

Abstract
BACKGROUND AND PURPOSE
The detailed molecular modulation of inward rectifier potassium channels (including the KIR2.3 channel) is not fully understood. The present study was designed to determine whether human KIR2.3 (KIR2.3) channels were regulated by protein tyrosine kinases (PTKs).
EXPERIMENTAL APPROACH
Whole-cell patch voltage-clamp, immunoprecipitation, Western blot analysis and site-directed mutagenesis were employed to determine the potential PTK phosphorylation of Kir2.3 current in HEK 293 cells stably expressing Kir2.3 gene.
KEY RESULTS
The broad-spectrum PTK inhibitor genistein (10 µM) and the selective epidermal growth factor (EGF) kinase inhibitor AG556 (10 µM) reversibly decreased KIR2.3 current and the effect was reversed by the protein tyrosine phosphatase inhibitor, orthovanadate (1 mM). Although EGF (100 ng·mL−1) and orthovanadate enhanced KIR2.3 current, this effect was antagonized by AG556. However, the Src-family tyrosine kinase inhibitor PP2 (10 µM) did not inhibit KIR2.3 current. Tyrosine phosphorylation of KIR2.3 channels was decreased by genistein or AG556, and was increased by EGF or orthovanadate. The decrease of tyrosine phosphorylation of KIR2.3 channels by genistein or AG556 was reversed by orthovanadate or EGF. Interestingly, the response of KIR2.3 channels to EGF or AG556 was lost in the KIR2.3 Y234A mutant channel.
CONCLUSION AND IMPLICATIONS
These results demonstrate that the EGF receptor tyrosine kinase up-regulates the KIR2.3 channel via phosphorylation of the Y234 residue of the WT protein. This effect may be involved in the endogenous regulation of cellular electrical activity.
Keywords: inward rectifier K+ channels, protein tyrosine kinase, EGFR tyrosine kinase, protein tyrosine phosphatase, tyrosine phosphorylation
Introduction
Inwardly rectifying potassium (KIR) channels (nomenclature follows Alexander et al., 2009) are characterised by passing current more readily in the inward direction and are expressed in many types of cells and play a critical role in regulating membrane potential and/or cell excitability (Murata et al., 2001; Bichet et al., 2003). KIR channels exhibit a subunit topology of two transmembrane domains surrounding a pore region (Murata et al., 2001). There are seven KIR subfamilies (KIR1.x to KIR7.x) identified by cloning (Bichet et al., 2003). The KIR2.x subfamily includes KIR2.1, KIR2.2, KIR2.3 and KIR2.4 channels (Makhina et al., 1994; Morishige et al., 1994; Perier et al., 1994; Raab-Graham et al., 1994; Ashen et al., 1995; Inagaki et al., 1995). It is particularly interesting that KIR2.3 channels are highly expressed in both heart and brain (Perier et al., 1994; Bichet et al., 2003). Unlike other members of the Kir2.x family, KIR2.3 channels are directly coupled to G proteins, which allows these channels to contribute to neurotransmission and cell–cell communications (Cohen et al., 1996). In addition, KIR2.3 channels are modulated by several intra- and extra-cellular signal molecules including ATP (Collins et al., 1996), protons (Coulter et al., 1995) and protein kinase C (Zhu et al., 1999a).
Protein tyrosine kinases (PTKs), including receptor PTKs, such as the epidermal growth factor receptor kinase (EGFR kinase) and non-receptor PTKs, such as the Src-family kinases, provide important intracellular signalling mechanisms (Hunter, 2000). In addition to the mediation of cellular events such as cell growth, differentiation, embryonic development, metabolism, immune system function and oncogenesis (Hunter, 2000), protein phosphorylation at tyrosine residues modulates ion channels (Levitan, 1994), including Ca2+ channels (Ogura et al., 1999), the voltage-gated Na+ channel (Ahern et al., 2005; Liu et al., 2007), volume-sensitive Cl- channels (Du et al., 2004b) as well as voltage-gated K+ channels (Davis et al., 2001; Tiran et al., 2003; Zhang et al., 2008; Dong et al., 2010). Protein kinases and phosphatases are believed to be the yin and yang of protein phosphorylation and signalling, that is, the level of protein phosphorylation is dependent on the balance between the kinases and phosphatases (Levitan, 1994; Hunter, 2000). It is not known whether KIR2.3 channels are regulated by the PTKs and protein tryrosine phosphatases (PTPs). The present study was therefore designed to determine whether and how KIR2.3 channels are regulated by PTKs and PTPs in HEK 293 cells stably expressing the Kir2.3 gene. We found that KIR2.3 channels were regulated by the interplay of EGFR kinase and PTPs at the Y234 residue of the channel.
Methods
Cell culture, mutagenesis and gene transfection
The pCDNA3.1/hKir2.3 plasmid was kindly provided by Dr Carol A Vandenberg (University of California at Santa Barbara, CA, USA.) (Perier et al., 1994), and transfected into HEK293 cells (ATCC, Manassas, VA, USA). The HEK 293 cell line stably expressed hKIR2.3 channels was established as previously described (Tang et al., 2007; Zhang et al., 2009), and cultured in Dulbecco's modified Eagle's medium (Invitrogen, Hong Kong, China) supplemented with 10% foetal bovine serum, 400 µg·mL−1 G418 (Invitrogen). The predicted potential tyrosine phosphorylation sites were examined using the software NetPhos 2.0 (http://www.cbs.dtu.dk/cgi-bin). The mutants of KIR2.3 channels were generated using a QuickChange site-directed mutagenesis kit following the manufacturer's instruction (Stratagene, La Jolla, CA, USA), and then confirmed with DNA sequencing, and the mutants Y234A, Y329A and Y333A were transiently expressed in HEK 293 cells using 10 µL of Lipofectamine 2000 with 4 µg of pCDNA3.1/hKIR2.3-mutant vector. Cells used for electrophysiology were seeded on a glass cover slip.
Solutions
Tyrode solution contained (mM) NaCl 140, KCl 5.4, MgCl2 1.0, CaCl2 1.8, HEPES 10.0 and glucose 10 (pH adjusted to 7.3 with NaOH). For whole-cell recordings, the pipette solution contained (mM) KCl 20, K- aspartate 110, MgCl2 1.0, HEPES 10, EGTA 5, GTP 0.1, Na2-phosphocreatine 5 and Mg-ATP 5 (pH adjusted to 7.2 with KOH).
Electrophysiology
Cells on a coverslip were transferred to a cell chamber (0.5 mL) mounted on the stage of an inverted microscope (Diaphot, Nikon, Tokyo, Japan), and were superfused at ∼2 mL·min−1 with Tyrode solution. Whole-cell currents were recorded at 22–23°C, as described previously (Tang et al., 2007; Zhang et al., 2009).
Immunoprecipitation and Western blot
The immunoprecipitation and Western blotting were performed as described previously (Zhang et al., 2008). Briefly, cells were treated, respectively, with 1 mM orthovanadate, 100 ng·mL−1 EGF, 10 µM genistein, 1 mM orthovanadate plus 10 µM genistein, 100 ng·mL−1 EGF plus 10 µM genistein, 10 µM AG556, 1 mM orthovanadate plus 10 µM AG556, and EGF plus 10 µM AG556 for 30 min at room temperature, and centrifuged at 4°C. The cell pellet was then lysed with the lysis buffer described previously (Zhang et al., 2008). Protein quantification of lysates was made using a protein assay reader (Bio-Rad Laboratories, Hercules, CA, USA), and diluted to equal concentrations. Proteins were immunoprecipitated overnight at 4°C using 2 µg of anti-KIR2.3 antibody (#sc-23632, Santa Cruz Biotech., Santa Cruz, CA, USA) and 100 µL of protein A agarose bead (#16–125, Millipore, Billerica, MA, USA). Immunoprecipitated proteins bound to pelleted protein A-beads were washed thoroughly in PBS, denatured in Laemmli sample buffer, separated using sodium dodecyl sulphate polyacrylamide gel electrophoresis and electroblotted onto nitrocellulose membranes. The immunoblots were probed with an anti-phosphotyrosine antibody (1:1000, P-Tyr-100 #9411, Cell Signalling Tech, Danvers, MA, USA) overnight at 4°C in a blocking solution containing 5% BSA in TBS and Tween 20, and subsequently treated with goat anti-mouse IgG-HRP antibody (1:5000, #sc-2005, Santa Cruz Biotech.) for 1 h at room temperature. Blots were developed with enhanced chemiluminescence (GE Healthcare, Hong Kong, China) and exposed on X-ray film (Fuji Photo Film GmbH, Düsseldorf, Germany). The blots were then stripped and reprobed with the anti-KIR2.3 antibody to determine total KIR2.3 channel proteins. The film was scanned, imaged by a Bio-Imaging System (Syngene, Cambridge, UK) and analysed via GeneTools software (Syngene).
Statistical analysis
The data are expressed as means ± SEM. Paired and/or unpaired Student's t-tests were used as appropriate to evaluate the statistical significance of differences between two group means, and anova was used for multiple groups. Values of P < 0.05 were considered to be statistically significant.
Materials
3-(4-Chlorophenyl) 1-(1,1-dimethylethyl)-1H-pyrazolo[3,4-d] pyrimidin-4-amine (PP2) was purchased from Tocris Bioscience (Bristol, UK). Other reagents were obtained from Sigma-Aldrich (St Louis, MO, USA). Stock solutions were made with dimethyl sulphoxide (DMSO) for genistein (100 mM), daidzein (100 mM), AG556 (100 mM), AG 1295 (20 mM), PP2 (10 mM). EGF was reconstituted using 10 mM acetic acid containing 0.1% BSA to 20 µg·mL−1 stock solution. The stocks were divided into aliquots and stored at −20°C. Sodium orthovanadate stock solution (200 mM) was made with distilled water and the pH adjusted to 9.0.
Results
Inhibition of KIR2.3 current by PTK inhibitors
Figure 1 shows the effects of the broad-spectrum PTK inhibitor genistein on KIR2.3 channels stably expressed in HEK 293 cells. Genistein (10 µM) inhibited voltage-dependent KIR2.3 current elicited by the voltage steps, as shown in the inset in a representative cell, and the inhibitory effect was fully reversed by washout (Figure 1A). Figure 1B displays the time course of KIR2.3 current recorded in a typical experiment with a 200-ms voltage step from −40 to −120 mV in the absence and presence of genistein, and with co-application of genistein and orthovanadate. The current was substantially suppressed by 10 µM genistein, and the inhibition was fully reversed by 1 mM orthovanadate. Similar results were obtained in assays of voltage-dependent KIR2.3 current (Figure 1C, n = 6). Figure 1D illustrates the current-voltage (I-V) relationships of KIR2.3 current activated by a 3 s ramp (0 to −120 mV, from a holding potential of −40 mV) during control, in the presence of genistein, genistein plus orthovanadate or Ba2+. The current was inhibited by 10 µM genistein, and the inhibitory action was antagonized by co-application of 1 mM orthovanadate. The cardiac inward rectifier K+ channel (IK1) blocker Ba2+ (Li and Dong, 2010) almost fully suppressed KIR2.3 current at 0.5 mM (n = 5). In a total of eight cells, genistein (10 µM) decreased KIR2.3 current (at −120 mV) by 27.5% (P < 0.01 vs. control), and the inhibition was reversed by 1 mM orthovanadate to 2.2% (P < 0.01 vs. genistein alone) (Figure 1E). These results suggest that the inhibitory effect of genistein on KIR2.3 current is related to PTK inhibition.
Figure 1.
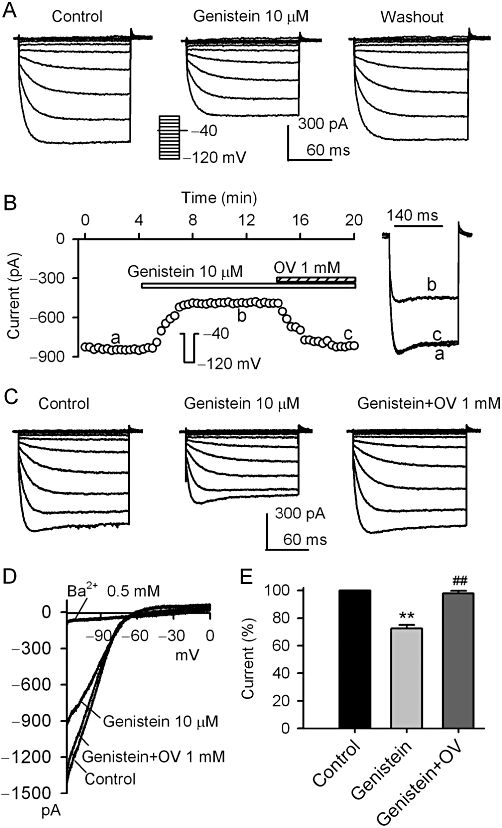
Inhibition of KIR2.3 current by genistein. (A) Voltage-dependent KIR2.3 current recorded in a representative cell with 200-ms voltage steps to between −120 to 0 mV from a holding potential of −40 mV (0.1 Hz, inset) in the absence and presence of 10 µM genistein. (B). Time-course of KIR2.3 current recorded in a representative cell with 200-ms voltage step to −120 mV from a holding potential of −40 mV in the absence and presence of genistein (10 µM), genistein plus 1 mM orthovanadate (OV). The current was measured from the zero level to steady-state current at end of voltage step. Original current traces at corresponding time points are shown in right of the panel. (C) Voltage dependent KIR2.3 current recorded in a typical experiment with the voltage protocol as shown in the inset of panel A during control, in the presence of 10 µM genistein and genistein plus 1 mM orthovanadate. (D) I–V relationships of KIR2.3 current recorded by a 3 s ramp (0 to −120 mV from a holding potential of −40 mV) in control, 10 µM genistein application, genistein plus 1 mM orthovanadate and 0.5 mM Ba2+. (E) Summary values of KIR2.3 current (−120 mV) as percent control, in cells before (control) or after 10 µM genistein and genistein plus 1 mM orthovanadate (n = 8, **P < 0.01 vs. control, ##P < 0.01 vs. genistein alone).
Figure 2 illustrates the effects of the selective EGFR tyrosine kinase inhibitor AG556 on KIR2.3 current. AG556 (10 µM) reversibly inhibited the voltage-dependent KIR2.3 current activated by the voltage protocol shown in the inset (Figure 2A, n = 5). Figure 2B displays the time course of KIR2.3 current in a typical experiment in the absence and presence of 10 µM AG556 and AG556 plus orthovanadate. The current was reduced by 10 µM AG556, and this inhibition was reversed by 1 mM orthovanadate. The reduction of voltage-dependent KIR2.3 current by AG556 was also antagonized by orthovanadate (Figure 2C, n = 6). Figure 2D illustrates the I-V relationships of KIR2.3 current activated by a 3 s ramp (0 to −120 mV from a holding potential of −40 mV) during control, in the presence of AG556, AG556 plus orthovanadate, or Ba2+. The current was inhibited by 10 µM AG556, and the inhibition was antagonized by 1 mM orthovanadate. Ba2+ (0.5 mM) almost fully suppressed KIR2.3 current (n = 5). In a total of nine cells, AG556 (10 µM) decreased KIR2.3 current (at −120 mV) by 25% (P < 0.01 vs. control), and the inhibition was reversed by 1 mM orthovanadate to 3.1% (P < 0.01 vs. genistein alone) (Figure 2E). These results suggest that the inhibition of KIR2.3 current by genistein or AG556 is related to the inhibition of EGFR kinase.
Figure 2.
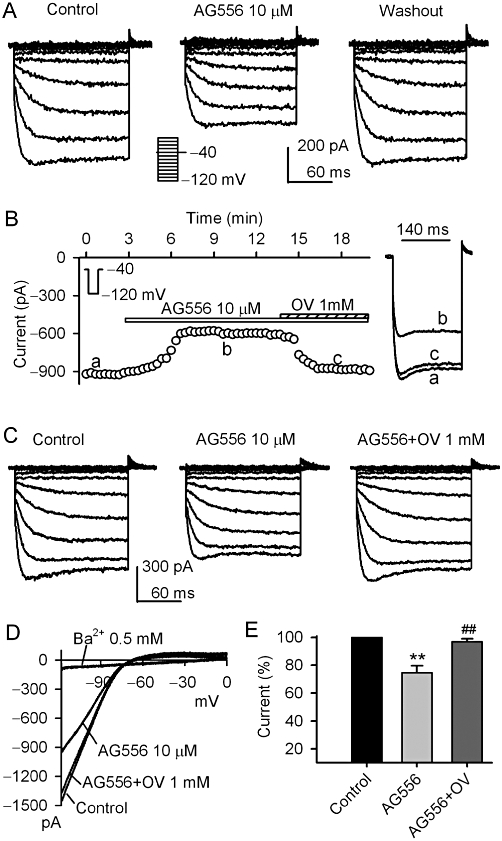
Effect of AG556 on KIR2.3 current. (A) Voltage-dependent KIR2.3 current recorded in a representative cell with the protocol shown in the inset in the absence and presence of 10 µM AG556. (B) Time-course of KIR2.3 current recorded in another cell in the absence and presence of 10 µM AG556, AG556 plus 1 mM orthovanadate (OV). Original current traces at corresponding time points are shown in right of the panel. (C) Voltage-dependent KIR2.3 current recorded in a representative cell during control, in the presence of 10 µM AG556 and AG556 plus 1 mM orthovanadate. D. I-V relationships of KIR2.3 current recorded by a ramp protocol in control, 10 µM AG556, AG556 plus 1 mM orthovanadate and 0.5 mM Ba2+. (E) Summary values of KIR2.3 current (−120 mV) as percent control, in cells before (control) or after, 10 µM AG556 and AG556 plus 1 mM orthovanadate (n = 8, **P < 0.01 vs. control, ##P < 0.01 vs. genistein alone).
Effects of EGF on KIR2.3 channels
To demonstrate whether EGF can up-regulate KIR2.3 current, 100 ng·mL−1 EGF was applied in bath solution. Figure 3A displays the time-course of KIR2.3 current recorded in a typical experiment using a voltage step from −40 to −120 mV (inset). The current was gradually increased after application of 100 ng·mL−1 EGF in the bath solution and the effect partially recovered on washout (n = 5). Figure 3B shows the voltage-dependent KIR2.3 current recorded in another cell with a voltage protocol shown in the inset in the absence and the presence of EGF. EGF (100 ng·mL−1) enhanced KIR2.3 current, and the effect partially recovered on washout (n = 7). This result suggests that the increase of Kir2.3 current by EGF is likely to be related to an enhanced tyrosine phosphorylation of the channel by EGFR kinase.
Figure 3.
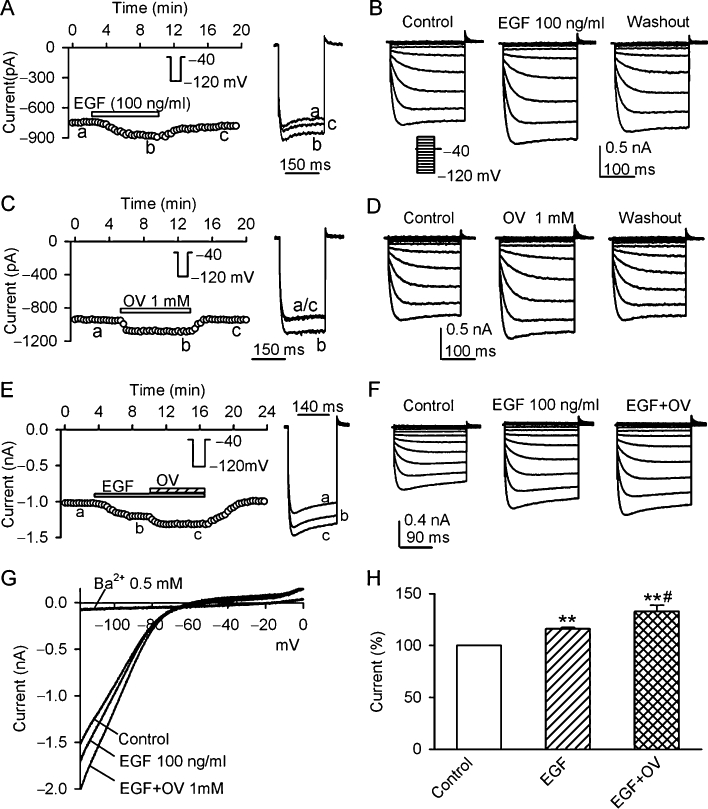
Effects of EGF and orthovanadate on KIR2.3 current. (A) Time-course of KIR2.3 current recorded in a representative cell with the voltage step as shown in the inset, in the absence and presence of 100 ng·mL−1 EGF. Original current traces at corresponding time points are shown in right of the panel. (B) Voltage-dependent KIR2.3 current traces recorded in a typical experiment with 200-ms voltage steps from −40 mV to between −120 and 0 mV under control conditions, in the presence of EGF (100 ng·mL−1) and washout. (C) Time-course of KIR2.3 current recorded in another cell with the voltage protocol shown in the inset, in the absence and presence of 1 mM orthovanadate (OV). Original current traces at corresponding time points are shown in right of the panel. (D) Voltage-dependent KIR2.3 current traces recorded with the voltage protocol as shown in the inset of panel B during control, in the presence of 1 mM orthovanadate and washout. (E) Time-course of KIR2.3 current recorded in a typical experiment in the absence and presence of EGF (100 ng·mL−1), EGF plus orthovanadate. Original current traces at corresponding time points are shown in right of the panel. (F) Voltage-dependent KIR2.3 current recorded in a representative cell during control, in the presence of 100 ng·mL−1 EGF and EGF plus 1 mM orthovanadate. (G). I-V relationships of KIR2.3 current recorded by a ramp protocol in control, EGF (100 ng·mL−1), EGF plus 1 mM orthovanadate and 0.5 Ba2+. (H) Summary values of KIR2.3 current, as percent control, in cells before (control) or after 100 ng·mL−1 EGF and EGF plus 1 mM orthovanadate. **P < 0.01 vs. control; #P < 0.01 vs. EGF alone (n = 6).
Effect of orthovanadate on KIR2.3 channels
If the enhancement of KIR2.3 current by EGF is mediated by the increased tyrosine phosphorylation of KIR2.3 channels, inhibition of PTPs might also increase the tyrosine phosphorylation and up-regulate the channel activity by reducing the dephosphorylation of the channel. Figure 3C shows the time-course of KIR2.3 current in another cell in the absence and presence of the PTP inhibitor orthovanadate (1 mM). Orthovanadate exhibited a reversible enhancement of KIR2.3 currents. The same effect was observed in voltage-dependent KIR2.3 current (Figure 3D, n = 6).
Figure 3E illustrates the time-course of KIR2.3 current recorded in a typical experiment, in which EGF (100 ng·mL−1) increased KIR2.3 current, and co-application of EGF with 1 mM orthovanadate produced an additional increase of the current. The same results were obtained in voltage-dependent KIR2.3 current (Figure 3F). Figure 3G shows the I–V relationships of KIR2.3 current activated by a ramp protocol during control, in the presence of 100 ng·mL−1 EGF, EGF plus 1 mM orthovanadate or 0.5 mM Ba2+. EGF (100 ng·mL−1) enhanced KIR2.3 current and this effect was further potentiated by co-application of EGF and 1 mM orthovanadate. Ba2+ almost fully inhibited KIR2.3 current Figure 3H summarizes the increases of KIR2.3 current at −120 mV with 100 ng·mL−1 EGF and EGF plus 1 mM orthovanadate. These results indicate that KIR2.3 current is up-regulated by tyrosine phosphorylation of the channel via activating EGFR kinase or inhibiting PTPs.
Interaction of EGF and orthovanadate with AG556
The increase of voltage-dependent KIR2.3 current by 100 EGF (Figure 4A) or 1 mM orthovanadate (Figure 4B) was fully antagonized by 10 µM AG556. Summary data from these experiments are shown in Figure 4C. These results suggest that KIR2.3 channels may be up-regulated by the increase of tyrosine phosphorylation via either activating EGFR kinase, as with EGF, by or reducing dephosphorylation by inhibiting PTPs, as with orthovanadate.
Figure 4.
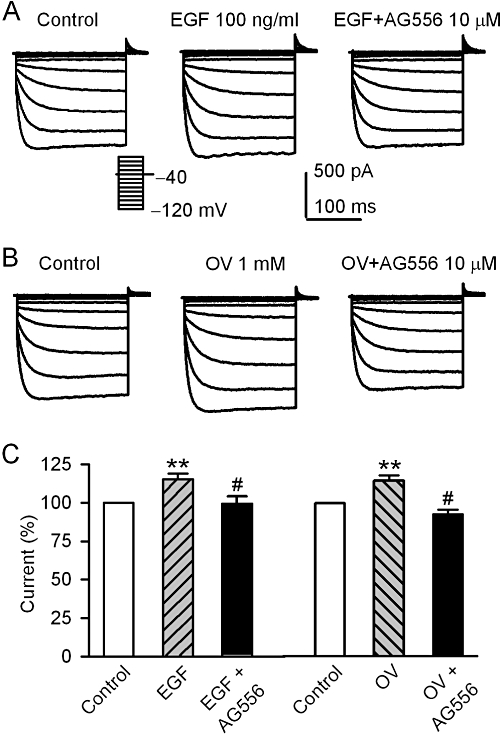
AG556 and EGF or orthovanadate effect on hKIR2.3 current. (A) Voltage-dependent KIR2.3 current recorded with the protocol as shown in the inset in a typical cell during control, in the presence of 100 ng·mL−1 EGF, EGF plus 10 µM AG556. (B) Voltage-dependent KIR2.3 current recorded in a representative cell during control, in the presence of 1 mM orthovanadate (OV), orthovanadate plus 10 µM AG556. (C) Summary values of KIR2.3 current, as percent control, in cells before (control) or after 100 ng·mL−1 EGF, EGF plus 10 µM AG556, 1 mM orthovanadate and orthovanadate plus 10 µM AG556. **P < 0.01 vs. control; #P < 0.01 vs. EGF alone or orthovanadate alone (n = 6).
Other PTK inhibitors on KIR2.3 current
Tyrphostin AG1295 (10 µM), a selective inhibitor of the platelet-derived growth factor receptor (PDGFR) kinase, and PP2 (10 µM), a membrane-permeable inhibitor of the Src-family PTKs, had no inhibitory effect on KIR2.3 current (Figure 5). These results suggest that the PDGFR kinase and Src-family kinases are not involved in the regulation of the KIR2.3 channels.
Figure 5.
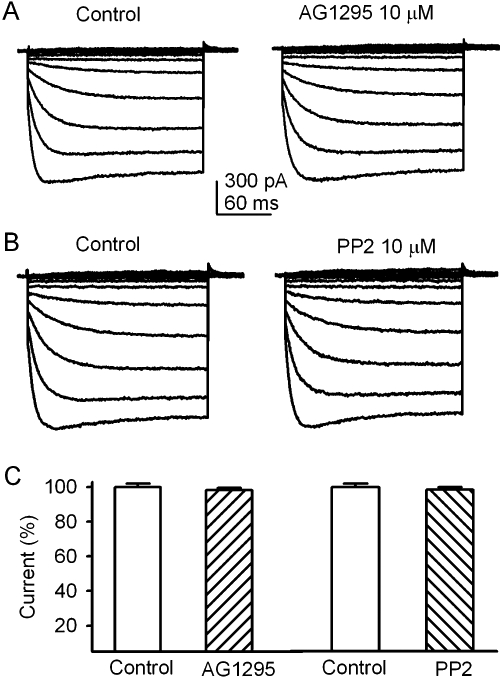
AG1295 and PP2 had no effect on KIR2.3 channels. (A) KIR2.3 current traces recorded with the voltage protocol as shown in inset of Figure 4A in a representative cell before (control) and after addition of 10 µM AG1295. (B) KIR2.3 current traces recorded in a typical experiment before (control) and after addition of 10 µM PP2. (C) Summary values of KIR2.3 current (−120 mV) as percent control, in cells before (control) or after 10 µM AG1295 (n = 5) or 10 µM PP2 (n = 5).
Tyrosine phosphorylation level of KIR2.3 channels
To demonstrate the tyrosine phosphorylation level of KIR2.3 channels, immunoprecipitation and Western blot analysis were employed in cells pre-treated with the agents already studied. EGF (100 ng·mL−1) or orthovanadate (1 mM) increased tyrosine phosphorylation of KIR2.3 channels, whereas genistein (10 µM) and AG556 (10 µM) reduced the level of phosphorylation. This reduction was substantially reversed by pre-treatment with 100 ng·mL−1 EGF or 1 mM orthovanadate (Figure 6A). The quantitative tyrosine phosphorylation of KIR2.3 channels after different treatments are summarised in Figure 6B. Phosphorylation of KIR2.3 channels was increased by 1 mM orthovanadate or 100 ng·mL−1 EGF (n = 4, P < 0.01 vs. control), and decreased by 10 µM genistein or 10 µM AG556 (P < 0.01). Orthovanadate and EGF reversed the decrease in phosphorylation by genistein (P < 0.01 vs. genistein alone) or by AG556 (P < 0.01 vs. AG556 alone). These results indicated that the KIR2.3 channel protein is phosphorylated by interaction of EGFR tyrosine kinase and PTPs.
Figure 6.
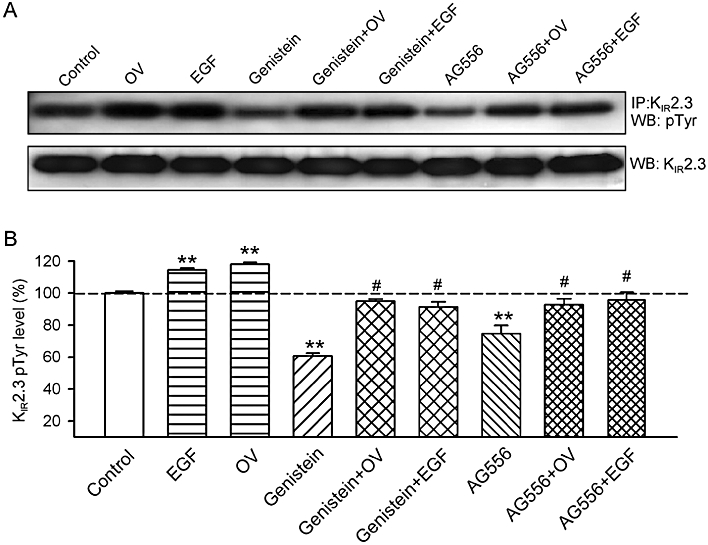
Tyrosine phosphorylation levels of KIR2.3 channels. (A) Upper panel, protein lysates were immunoprecipitated with anti-KIR2.3 antibody, and then Western blots were prepared and probed with the anti-phosphotyrosine (Tyr(P)) antibody. Lower panel, Western blots of cell lysates, probed with the anti-KIR2.3 channel antibody. (B) Histogram summarizes the mean values of the relative levels of tyrosine-phosphorylated KIR2.3 channel protein analysed by densitometry. The amount of protein from the immunoprecipitation (as in A) was normalized to those from the Western blots. Phosphorylated protein levels (n = 5 experiments) are expressed as a percentage of the vehicle control. **P < 0.01, *P < 0.05 vs. vehicle control, #P < 0.05 vs. genistein or AG556 alone.
PTK phosphorylation sites of KIR2.3 channels
To detect the potential tyrosine phosphorylation site(s) of KIR2.3 channels, three mutants (Y234A, Y329A and Y333A) were generated using a site-directed mutagenesis technique and their responses to EGF or the EGFR kinase inhibitor AG556 were examined. The currents recorded with a 200-ms voltage step to −110 from −40 mV in HEK 293 cells transfected with corresponding genes are illustrated in Figure 7. The mutant Y234A channel was not affected by 100 ng·mL−1 EGF, unlike the WT KIR2.3 channels or the other mutant channels Y329A and Y333A (Figure 7A; summary data in Figure 7B). The selective EGFR kinase inhibitor AG556 reduced the current amplitude of WT KIR2.3, Y329A and Y333A mutant channels, but not that carried by the Y234A mutant channel (Figure 7C, 7D).
Figure 7.
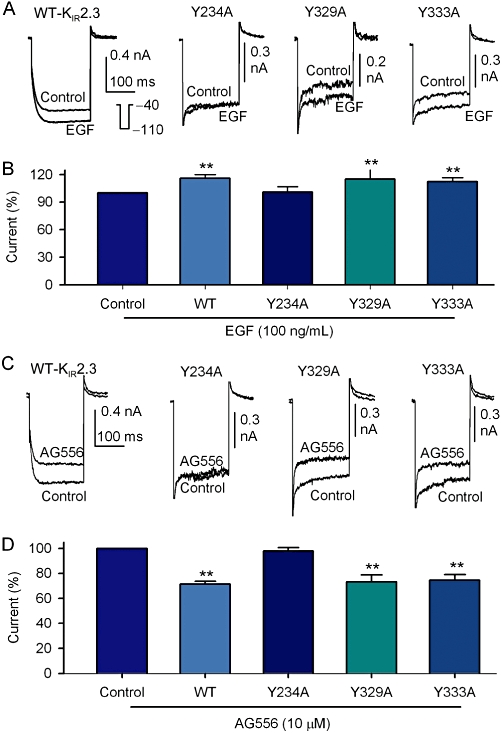
Single point mutations of KIR2.3 channels. (A) KIR2.3 current recorded in HEK 293 cells expressing WT Kir2.3, Y234A, Y329A and Y333A mutants with a 200-ms voltage step to −110 from −40 mV (inset) before (control) and after application of 100 ng·mL−1 EGF (5 min). (B) Summary values of KIR2.3 current, as percent control, in cells expressing WT (n = 13), Y234A (n = 5), Y329A (n = 5) and Y339A (n = 13) mutant channels in response to 100 ng·mL−1 EGF. (C) KIR2.3 current recorded in HEK 293 cells expressing WT Kir2.3, Y234A, Y329A and Y333A mutants with voltage protocol shown in the inset of panel A before (control) and after application of 10 µM AG556 (5 min). (D) Summary values of KIR2.3 current, as percent control, in cells expressing WT (n = 6), Y234A (n = 5), Y329A (n = 5) and Y339A (n = 5) mutants in response to 10 µM AG556, **P < 0.01 vs. control or Y234A mutant.
Tyrosine phosphorylation level of the mutant Y234A channel protein
To further confirm whether the protein tyrosine phosphorylation of KIR2.3 channels was at the site of Y234, we determined the tyrosine phosphorylation of the mutant Y234A protein in HEK 293 cells, stably expressing the mutant gene (Figure 8A and B). Phosphorylation of the Y234A channel, in response to EGF and/or orthovanadate, was almost totally lost, whereas the WT KIR2.3 channels and the mutants Y329A and Y333A showed the expected increased phosphorylation with EGF or orthovanadate. These results indicate that the function of the KIR2.3 channel protein is regulated by the interplay of EGFR kinase and PTPs via tyrosine phosphorylation at the Y234 residue, as illustrated by Figure 8C.
Figure 8.
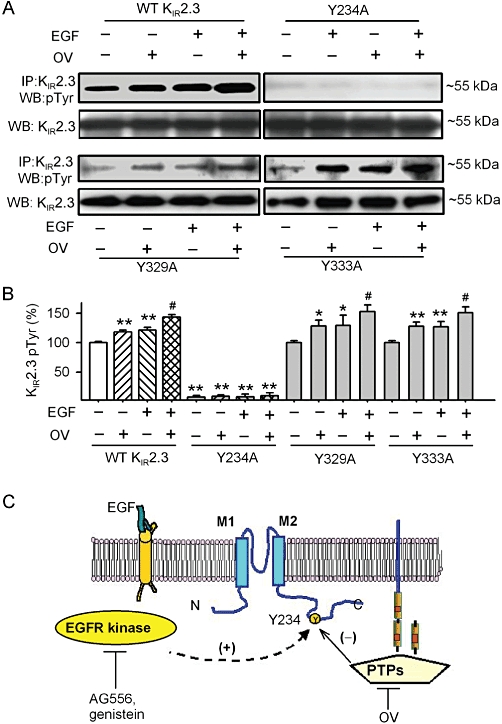
Tyrosine phosphorylation of the mutant Y234A channels and a model scheme for the regulation of KIR2.3 channels by EGFR tyrosine kinase and PTPs. (A) Tyrosine phosphorylation of KIR2.3 channels was detected with immunoprecipitation and Western blot in WT and mutant KIR2.3 channels. (B) Histogram summarizes the mean values of the relative levels of tyrosine phosphorylated KIR2.3 channel protein analysed by densitometry. The amount of protein from the immunoprecipitation (as in A) was normalized to those from the Western blots. Relative phosphorylated protein level (n = 4 experiments) is expressed as a percentage of the vehicle control. *P < 0.05, **P < 0.01 vs. vehicle control (without EGF or orthovanadate) in WT KIR2.3; #P < 0.05 vs. EGF or OV. (C) Tyrosine phosphorylation site on KIR2.3 channels and signalling molecules are postulated. The tyrosine of the KIR2.3 protein is phosphorylated by EGFR kinase and dephosphorylated by PTPs. Tyrosine phosphorylation at Y234 by one or more steps (dashed line) favours KIR2.3 channel opening, whereas dephosphorylation favors closure. AG556 or genistein inhibited EGFR tyrosine kinase and reduced phosphorylation, while orthovanadate (OV) inhibited PTPs and decreased dephosphorylation of the channel.
Discussion
It is well recognized that the activation of ion channels is regulated by tonic activity of several protein kinases and phosphatases to catalyse reversible phosphorylation and dephosphorylation events (Levitan, 1994). We and others have previously demonstrated that PTKs regulate several types of ion channels (Davis et al., 2001; Du et al., 2004b; Gao et al., 2004; Zhang et al., 2008), including L-type Ca2+ channel (Ogura et al., 1999), voltage-gated Na+ channel (Ashen et al., 1995; Liu et al., 2007), volume-sensitive Cl- channel (Du et al., 2004b) and cardiac K+ channels (Gao et al., 2004; Missan et al., 2006; Zhang et al., 2008; Dong et al., 2010) in different types of cells. The present study has demonstrated that the KIR2.3 channels are regulated by the EGFR tyrosine kinase. Although a recent study reported that genistein showed a non-specific inhibitory effect on rat KIR2.3 channels expressed in Xenopus oocytes and HEK 293 cells with an IC50 of 16.9–19.4 µM (Zhao et al., 2008), which is close to that in the present study (IC50: 14.3 µM, n = 5, data not shown), the results from the present study strongly support the notion that genistein at a low concentration of 10 µM reduced tyrosine phosphorylation of KIR2.3 channels, because the reduction of KIR2.3 current by 10 µM genistein, like that after the selective EGFR kinase inhibitor, AG556 (10 µM), was almost fully reversed by the PTP inhibitor, orthovanadate (Figure 1). The IC50 of AG556 for maximum inhibition of KIR2.3 currents was 8.9 µM in the present observation (n = 5, data not shown).
On the other hand, an earlier study reported that rat KIR2.1 channels were inhibited by the PTP inhibitor perorthovanadate or the PTK activator EGF or insulin (Wischmeyer et al., 1998). In addition, KIR2.1 channels expressed in Xenopus oocytes were reduced by tyrosine (v-Src) decaging, which is related to the interaction of clathrin adaptor protein and tyrosine-based sorting motif of KIR2.1 and also membrane endotocytosis (Tong et al., 2001). Moreover, Du and colleagues reported that the recombinant KIR channels including KIR2.1 and KIR2.3 channels co-expressed with muscarinic M1 and EGF receptors in Xenopus oocytes were up-regulated by phosphatidylinositol 4,5-bisphosphate and inhibited by acetylcholine or EGF (Du et al., 2004a). However, this contrasts with our present observations, in which EGFR kinase activation up-regulated, while EGFR kinase inhibition decreased KIR2.3 currents expressed in HEK293 cells, via direct tyrosine phosphorylation of the channel. It is unclear whether these differences result from the different expression system or subtle variation of channel types and/or different experimental conditions.
The important finding of the present observation was that KIR2.3 current was reversibly augmented by either activating EGFR kinase with EGF or inhibiting PTPs with orthovanadate, and the EGF-induced activation of KIR2.3 channels was potentiated by orthovanadate (Figure 3). The increase of KIR2.3 current by EGF or orthovanadate was fully reversed by the EGFR kinase inhibitor AG556 (Figure 4). These results strongly support the notion that KIR2.3 channels are regulated by the interplay of EGFR kinase and PTPs. However, we did not find inhibition (Figure 5) of KIR2.3 current by the PDGFR kinase inhibitor AG1295 (10 µM, 20 times higher than IC50 for inhibiting PDGFR tyrosine kinase) (Naraghi et al., 2000; Zhang et al., 2008) or the inhibitor of Src-family kinases PP2 (10 µM, 50 times higher than IC50 for inhibiting Src-kinases) (Zhu et al., 1999b), indicating that these tyrosine kinases are not involved in the regulation of KIR2.3 channels.
The simplest explanation for the actions of genistein, AG556, EGF, and orthovanadate and their interaction is that phosphorylation and/or dephosphorylation of a critical tyrosine residue (Y234) on KIR2.3 channels or a signalling molecule, directly or indirectly, increases or reduces the channel activity. In this model (Figure 8C), activation of EGFR kinase by EGF increases KIR2.3 channel activity by enhancing tyrosine phosphorylation of the channel, whereas inhibition of EGFR kinase by AG556 or genistein decreases the channel activity by reducing tyrosine phosphorylation. In addition, inhibition of PTPs with orthovanadate increases KIR2.3 channel activity by allowing its unopposed phosphorylation. This may also explain why orthovanadate antagonized the effect of AG556 or genistein. Moreover, when the Y234 residue of the KIR2.3 channel was replaced by alanine, the mutant channel was no longer sensitive to stimulation by EGF or inhibition by the EGFR kinase inhibitor AG556 (Figure 7).
The regulation of KIR2.3 channels by the interplay of EGFR kinase and PTPs is further supported by the results from immunoprecipitation and Western blot analysis. Tyrosine phosphorylation of KIR2.3 channel protein was enhanced by EGF or orthovanadate, while being reduced by genistein or AG556. The reduced phosphorylation of KIR2.3 channels by genistein or AG556 was reversed by EGF or orthovanadate (Figure 6). The strongest evidence was the dramatic reduction of tyrosine phosphorylation and the lack of response to EGF and/or orthovanadate in the mutant Y234A channel, but not in the other mutant channels, Y329A and Y333A (Figure 8A and B).
Alterations in the electrical properties of the KIR2.3 mutant channels were observed, leading to a rapid onset of the currents with voltage changes and a fast inactivation component (Figure 7A and C). These alterations in the mutants is more likely to be related to channel general function, rather than a specific action on tyrosine phosphorylation, because the mutant Y329A and Y333A channels exhibited the same electrical responses to the EGFR kinase inhibitor AG556 and the activator EGF (Figure 7), and also similar phosphorylation responses to those of the WT KIR2.3 channels (Figure 8).
A number of endogenous signalling molecules, including ATP (Collins et al., 1996), protein kinase C (Zhu et al., 1999a), G-protein βγ subunits (Cohen et al., 1996) and protons (Coulter et al., 1995; Munoz et al., 2007) have been reported to inhibit KIR2.3 channels. The present study provides the novel evidence that EGFR kinase activated the KIR2.3 channels. Because KIR channels play a critical role in setting the resting membrane potential and firing pattern in a variety of cell types (Murata et al., 2001; Bichet et al., 2003), activation of KIR2.3 channels may be important for decreasing cellular excitability, and this is most likely to be of particular significance in tissues of the brain and heart where KIR2.3 channels are abundantly expressed (Perier et al., 1994).
Collectively, the present study has provided the novel evidence that KIR2.3 channels are regulated by the interaction of EGFR kinase (but not PDGFR kinase and Src-family kinases) and PTPs. PTPs negatively, while EGFR kinase positively, modulate these channel stably expressed in HEK 293 cells via phosphorylation of the Y234 residue of the channel. Such effects may be involved in the endogenous regulation of cellular electrical activity.
Acknowledgments
This work was supported in part by a General Research Fund (HKU 760306 M) from Research Grant Council of Hong Kong and a grant from Sun Chieh Yeh Heart Foundation of Hong Kong. The authors greatly appreciate the generous offer of pcDNA3.1/hKIR2.3 plasmid for the present study by Dr Carol A Vandenberg, University of California Santa Barbara. De-Yong Zhang and Yan-Hui Zhang are supported by postgraduate studentships from the University of Hong Kong.
Glossary
Abbreviations
- EGF
epidermal growth factor
- EGFR
epidermal growth factor receptor
- PDGFR
platelet-derived growth factor receptor
- PTKs
protein tyrosine kinases
- PTPs
protein tyrosine phosphatases
Conflict of interest
None is declared.
References
- Ahern CA, Zhang JF, Wookalis MJ, Horn R. Modulation of the cardiac sodium channel NaV1.5 by Fyn, a Src family tyrosine kinase. Circ Res. 2005;96:991–998. doi: 10.1161/01.RES.0000166324.00524.dd. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edn. Br J Pharmacol. 2009;158(Suppl 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashen MD, O'Rourke B, Kluge KA, Johns DC, Tomaselli GF. Inward rectifier K+ channel from human heart and brain: cloning and stable expression in a human cell line. Am J Physiol. 1995;268:H506–H511. doi: 10.1152/ajpheart.1995.268.1.H506. [DOI] [PubMed] [Google Scholar]
- Bichet D, Haass FA, Jan LY. Merging functional studies with structures of inward-rectifier K(+) channels. Nat Rev Neurosci. 2003;4:957–967. doi: 10.1038/nrn1244. [DOI] [PubMed] [Google Scholar]
- Cohen NA, Sha Q, Makhina EN, Lopatin AN, Linder ME, Snyder SH, et al. Inhibition of an inward rectifier potassium channel (Kir2.3) by G-protein beta gamma subunits. J Biol Chem. 1996;271:32301–32305. doi: 10.1074/jbc.271.50.32301. [DOI] [PubMed] [Google Scholar]
- Collins A, German MS, Jan YN, Jan LY, Zhao B. A strongly inwardly rectifying K+ channel that is sensitive to ATP. J Neurosci. 1996;16:1–9. doi: 10.1523/JNEUROSCI.16-01-00001.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulter KL, Pe'rier F, Redeke CM, Vandenberg CA. Identification and molecular localization of a pH-sensing domain for the inward rectifier potassium channel HIR. Neuron. 1995;15:1157–1168. doi: 10.1016/0896-6273(95)90103-5. [DOI] [PubMed] [Google Scholar]
- Davis MJ, Wu X, Nurkiewicz TR, Kawasaki J, Gui P, Hill MA, et al. Regulation of ion channels by protein tyrosine phosphorylation. Am J Physiol Heart Circ Physiol. 2001;281:H1835–H1862. doi: 10.1152/ajpheart.2001.281.5.H1835. [DOI] [PubMed] [Google Scholar]
- Dong MQ, Sun HY, Tang Q, Tse HF, Lau CP, Li GR. Regulation of human cardiac KCNQ1/KCNE1 channel by epidermal growth factor receptor kinase. Biochim Biophys Acta. 2010;1798:995–1001. doi: 10.1016/j.bbamem.2010.01.010. [DOI] [PubMed] [Google Scholar]
- Du X, Zhang H, Lopes C, Mirshahi T, Rohacs T, Logothetis DE. Characteristic interactions with phosphatidylinositol 4,5-bisphosphate determine regulation of Kir channels by diverse modulators. J Biol Chem. 2004a;279:37271–37281. doi: 10.1074/jbc.M403413200. [DOI] [PubMed] [Google Scholar]
- Du XL, Gao Z, Lau CP, Chiu SW, Tse HF, Baumgarten CM, et al. Differential effects of tyrosine kinase inhibitors on volume-sensitive chloride current in human atrial myocytes: evidence for dual regulation by Src and EGFR kinases. J Gen Physiol. 2004b;123:427–439. doi: 10.1085/jgp.200409013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z, Lau CP, Wong TM, Li GR. Protein tyrosine kinase-dependent modulation of voltage-dependent potassium channels by genistein in rat cardiac ventricular myocytes. Cell Signal. 2004;16:333–341. doi: 10.1016/j.cellsig.2003.08.003. [DOI] [PubMed] [Google Scholar]
- Hunter T. Signaling – 2000 and beyond. Cell. 2000;100:113–127. doi: 10.1016/s0092-8674(00)81688-8. [DOI] [PubMed] [Google Scholar]
- Inagaki N, Tsuura Y, Namba N, Masuda K, Gonoi T, Horie M, et al. Cloning and functional characterization of a novel ATP-sensitive potassium channel ubiquitously expressed in rat tissues, including pancreatic islets, pituitary, skeletal muscle, and heart. J Biol Chem. 1995;270:5691–5694. doi: 10.1074/jbc.270.11.5691. [DOI] [PubMed] [Google Scholar]
- Levitan IB. Modulation of ion channels by protein phosphorylation and dephosphorylation. Annu Rev Physiol. 1994;56:193–212. doi: 10.1146/annurev.ph.56.030194.001205. [DOI] [PubMed] [Google Scholar]
- Li GR, Dong MQ. Pharmacology of cardiac potassium channels. Adv Pharmacol. 2010;59:93–134. doi: 10.1016/S1054-3589(10)59004-5. [DOI] [PubMed] [Google Scholar]
- Liu H, Sun HY, Lau CP, Li GR. Regulation of voltage-gated cardiac sodium current by epidermal growth factor receptor kinase in guinea pig ventricular myocytes. J Mol Cell Cardiol. 2007;42:760–768. doi: 10.1016/j.yjmcc.2006.10.013. [DOI] [PubMed] [Google Scholar]
- Makhina EN, Kelly AJ, Lopatin AN, Mercer RW, Nichols CG. Cloning and expression of a novel human brain inward rectifier potassium channel. J Biol Chem. 1994;269:20468–20474. [PubMed] [Google Scholar]
- Missan S, Linsdell P, McDonald TF. Tyrosine kinase and phosphatase regulation of slow delayed-rectifier K+ current in guinea-pig ventricular myocytes. J Physiol. 2006;573:469–482. doi: 10.1113/jphysiol.2005.104422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishige K, Takahashi N, Jahangir A, Yamada M, Koyama H, Zanelli JS, et al. Molecular cloning and functional expression of a novel brain-specific inward rectifier potassium channel. FEBS Lett. 1994;346:251–256. doi: 10.1016/0014-5793(94)00483-8. [DOI] [PubMed] [Google Scholar]
- Munoz V, Vaidyanathan R, Tolkacheva EG, Dhamoon AS, Taffet SM, Anumonwo JM. Kir2.3 isoform confers pH sensitivity to heteromeric Kir2.1/Kir2.3 channels in HEK293 cells. Heart Rhythm. 2007;4:487–496. doi: 10.1016/j.hrthm.2006.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata Y, Okado H, Katsuyama Y, Okamura Y, Kubo Y. Primary structure, developmental expression and functional properties of an inward rectifier K+ channel of the tunicate. Receptors Channels. 2001;7:387–399. [PubMed] [Google Scholar]
- Naraghi S, Khoshyomn S, DeMattia JA, Vane DW. Receptor tyrosine kinase inhibition suppresses growth of pediatric renal tumor cells in vitro. J Pediatr Surg. 2000;35:884–890. doi: 10.1053/jpsu.2000.6907. [DOI] [PubMed] [Google Scholar]
- Ogura T, Shuba LM, McDonald TF. L-type Ca2+ current in guinea pig ventricular myocytes treated with modulators of tyrosine phosphorylation. Am J Physiol. 1999;276:H1724–H1733. doi: 10.1152/ajpheart.1999.276.5.H1724. [DOI] [PubMed] [Google Scholar]
- Perier F, Radeke CM, Vandenberg CA. Primary structure and characterization of a small-conductance inwardly rectifying potassium channel from human hippocampus. Proc Natl Acad Sci USA. 1994;91:6240–6244. doi: 10.1073/pnas.91.13.6240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raab-Graham KF, Radeke CM, Vandenberg CA. Molecular cloning and expression of a human heart inward rectifier potassium channel. Neuroreport. 1994;5:2501–2505. doi: 10.1097/00001756-199412000-00024. [DOI] [PubMed] [Google Scholar]
- Tang Q, Jin MW, Xiang JZ, Dong MQ, Sun HY, Lau CP, et al. The membrane permeable calcium chelator BAPTA-AM directly blocks human ether a-go-go-related gene potassium channels stably expressed in HEK 293 cells. Biochem Pharmacol. 2007;74:1596–1607. doi: 10.1016/j.bcp.2007.07.042. [DOI] [PubMed] [Google Scholar]
- Tiran Z, Peretz A, Attali B, Elson A. Phosphorylation-dependent regulation of Kv2.1 Channel activity at tyrosine 124 by Src and by protein-tyrosine phosphatase epsilon. J Biol Chem. 2003;278:17509–17514. [Google Scholar]
- Tong Y, Brandt GS, Li M, Shapovalov G, Slimko E, Karschin A, et al. Tyrosine decaging leads to substantial membrane trafficking during modulation of an inward rectifier potassium channel. J Gen Physiol. 2001;117:103–118. doi: 10.1085/jgp.117.2.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wischmeyer E, Doring F, Karschin A. Acute suppression of inwardly rectifying Kir2.1 channels by direct tyrosine kinase phosphorylation. J Biol Chem. 1998;273:34063–34068. doi: 10.1074/jbc.273.51.34063. [DOI] [PubMed] [Google Scholar]
- Zhang DY, Lau CP, Wang Y, Tse HF, Li GR. Both EGFR kinase and Src-related tyrosine kinases regulate human ether-a-go-go-related gene potassium channels. Cell Signal. 2008;20:1815–1821. doi: 10.1016/j.cellsig.2008.06.006. [DOI] [PubMed] [Google Scholar]
- Zhang DY, Lau CP, Li GR. Human Kir2.1 channel carries a transient outward potassium current with inward rectification. Pflugers Arch. 2009;457:1275–1285. doi: 10.1007/s00424-008-0608-0. [DOI] [PubMed] [Google Scholar]
- Zhao Z, Liu B, Zhang G, Jia Z, Jia Q, Geng X, et al. Molecular basis for genistein-induced inhibition of Kir2.3 currents. Pflugers Arch. 2008;456:413–423. doi: 10.1007/s00424-007-0391-3. [DOI] [PubMed] [Google Scholar]
- Zhu G, Qu Z, Cui N, Jiang C. Suppression of Kir2.3 activity by protein kinase C phosphorylation of the channel protein at threonine 53. J Biol Chem. 1999a;274:11643–11646. doi: 10.1074/jbc.274.17.11643. [DOI] [PubMed] [Google Scholar]
- Zhu X, Kim JL, Newcomb JR, Rose PE, Stover DR, Toledo LM, et al. Structural analysis of the lymphocyte-specific kinase Lck in complex with non-selective and Src family selective kinase inhibitors. Structure. 1999b;7:651–661. doi: 10.1016/s0969-2126(99)80086-0. [DOI] [PubMed] [Google Scholar]