J. 2002;144:1036–1043. doi: 10.1067/mhj.2002.129312. [DOI] [PubMed] [Google Scholar]
- Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science. 1986;232:34–47. doi: 10.1126/science.3513311. [DOI] [PubMed] [Google Scholar]
- Capuzzi DM, Morgan JM, Weiss RJ, Chitra RR, Hutchinson HG, Cressman MD. Beneficial effects of rosuvastatin alone and in combination with extended-release niacin in patients with a combined hyperlipidemia and low high-density lipoprotein cholesterol levels. Am J Cardiol. 2003;91:1304–1310. doi: 10.1016/s0002-9149(03)00318-7. [DOI] [PubMed] [Google Scholar]
- Chiesa G, Parolini C, Canavesi M, Colombo N, Sirtori CR, Fumagalli R, et al. Human apolipoproteins A-I and A-II in cell cholesterol efflux: studies with transgenic mice. Arterioscler Thromb Vasc Biol. 1998;18:1417–1423. doi: 10.1161/01.atv.18.9.1417. [DOI] [PubMed] [Google Scholar]
- Dallinga-Thie GM, van Tol A, Dullaart RP. Plasma pre beta-HDL formation is decreased by atorvastatin treatment in type 2 diabetes mellitus: role of phospholipid transfer protein. Biochim Biophys Acta. 2009;1791:714–718. doi: 10.1016/j.bbalip.2009.03.008. [DOI] [PubMed] [Google Scholar]
- Davidson M, McKenney J, Stein E, Schrott H, Bakker-Arkema R, Fayyad R, et al. Comparison of one-year efficacy and safety of atorvastatin versus lovastatin in primary hypercholesterolemia. Atorvastatin Study Group I. Am J Cardiol. 1997;79:1475–1481. doi: 10.1016/s0002-9149(97)00174-4. [DOI] [PubMed] [Google Scholar]
- Davidson M, Ma P, Stein EA, Gotto AM, Jr, Raza A, Chitra R, et al. Comparison of effects on low-density lipoprotein cholesterol and high-density lipoprotein cholesterol with rosuvastatin versus atorvastatin in patients with type IIa or IIb hypercholesterolemia. Am J Cardiol. 2002a;89:268–275. doi: 10.1016/s0002-9149(01)02226-3. [DOI] [PubMed] [Google Scholar]
- Davidson MH, McGarry T, Bettis R, Melani L, Lipka LJ, LeBeaut AP, et al. Ezetimibe coadministered with simvastatin in patients with primary hypercholesterolemia. J Am Coll Cardiol. 2002b;40:2125–2134. doi: 10.1016/s0735-1097(02)02610-4. [DOI] [PubMed] [Google Scholar]
- Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. Executive Summary of The Third Report of The National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, And Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III) JAMA. 2001;285:2486–2497. doi: 10.1001/jama.285.19.2486. [DOI] [PubMed] [Google Scholar]
- van Gent T, van Tol A. Automated gel permeation chromatography of plasma lipoproteins by preparative fast protein liquid chromatography. J Chromatogr. 1990;525:433–441. doi: 10.1016/s0378-4347(00)83420-9. [DOI] [PubMed] [Google Scholar]
- Guerin M, Lassel TS, Le Goff W, Farnier M, Chapman MJ. Action of atorvastatin in combined hyperlipidemia: preferential reduction of cholesteryl ester transfer from HDL to VLDL1 particles. Arterioscler Thromb Vasc Biol. 2000;20:189–197. doi: 10.1161/01.atv.20.1.189. [DOI] [PubMed] [Google Scholar]
- Gordon DJ, Rifkind BM. High-density lipoprotein – the clinical implications of recent studies. N Engl J Med. 1989;321:1311–1316. doi: 10.1056/NEJM198911093211907. [DOI] [PubMed] [Google Scholar]
- de Haan W, van der Hoogt CC, Westerterp M, Hoekstra M, Dallinga-Thie GM, Princen HM, et al. Atorvastatin increases HDL cholesterol by reducing CETP expression in cholesterol-fed APOE*3-Leiden.CETP mice. Atherosclerosis. 2008;197:57–63. doi: 10.1016/j.atherosclerosis.2007.08.001. [DOI] [PubMed] [Google Scholar]
- Havel RJ, Eder HA, Bragdon JH. The distribution and chemical composition of ultracentrifugally separated lipoproteins in human serum. J Clin Invest. 1955;34:1345–1353. doi: 10.1172/JCI103182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayek T, Ito Y, Azrolan N, Verdery RB, Aalto-Setala K, Walsh A, et al. Dietary fat increases high density lipoprotein (HDL) levels both by increasing the transport rates and decreasing the fractional catabolic rates of HDL cholesterol ester and apolipoprotein (Apo) A-I. Presentation of a new animal model and mechanistic studies in human Apo A-I transgenic and control mice. J Clin Invest. 1993;91:1665–1671. doi: 10.1172/JCI116375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunninghake DB, Stein EA, Bays HE, Rader DJ, Chitra RR, Simonson SG, et al. Rosuvastatin improves the atherogenic and atheroprotective lipid profiles in patients with hypertriglyceridemia. Coron Artery Dis. 2004;15:115–123. doi: 10.1097/00019501-200403000-00008. [DOI] [PubMed] [Google Scholar]
- Ikewaki K, Terao Y, Ozasa H, Nakada Y, Tohyama J, Inoue Y, et al. Effects of atorvastatin on nuclear magnetic resonance-defined lipoprotein subclasses and inflammatory markers in patients with hypercholesterolemia. J Atheroscler Thromb. 2009;16:51–56. doi: 10.5551/jat.e563. [DOI] [PubMed] [Google Scholar]
- Jones PH, Davidson MH, Stein EA, Bays HE, McKenney JM, Miller E, et al. Comparison of the efficacy and safety of rosuvastatin versus atorvastatin, simvastatin, and pravastatin across doses (STELLAR* Trial) Am J Cardiol. 2003;92:152–160. doi: 10.1016/s0002-9149(03)00530-7. [DOI] [PubMed] [Google Scholar]
- Klein A, Deckert V, Schneider M, Dutrillaux F, Hammann A, Athias A, et al. Alpha-tocopherol modulates phosphatidylserine externalization in erythrocytes: relevance in phospholipid transfer protein-deficient mice. Arterioscler Thromb Vasc Biol. 2006;26:2160–2167. doi: 10.1161/01.ATV.0000235699.98024.11. [DOI] [PubMed] [Google Scholar]
- Maejima T, Yamazaki H, Aoki T, Tamaki T, Sato F, Kitahara M, et al. Effect of pitavastatin on apolipoprotein A-I production in HepG2 cell. Biochem Biophys Res Commun. 2004;324:835–839. doi: 10.1016/j.bbrc.2004.09.122. [DOI] [PubMed] [Google Scholar]
- Martin G, Duez H, Blanquart C, Berezowski V, Poulain P, Fruchart JC, et al. Statin-induced inhibition of the Rho-signaling pathway activates PPARalpha and induces HDL apoA-I. J Clin Invest. 2001;107:1423–1432. doi: 10.1172/JCI10852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McTaggart F, Jones P. Effects of statins on high-density lipoproteins: a potential contribution to cardiovascular benefit. Cardiovasc Drugs Ther. 2008;22:321–338. doi: 10.1007/s10557-008-6113-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols AV, Krauss RM, Musliner TA. Nondenaturing polyacrylamide gradient gel electrophoresis. Methods Enzymol. 1986;128:417–431. doi: 10.1016/0076-6879(86)28084-2. [DOI] [PubMed] [Google Scholar]
- NRC (National Research Council), Institute of Laboratory Animal Research, Commission on Life Sciences. Guide for the care and use of laboratory animals. National Academy Press: Washington DC; 1996. [Google Scholar]
- Ooi EM, Watts GF, Nestel PJ, Sviridov D, Hoang A, Barrett PH. Dose-dependent regulation of high-density lipoprotein metabolism with rosuvastatin in the metabolic syndrome. J Clin Endocrinol Metab. 2008;93:430–437. doi: 10.1210/jc.2007-0854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin S, Koga T, Ganji SH, Kamanna VS, Kashyap ML. Rosuvastatin selectively stimulates apolipoprotein A-I but not apolipoprotein A-II synthesis in Hep G2 cells. Metabolism. 2008;57:973–979. doi: 10.1016/j.metabol.2008.02.014. [DOI] [PubMed] [Google Scholar]
- Rotllan N, Llaverias G, Julve J, Jauhiainen M, Calpe-Berdiel L, Hernandez C, et al. Differential effects of gemfibrozil and fenofibrate on reverse cholesterol transport from macrophages to feces in vivo. Biochim Biophys Acta. 2011;1811:104–110. doi: 10.1016/j.bbalip.2010.11.006. [DOI] [PubMed] [Google Scholar]
- Rubin EM, Ishida BY, Clift SM, Krauss RM. Expression of human apolipoprotein A-I in transgenic mice results in reduced plasma levels of murine apolipoprotein A-I and the appearance of two new high density lipoprotein size subclasses. Proc Natl Acad Sci U S A. 1991;88:434–438. doi: 10.1073/pnas.88.2.434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz JR, Gong EL, McCall MR, Nichols AV, Clift SM, Rubin EM. Expression of human apolipoprotein A-II and its effect on high density lipoproteins in transgenic mice. J Biol Chem. 1992;267:21630–21636. [PubMed] [Google Scholar]
- Srivastava RA, He S, Newton RS. Differential regulation of human apolipoprotein AI and high-density lipoprotein by fenofibrate in hapoAI and hapoAI-CIII-AIV transgenic mice. Biochim Biophys Acta. 2011;1811:76–83. doi: 10.1016/j.bbalip.2010.11.004. [DOI] [PubMed] [Google Scholar]
- Vega GL, Grundy SM. Effect of statins on metabolism of apo-B-containing lipoproteins in hypertriglyceridemic men. Am J Cardiol. 1998;81:36B–42B. doi: 10.1016/s0002-9149(98)00036-8. [DOI] [PubMed] [Google Scholar]
- Verges B, Florentin E, Baillot-Rudoni S, Petit JM, Brindisi MC, Pais de Barros JP, et al. Rosuvastatin 20 mg restores normal HDL-apoA-I kinetics in type 2 diabetes. J Lipid Res. 2009;50:1209–1215. doi: 10.1194/jlr.P800040-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson R, Lee D, Hagaman J, Maeda N. Marked reduction of high density lipoprotein cholesterol in mice genetically modified to lack apolipoprotein A-I. Proc Natl Acad Sci U S A. 1992;89:7134–7138. doi: 10.1073/pnas.89.15.7134. [DOI] [PMC free article] [PubMed] [Google Scholar]