

Abstract
BACKGROUND AND PURPOSE
P2Y1, P2Y2, P2Y4, P2Y12 and P2Y13 receptors for nucleotides have been reported to mediate presynaptic inhibition, but unequivocal evidence for facilitatory presynaptic P2Y receptors is not available. The search for such receptors was the purpose of this study.
EXPERIMENTAL APPROACH
In primary cultures of rat superior cervical ganglion neurons and in PC12 cell cultures, currents were recorded via the perforated patch clamp technique, and the release of [3H]-noradrenaline was determined.
KEY RESULTS
ADP, 2-methylthio-ATP and ATP enhanced stimulation-evoked 3H overflow from superior cervical ganglion neurons, treated with pertussis toxin to prevent the signalling of inhibitory G proteins. This effect was abolished by P2Y1 antagonists and by inhibition of phospholipase C, but not by inhibition of protein kinase C or depletion of intracellular Ca2+ stores. ADP and a specific P2Y1 agonist caused inhibition of Kv7 channels, and this was prevented by a respective antagonist. In neurons not treated with pertussis toxin, 3H overflow was also enhanced by a specific P2Y1 agonist and by ADP, but only when the P2Y12 receptors were blocked. ADP also enhanced K+-evoked 3H overflow from PC12 cells treated with pertussis toxin, but only in a clone expressing recombinant P2Y1 receptors.
CONCLUSIONS AND IMPLICATIONS
These results demonstrate that presynaptic P2Y1 receptors mediate facilitation of transmitter release from sympathetic neurons most likely through inhibition of Kv7 channels.
Keywords: P2Y1 receptors, noradrenaline release, presynaptic facilitation, phospholipase C, Kv7 channels
Introduction
ATP is released together with noradrenaline from sympathetic nerve terminals and contributes to the sympatho-effector transmission (von Kugelgen and Starke, 1991). This neurotransmission is tightly controlled by a large number of different presynaptic receptors, which also include autoreceptors for noradrenaline as well as nucleotides (Boehm and Kubista, 2002). Amongst the members of the adrenoceptor family, presynaptic α2A and α2C receptors mediate autoinhibition of noradrenaline release, whereas β2 receptors mediate facilitation (Boehm and Kubista, 2002). Within the family of P2 receptors, ionotropic P2X receptors mediate facilitation of transmitter release, whereas metabotropic P2Y receptors were found to mediate inhibition only (Sperlagh et al., 2007; Dorostkar and Boehm, 2008; Goncalves and Queiroz, 2008). In this respect, the family of P2Y receptors appear to differ from other GPCRs: most neurotransmitters and/or mediators, such as acetylcholine, adenosine, histamine, noradrenaline and prostaglandins are known to cause presynaptic inhibition as well as facilitation of sympathetic transmitter release, the two opposing actions being mediated by two different GPCRs (Boehm and Kubista, 2002). In general, presynaptic GPCRs linked to Gs or Gq type G-proteins mediate facilitation of noradrenaline release, whereas receptors linked to Gi/o-proteins mediate inhibition, although there are exceptions to this rule (Kubista and Boehm, 2006).
Within the family of G-protein-coupled P2Y receptors, at least eight different subtypes have been identified (P2Y1, P2Y2, P2Y4, P2Y6, P2Y11, P2Y12, P2Y13, P2Y14). In heterologous expression systems, all P2Y receptor subtypes, with the exception of P2Y12, couple to PLC via Gq and mediate increases in inositol phosphates; via Gi/o, P2Y12, P2Y13 and P2Y14 mediate inhibition, while P2Y11 mediates activation of adenylyl cyclases (Abbracchio et al., 2006; Burnstock, 2006). Accordingly, one might expect all P2Y but P2Y12 receptors to act as facilitatory presynaptic P2Y receptors. However, with respect to the sub-classification of presynaptic P2Y receptors, the information available is limited, as most subtype selective ligands have been developed quite recently (Jacobson and Boeynaems, 2010). In fact, P2Y1, P2Y2, P2Y4, P2Y12 and P2Y13 receptors have all been implicated in the presynaptic inhibition of transmitter release, but unequivocal evidence for facilitatory presynaptic P2Y receptors is lacking (Goncalves and Queiroz, 2008).
In sympathetically innervated tissues, such as the rat vas deferens, ADP and 2-methylthio-ADP inhibit [3H]-noradrenaline release, and this is prevented by 2-methylthio-AMP, an antagonist at P2Y12 and P2Y13 receptors (Queiroz et al., 2003) Likewise, in PC12 cells and rat superior cervical ganglion (SCG) neurons, inhibitory presynaptic P2Y receptors were blocked by 2-methylthio-AMP and by the P2Y12 antagonist cangrelor (Kulick & von Kugelgen, 2002; Lechner et al., 2004). More recently, P2Y12 together with P2Y1 receptors were shown to mediate autoinhibition in sympathetically innervated tissues (Quintas et al., 2009). Along the same line, evidence has been presented that P2Y1, P2Y12 and P2Y13 receptors mediate inhibition of noradrenaline release in the central nervous system (Csolle et al., 2008; Heinrich et al., 2008). For this study, rat SCG neurons in primary cell culture as well as PC12 cells were used to search for facilitatory presynaptic P2Y receptors. The results reveal that activation of presynaptic P2Y1 receptors leads to an increase in sympathetic transmitter release through activation of PLC.
Methods
Cell cultures
Primary cultures of dissociated SCG neurons from neonatal rats were prepared as described previously (Boehm, 1999). Newborn Sprague–Dawley rats were kept and killed 3 to 10 days after birth by decapitation in full accordance with all rules of the Austrian animal protection law and the Austrian animal experiment bylaws. Ganglia were removed immediately after decapitation of the animals, cut into three to four pieces and incubated in collagenase (1.5 mg·mL−1; Sigma, Vienna, Austria) and dispase (3.0 mg·mL−1; Boehringer Mannheim, Vienna, Austria) for 20 min at 36°C. Subsequently, they were further incubated in trypsin (0.25% trypsin; Worthington, Lakewood, NJ) for 15 min at 36°C, dissociated by trituration and resuspended in Dulbeccos modified Eagle's Medium (Invitrogen, Lofer, Austria) containing 2.2 g·L−1 glucose, 10 mg·L−1 insulin, 25 000 IU·L−1 penicillin and 25 mg·L−1 streptomycin (Invitrogen), 50 µg·L−1 nerve growth factor (R&D Systems Inc., Minneapolis, MN) and 5% fetal calf serum (Invitrogen). Finally, all cells were seeded onto 5 mm plastic discs for radiotracer release experiments and onto 35 mm culture dishes for electrophysiological experiments. The cultures were stored for 4 to 8 days in a humidified 5% CO2 atmosphere at 36°C. On days 1 and 4 after dissociation, the medium was exchanged entirely.
PC12 cells were obtained from the European Collection of Cell Cultures (ECACC; Salisbury, UK) and kept in OptiMEM (Life Technologies, Vienna, Austria) supplemented with 0.2 mM L-glutamine (HyClone, Aalst, Belgium), 25 000 IU·L−1 penicillin and 25 mg·L−1 streptomycin (Sigma), 5% fetal calf serum and 10% horse serum (both Life Technologies). Once per week, cell cultures were split, and the medium was exchanged twice weekly. To investigate the release of previously incorporated [3H]-noradrenaline under continuous superfusion, PC12 cells were plated onto 5 mm discs, as described for the SCG neurons above. All tissue culture plastic was coated with rat tail collagen (Biomedical Technologies Inc., Stoughton, MA, USA).
PC12 cell clones stably expressing the rat P2Y1 receptor linked to the green fluorescent protein (P2Y1-GFP) were generated as described previously; here, cells of clone 8 were used (Moskvina et al., 2003).
Determination of [3H]-noradrenaline release
[3H]-noradrenaline uptake and superfusion of SCG neurons and PC12 cells was performed as described previously (Lechner et al., 2004). The plastic discs with dissociated neurons or PC12 cells were incubated in 0.05 µM [3H]-noradrenaline ((–)-[ring-2,5,6-3H]-noradrenaline, specific activity 1.369 TBq·mmol−1; Perkin Elmer, Vienna, Austria) in culture medium supplemented with 1 mM ascorbic acid at 37°C for 1 h. Thereafter, the culture discs were transferred to small chambers and superfused with a buffer containing (mM) NaCl (120), KCl (3.0), CaCl2 (2.0), MgCl2 (2.0), glucose (20), HEPES (10), fumaric acid (0.5), Na-pyruvate (5.0), ascorbic acid (0.57) and desipramine (0.001), adjusted to pH 7.4 with NaOH. Superfusion was performed at 25°C at a rate of about 1.0 mL·min−1. Collection of 4 min fractions of superfusate was started after a 60 min washout period during which excess radioactivity had been removed.
Depolarization-dependent tritium overflow was triggered either by 36 monophasic rectangular electrical pulses (0.5 ms, 60 mA, 66 V·cm−1) delivered at 0.3 Hz or by the inclusion of 25 mM KCl (NaCl was reduced accordingly to maintain isotonicity) in the buffer for periods of 120 s. These stimulations were started after 72 (S1) and 92 min (S2) of superfusion. Nucleotides and nucleotide receptor agonists or antagonists were included in the buffer from minute 88 onwards (see Figure 1). Tetrodotoxin (TTX), whenever appropriate, was included in the buffer after 50 min of superfusion (i.e. 10 min prior to the start of sample collection). The radioactivity remaining in the cells after the completion of experiments was extracted by immersion of the discs in 2% (v/v) perchloric acid followed by sonication. Radioactivity in extracts and collected fractions was determined by liquid scintillation counting (Packard Tri-Carb 2800 TR) with a counting efficiency of 63%. Radioactivity released in response to electrical field stimulation from rat sympathetic neurons after labelling with tritiated noradrenaline under conditions similar to those of the present study had previously been shown to consist predominantly of the authentic transmitter and to contain only small amounts (≤15%) of metabolites (Schwartz and Malik, 1993). Hence, the outflow of tritium measured in this study was assumed to reflect the release of noradrenaline and not that of metabolites.
Figure 1.
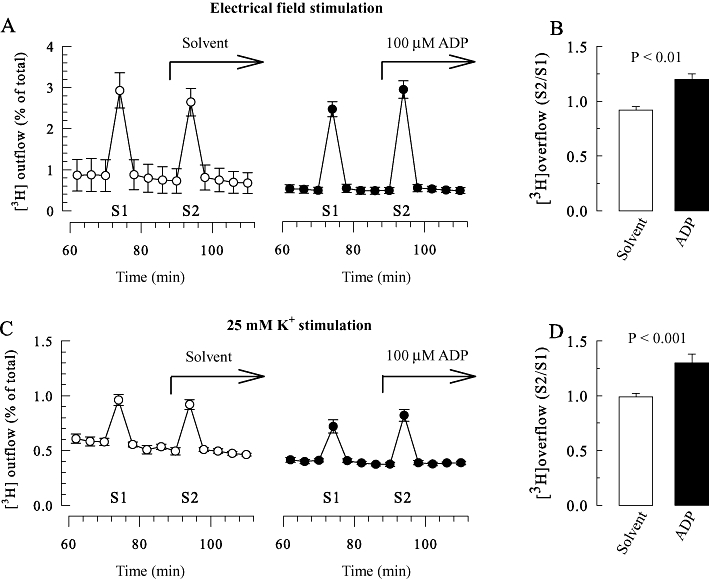
Enhancement of [3H]-noradrenaline release from rat SCG neurons treated with pertussis toxin by ADP. SCG cell cultures were treated with pertussis toxin (PTX; 100 ng·mL−1 for 24 h) and were labelled with [3H]-noradrenaline and superfused. When appropriate (C and D), 0.1 µM TTX was present from minute 50 of superfusion onwards. Subsequent to a 60 min washout period, 4 min fractions of superfusate were collected. Tritium overflow was stimulated twice (S1 after 72 min and S2 after 92 min of superfusion) by 2 min exposures to either electrical pulses (A and B) or 25 mM KCl (C and D). Either 100 µM ADP or the appropriate solvent were present from minute 88 onwards, as indicated by the arrows in (A and C). (A and C) Exemplary time courses of fractional 3H outflow as a percentage of the total radioactivity in the cells (n = 3). (B and D) The S2/S1 ratios of tritium overflow evoked by electrical stimulation (B) or 25 mM K+ (D) in the presence of either solvent or 100 µM ADP (n = 11–12); the P-values for the statistical significance of differences (Student's unpaired t-test) are indicated above the bars.
The spontaneous (unstimulated) rate of 3H efflux was obtained by expressing the radioactivity retrieved during a collection period as percentage of the total radioactivity in the cultures at the beginning of this period. Stimulation-evoked tritium overflow was calculated as the difference between the total tritium outflow during and after stimulation and the estimated basal outflow that was assumed to follow a linear time course throughout experiments. Therefore, basal outflow during periods of stimulation was assumed to equate to the arithmetic mean of the samples preceding and those following stimulation, respectively. Differences between total and estimated basal outflow during periods of stimulation were expressed as percentages of total radioactivity in the cultures at the onset of stimulation (% of total radioactivity; S%). The amount of radioactivity in the cultures at the beginning of each collection period is calculated by summing up the radioactivity remaining in the cells at the end of experiments and that retrieved during the respective and all subsequent collection periods.
As the amount of depolarization- or drug-induced tritium overflow may vary considerably between different cultures (Scholze et al., 2002), the effects of nucleotides on depolarization-dependent release were evaluated by determining changes in the ratio of tritium overflow evoked during the two periods of electrical or K+ stimulation (S2/S1). When cultures had been subjected to a certain treatment (e.g. cholera toxin or U73122), control experiments were also performed in sister cultures that had not been exposed to that treatment (i.e. remained ‘untreated’). In order to directly compare the effects of ADP observed in the absence with those observed in the presence of antagonists, ADP was applied either alone or in combination with the appropriate antagonist. As controls, either the antagonist alone or solvent was applied. Thereafter, the S2/S1 ratio obtained with ADP was expressed as percentage of the corresponding S2/S1 value obtained in its absence (S2/S1, % of control).
Electrophysiology
Currents through Kv7 channels in SCG neurons, so called M currents (IM), were determined using the perforated patch clamp technique as described previously (Lechner et al., 2003). Currents were recorded at room temperature (20–24°C) from single SCG neurons in vitro using an Axopatch 200B amplifier and the pCLAMP 8.0 hardware and software (Molecular Devices, Sunnyvale, CA). Signals were low-pass filtered at 5 kHz, digitized at 10 to 50 kHz and stored on an IBM compatible computer. Traces were analysed off-line by the Clampfit 8.1 programme (Molecular Devices). Patch electrodes were pulled (Flaming-Brown puller, Sutter Instruments, Novato, CA) from borosilicate glass capillaries (Science Products, Frankfurt/Main, Germany), front-filled with a solution consisting of (mM) K2SO4 (75), KCl (55), MgCl2 (8) and HEPES (10), adjusted to pH 7.3 with KOH. Electrodes were then back-filled with the same solution containing 200 µg·mL−1 amphotericin B (in 0.8% DMSO), which yielded tip resistances of 2 to 3 MΩ. The bathing solution contained (mM) NaCl (140), KCl (3.0), CaCl2 (2.0), MgCl2 (2.0), glucose (20), HEPES (10), adjusted to pH 7.4 with NaOH. TTX (0.5 µM) was included to suppress voltage-activated Na+ currents. ADP and all other drugs were applied via a DAD-12 drug application device (Adams & List, Westbury, NY), which permits a complete exchange of solutions surrounding the cells under investigation within less than 100 ms (Boehm, 1999). To investigate IM, cells were held at a potential of −30 mV, and three times per minute 1 s hyperpolarizations to −55 mV were applied to deactivate the Kv7 channels; the difference between current amplitudes 20 ms after the onset of hyperpolarizations and 20 ms prior to re-depolarization was taken as a measure for IM. Amplitudes obtained during the application of test drugs (b) were compared with those measured before (a) and after (c) application of these drugs by calculating 200b / (a + c) = % of control or 100 − (200b / [a + c]) = % inhibition (Boehm, 1998).
Statistics
Statistical significance of differences between two groups was evaluated by Student's unpaired t-tests; when electrophysiological results with nucleotides were obtained in the absence and presence of antagonists in each cell, Student's paired t-tests were employed instead (Figure 6). For comparisons between multiple groups, one-way ANOVA followed by Bonferroni's multiple comparison corrections were used. P-values < 0.05 were considered as indicating statistical significance.
Figure 6.
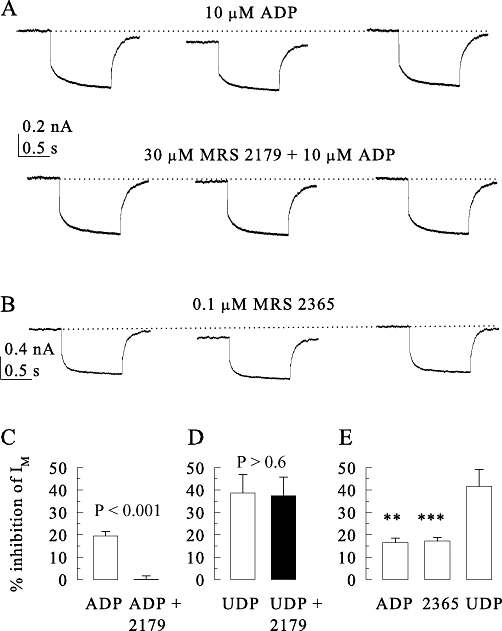
P2Y1 and P2Y6 receptors mediate inhibition of currents through Kv7 channels (IM) in SCG neurons. Currents were activated at −30 mV and quantified by determining the amplitudes of the slow current de-activation relaxations during 1 s hyperpolarizing voltage steps to −55 mV. These IM amplitudes were determined in the presence of ADP (10 µM), UDP (10 µM) or MRS2365 (0.1 µM) applied either alone or together with MRS2179 (30 µM). (A) Original current traces recorded from one neuron before, during and after the exposure to ADP or ADP plus MRS2179. (B) Original current traces recorded from another neuron before, during and after the exposure to MRS2365. (C) Summarizes the inhibition of IM by ADP in the absence or presence of MRS2179 (n = 9). (D) Summarizes the inhibition of IM by UDP in the absence or presence of MRS2179 (n = 10). (E) Summarizes the inhibition of IM by ADP, UDP or MRS2365 (n = 6 to 12). P-values for the significances of differences between the results obtained in the absence and presence of MRS2179 are indicated above the bars; **, *** indicate significant differences versus the inhibition by UDP at P < 0.01 and P < 0.001, respectively.
Materials
(–)-[Ring-2,5,6-3H]-noradrenaline was obtained from PerkinElmer (Vienna, Austria); amphotericin B, ADP, ATP, 2-methylthio-ATP (2-MeSATP), U73122 (1-[6-[((17β)-3-methoxyestra-1,3,5[10]-trien-17-yl)amino]hexyl]-1H-pyrrole-2,5-dione), thapsigargin, H-7 (1-(5-isoquinolinylsulphonyl)-2-methylpiperazine) as well as cholera and pertussis toxin (PTX) were from Sigma; TTX from Latoxan (Rosans, France); MRS 2179 (2′-deoxy-N6-methyladenosine 3′,5′-bisphosphate tetrasodium salt) and MRS 2365 ([[(1R,2R,3S,4R,5S)-4-[6-amino-2-(methylthio)-9H-purin-9-yl]-2,3-dihydroxybicyclo[3.1.0]hex-1-yl]methyl] diphosphoric acid mono ester trisodium salt) were from Tocris (Bristol, UK); bulk chemicals were from Merck (Vienna, Austria). Cangrelor was a kind gift from The Medicines Company (Parsippany, NJ). Water-insoluble drugs were first dissolved in DMSO and then diluted into buffer to yield final DMSO concentrations of up to 0.1%, which were also included in control solutions. At these concentrations, DMSO did not affect any of the parameters investigated (Lechner et al., 2003).
Results
Enhancement of stimulation-evoked noradrenaline release from SCG neurons treated with pertussis toxin by ADP
ADP, at a concentration of 100 µM, has been found to reduce noradrenaline release from rat SCG neurons triggered by 30 mM K+; however, in neurons treated with pertussis toxin to prevent the signalling via inhibitory G-proteins, the nucleotide tended to enhance stimulation-evoked release (Lechner et al., 2004). Therefore, the effect of ADP was investigated in SCG cultures treated with pertussis toxin (100 ng·mL−1) for 24 h and labelled with [3H]-noradrenaline. In these experiments, 100 µM ADP clearly enhanced tritium overflow triggered by electrical field stimulation, but left spontaneous 3H outflow unaltered (Figure 1A and B). The lack of change in spontaneous outflow suggests that ADP did not trigger action potential-dependent exocytosis, as does the activation of other Gq-coupled receptors, such as B2 bradykinin receptors (Scholze et al., 2002) or M1 muscarinic cholinoceptors (Lechner et al., 2003). To confirm that the effect of ADP was not due to enhanced action potential firing, experiments were repeated in the presence of the Na+ channel blocker TTX (0.1 µM). As TTX prevents electrically evoked noradrenaline release from SCG neurons (Boehm, 1999), tritium overflow was stimulated by exposing the cultures to 25 mM KCl for 2 min. Under these conditions, ADP also increased stimulation-evoked tritium overflow (Figure 1C and D) and left the spontaneous outflow unaltered. Thus, the facilitation effect induced by ADP does not require action potential propagation.
The enhancement of stimulation-evoked noradrenaline release from pertussis toxin-treated SCG neurons is mediated by P2Y1 receptors
Amongst the P2Y receptors, P2Y1, P2Y12 and P2Y13 are the primary binding sites for ADP (von Kugelgen, 2006). To differentiate between these three, ADP, ATP and 2-MeSATP were chosen as agonists, the latter being a preferred agonist of P2Y1, but not of P2Y12 and P2Y13, receptors of the rat (von Kugelgen, 2006). All these nucleotides enhanced the electrically evoked 3H overflow from cultures treated with pertussis toxin; from the resulting concentration–response curves, 2-MeSATP was found to be more potent than ADP and ATP, which were about equipotent (Figure 2A). When considering the effects of ATP at concentrations higher than 3 µM, one must not forget the concomitant activation of P2X receptors, which also leads to noradrenaline release from SCG neurons (Boehm, 1999). Nevertheless, the rank order of agonist potency 2-MeSATP > ATP = ADP indicates that P2Y1 receptors are involved in this effect, even though the concentrations of these nucleotides required to produce this effect were relatively high.
Figure 2.
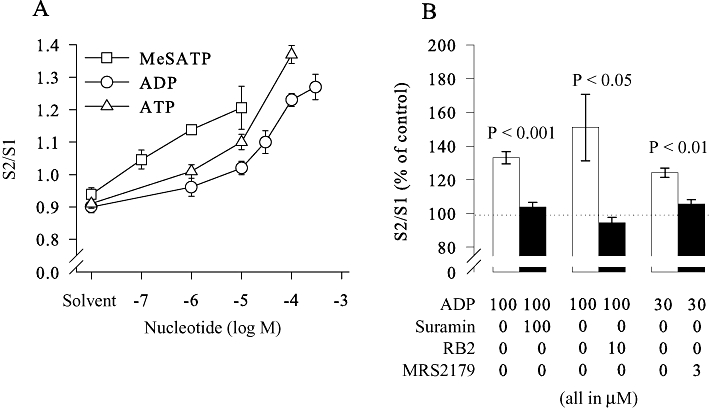
Pharmacological characterization of the receptor mediating the enhancement of [3H]-noradrenaline release. SCG cell cultures were treated with pertussis toxin (PTX; 100 ng·mL−1 for 24 h) and were labelled with [3H]-noradrenaline and superfused. Subsequent to a 60 min washout period, 4 min fractions of superfusate were collected, and tritium overflow was evoked by electrical field stimulation as shown in Figure 1A. (A) The concentration-dependent increase in the S2/S1 ratio of tritium overflow caused by ADP, ATP or 2-MesATP (n = 6 to 13). Nucleotides used at the concentrations indicated or the appropriate solvent were present from minute 88 onwards. (B) The increase in the S2/S1 ratio caused by the indicated concentrations of ADP as percentage of control in the absence or presence of the indicated concentrations (in µM) of suramin (n = 8–9), reactive blue 2 (RB2; n = 10–12) or MRS 2179 (n = 8–9). P-values for the significance of differences between the results obtained in the absence and presence of antagonists are indicated above the bars.
To corroborate the results obtained with the agonistic nucleotides, suramin, reactive blue 2 and MRS 2179 were employed as antagonists and applied together with ADP. While the former two block all three ADP-sensitive P2Y receptors (von Kugelgen, 2006), MRS 2179 is selective for P2Y1 (Boyer et al., 1998). All three antagonists abolished the facilitation of tritium overflow induced by ADP (Figure 2B), thereby confirming that this effect was mediated by P2Y1 receptors.
Enhancement of stimulation-evoked noradrenaline release from SCG neurons not treated with pertussis toxin
The above data indicate that ADP has the ability to enhance stimulation-evoked noradrenaline release when the signalling via inhibitory G proteins is blocked by pertussis toxin. To reveal whether the facilitation by ADP may also occur in neurons with functional Gi/o proteins, experiments were repeated in cultures not treated with PTX; 100 µM ADP caused a significant reduction of electrically-evoked tritium overflow in these cultures (Figure 3A), as described previously where this inhibition of noradrenaline release was suggested to involve P2Y12 receptors (Lechner et al., 2004). Therefore, experiments were performed in the presence of the P2Y12 antagonist cangrelor (Jacobson and Boeynaems, 2010). Cangrelor (10 µM), when applied alone, did not cause obvious changes in tritium outflow (Figure 3C). However, when ADP was applied together with cangrelor, it caused a significant increase in tritium overflow (Figure 3C and D). Hence, the facilitation by ADP can be observed as soon as P2Y12 receptors are blocked.
Figure 3.
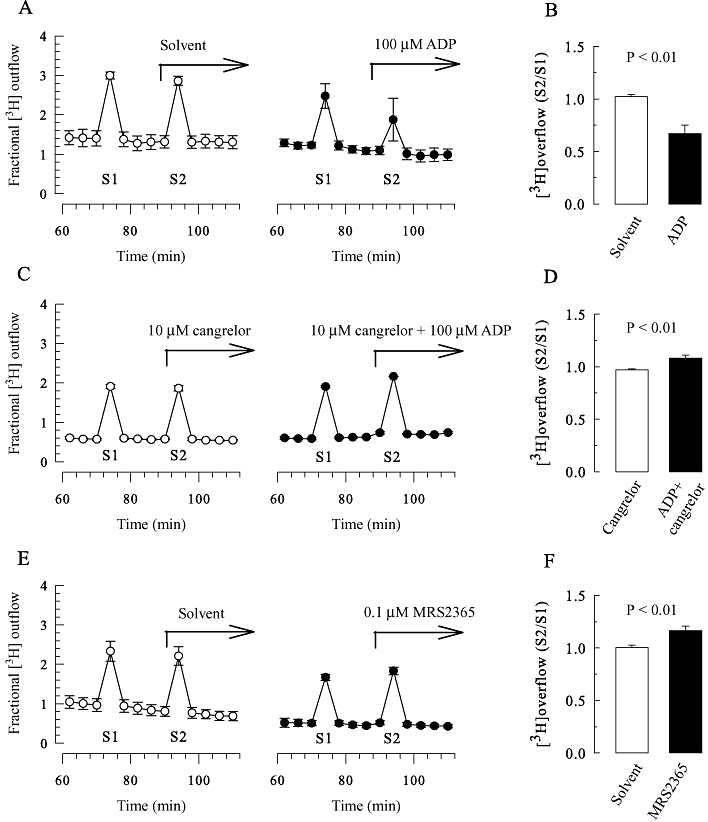
Modulation of [3H]-noradrenaline release from SCG neurons by ADP, MRS 2365 and cangrelor. SCG cell cultures were labelled with [3H]-noradrenaline and superfused. Subsequent to a 60 min washout period, 4 min fractions of superfusate were collected, and tritium overflow was evoked by electrical field stimulation as shown in Figure 1A. (A, C and E) Exemplary time courses of fractional 3H outflow as a percentage of the total radioactivity in the cells (n = 3); 100 µM ADP, 10 µM cangrelor, 0.1 µM MRS 2365 or the appropriate solvent were present from minute 88 onwards as indicated by the arrows. (B) S2/S1 ratios obtained in the presence of either solvent or 100 µM ADP (n = 11). (D) S2/S1 ratios obtained in the presence of either 10 µM cangrelor or 10 µM cangrelor plus 100 µM ADP (n = 6). (F) S2/S1 ratios obtained in the presence of either solvent or 0.1 µM MRS 2365 (n = 12); the P-values for the statistical significances of differences (Student's unpaired t-test) are indicated above the bars.
To reveal whether this facilitatory effect of ADP is mediated by P2Y1 receptors, the selective and potent P2Y1 receptor agonist MRS2365 (Chhatriwala et al., 2004) was employed instead of ADP. At a concentration of 0.1 µM, MRS2365 significantly enhanced electrically-evoked tritium overflow (Figure 3E and F). Thus, the facilitation of tritium overflow can also be seen in cultures not treated with pertussis toxin if either the P2Y12 antagonist cangrelor or the selective P2Y1 agonist MRS2365 is used. Nevertheless, all future experiments were performed in cultures treated with pertussis toxin to avoid excessive use of these specific P2Y receptor ligands.
The enhancement of stimulation-evoked noradrenaline release from SCG neurons by ADP involves PLC
As the facilitation by ADP was observed in cultures treated with pertussis toxin, this effect cannot be mediated by inhibitory G-proteins. However, the facilitation of noradrenaline release via presynaptic GPCRs may involve stimulating Gs-proteins (Kubista and Boehm, 2006). To test for this alternative, cultures were treated not only with pertussis toxin, but also with cholera toxin (100 ng·mL−1), both for 24 h; this strategy eliminates αs G-protein subunits from primary cultures of sympathetic neurons (Boehm et al., 1996). However, the facilitation of electrically evoked 3H overflow was the same in cultures treated with pertussis toxin only as in those treated with pertussis toxin plus cholera toxin (Figure 4A). Thus, the facilitatory effects of ADP do not involve Gs proteins.
Figure 4.
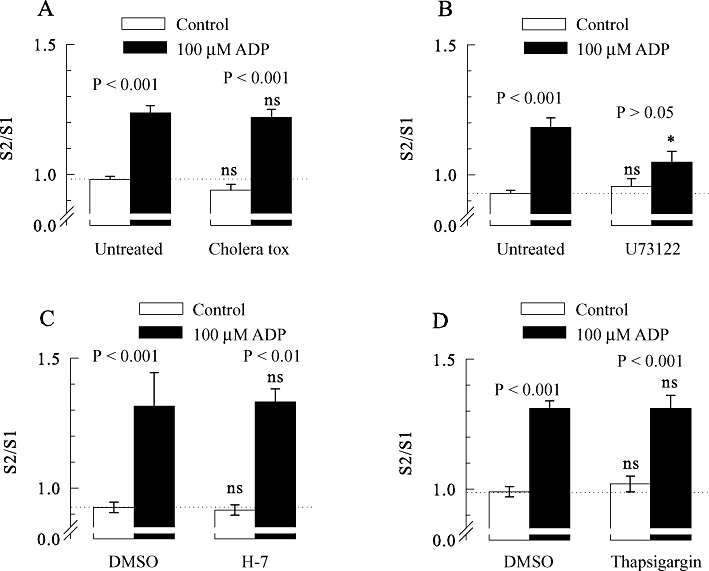
The enhancement of [3H]-noradrenaline release from rat SCG neurons by ADP involves phospholipase C, but not Gs-proteins, PKC or Ca2+-ATPase. All SCG cell cultures were treated with pertussis toxin (PTX; 100 ng·mL−1 for 24 h). In addition, some cultures were treated with cholera toxin or remained otherwise untreated. Thereafter, the cultures were labelled with [3H]-noradrenaline in the absence (untreated) or presence of 3 µM U73122 and were then superfused. Subsequent to a 60 min washout period, 4 min fractions of superfusate were collected, tritium overflow was evoked by electrical field stimulation, and ADP was applied as shown in Figure 1A. When appropriate, 10 µM H-7, 0.3 µm thapsigargin, or 0.1% DMSO were present from minute 50 of superfusion onwards. (A) The S2/S1 ratios of tritium overflow in the absence (control) or presence of 100 µM ADP in either untreated or cholera toxin-treated neurons (n = 10–12). (B) The S2/S1 ratios of tritium overflow in the absence or presence of 100 µM ADP in either untreated or U73122-treated neurons (n = 9). (C) The S2/S1 ratios of tritium overflow in the absence (control) or presence of 100 µM ADP applied either in a solution containing DMSO, or in a solution containing H-7 (n = 9). (D) The S2/S1 ratios of tritium overflow in the absence (control) or presence of 100 µM ADP applied either in a solution containing DMSO or in a solution containing thapsigargin (n = 9). P-values for the significance of differences between the results obtained in the absence and presence of ADP are indicated above the bars; * indicates a significant difference versus the corresponding result obtained in untreated cultures at P < 0.05; ns indicates no significant difference versus results obtained in either untreated cultures (A and B) or in the presence of DMSO (C and D).
P2Y1 receptors are most commonly linked to proteins of the Gq family and thereby to PLC (Abbracchio et al., 2006). To test for a role of these latter enzymes, cultures were treated with 3 µM U73122, which irreversibly blocks signalling via PLC in SCG neurons (Bofill-Cardona et al., 2000). In neurons treated with U73122 and pertussis toxin, ADP failed to significantly enhance electrically evoked tritium overflow. However, in sister cultures treated with pertussis toxin only, ADP clearly caused facilitation (Figure 4B). Thus, the facilitatory effects of the nucleotide involve activation of PLC.
The enhancement of stimulation-evoked noradrenaline release from SCG neurons does not involve PKC or increases in intracellular Ca2+
Activation of presynaptic receptors linked to PLC can lead to facilitation of noradrenaline release through increases in intracellular Ca2+ and subsequent activation of protein kinase C (Kubista and Boehm, 2006). To test for a role of the latter mechanism, experiments were performed in the presence and absence of 10 µM H-7, an inhibitor of PKA, PKC and PKG, with affinities for these enzymes in the low micromolar range (Hidaka et al., 1984). However, the facilitation of electrically-evoked release was the same in the absence and presence of this broad spectrum kinase inhibitor (Figure 4C).
Increases in intracellular Ca2+ may facilitate transmitter release independently of protein kinases (Kubista and Boehm, 2006). Therefore, the intracellular Ca2+ stores of the SCG neurons were depleted by the Ca2+-ATPase inhibitor thapsigargin (Bofill-Cardona et al., 2000), and the facilitatory effect of ADP was assessed again. However, the facilitation of electrically-evoked 3H overflow by ADP was the same whether 0.3 µM thapsigargin were present or not (Figure 4D). Hence, the effect of ADP is independent of increases in intracellular Ca2+ and activation of protein kinases A, C and G.
Recombinant P2Y1 receptors mediate the enhancement of stimulation-evoked noradrenaline release from PC12 cells treated with pertussis toxin
PC12 cells, in contrast to SCG neurons, do not express endogenous P2Y1 receptors (Moskvina et al., 2003), but both types of cells do express P2Y12 and P2Y13 receptors (Lechner et al., 2004). To investigate whether P2Y1 receptors might mediate an enhancement of transmitter release in a neuronal background other than SCG neurons, either non-transfected PC12 cells or a PC12 cell clone stably expressing rat P2Y1-GFP (Moskvina et al., 2003) were compared with respect to the modulation of stimulation-evoked noradrenaline release by ADP. In agreement with previous results (Lechner et al., 2004), ADP (10 µM) reduced K+-evoked tritium overflow from non-transfected PC12 cells by about 50%; however, when these cells had been treated with pertussis toxin (100 ng·mL−1 for 24 h), ADP failed to cause any significant change (Figure 5A). In PC12 cells expressing P2Y1 receptors, for comparison, 10 µM ADP reduced 3H overflow by only 15% and when the cells had been exposed to pertussis toxin ADP caused a significant enhancement of K+-evoked overflow (Figure 5B). Thus, the expression of P2Y1 receptors in PC12 cells is sufficient to counteract the inhibition of transmitter release induced by ADP and instead ADP enhanced the electrically-evoked release of noradrenaline in the pertussis toxin-treated cells.
Figure 5.
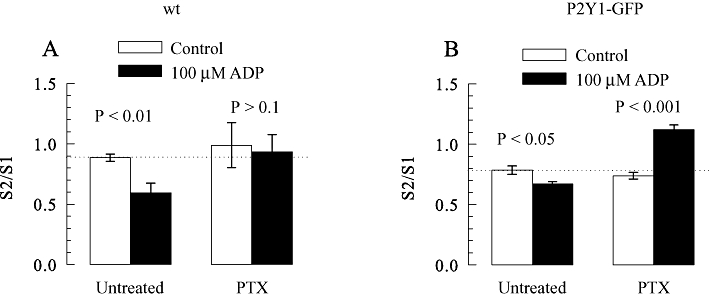
Enhancement of [3H]-noradrenaline release from PC12 cell expressing P2Y1 receptors by ADP. PC12 cell cultures were treated with pertussis toxin (PTX; 100 ng·mL−1 for 24 h) or remained untreated, were labelled with [3H]-noradrenaline and superfused. Subsequent to a 60 min washout period, 4 min fractions of superfusate were collected. Tritium overflow was stimulated twice (S1 after 72 min and S2 after 92 min of superfusion) by 2 min exposures 25 mM KCl. ADP (10 µM) was applied as shown in Figure 1B. (A) The S2/S1 ratios of tritium overflow from non transfected (wild type; wt) PC12 cells in the absence (control) or presence of 10 µM ADP (n = 6). (B) The S2/S1 ratios of tritium overflow from PC12 cells expressing rat P2Y1-GFP in the absence (control) or presence of 10 µM ADP (n = 6). P-values for the significance of differences between the results obtained in the absence and presence of ADP are indicated above the bars.
P2Y1 receptors mediate inhibition of Kv7 channels by ADP
Recombinant P2Y1 receptors mediate inhibition of currents through Kv7 channels (IM) in PC12 cells (Moskvina et al., 2003). Moreover, ADP and UDP have been found to inhibit IM in rat SCG neurons, but this effect was suggested to involve P2Y6 receptors (Boehm, 1998). More recently, evidence has been presented for inhibition of Kv7 channels of SCG neurons mediated by P2Y1 receptors (Filippov et al., 2010). Here, we re-evaluated the receptors mediating the inhibition of Kv7 channels by ADP in SCG neurons. In line with previous results, 10 µM ADP reduced IM relaxation amplitudes, and this effect was entirely reversible (Figure 6A). To determine whether P2Y1 receptors might mediate this effect, ADP was also applied in the presence of MRS 2179, which abolished the inhibition by ADP (Figure 6A and C). To verify that the antagonism by MRS 2179 (30 µM) was specific for the action of ADP, 10 µM UDP was also used to inhibit IM; this latter effect, however, remained unaltered in the presence of MRS 2179 (Figure 6D).
To confirm the results obtained with the P2Y1 antagonist, the selective P2Y1 agonist MRS 2365 was employed again. As expected, 0.1 µM MRS 2365 also reduced IM in an entirely reversible manner (Figure 6B), and the inhibition was the same as that induced by 10 µM ADP (Figure 6E). Thus, in rat SCG neurons, P2Y1 receptors, in addition to P2Y6, mediate inhibition of Kv7 channels.
Discussion
P2Y1 receptors are widely distributed in the central and peripheral nervous system and mediate a plethora of effects including the modulation of voltage- and transmitter-gated ion channels (Hussl and Boehm, 2006). However, evidence for presynaptic P2Y1 receptors is scarce and, if available, only favours inhibitory presynaptic P2Y1 receptors, as suggested for peripheral sensory neurons (Gerevich et al., 2004), for hippocampal neurons (Rodrigues et al., 2005; Csolle et al., 2008), for spinal cord neurons (Heinrich et al., 2008) and also for sympathetic neurons (Quintas et al., 2009). In general, presynaptic P2Y receptors have been shown to mediate inhibition, but not facilitation, of transmitter release (Goncalves and Queiroz, 2008). Hence, the results of this study provide the first evidence for an enhancement of transmitter release via presynaptic P2Y1 receptors, thereby providing a demonstration of facilitatory presynaptic P2Y receptors.
In postganglionic sympathetic neurons, multifarious evidence for inhibitory and facilitatory presynaptic P2 receptors has been presented. The pharmacological data support the idea that the nucleotide-dependent presynaptic facilitation involves P2X receptors, whereas the inhibition involves P2Y receptors (Boehm and Kubista, 2002; Sperlagh et al., 2007), most likely P2Y12 (Queiroz et al., 2003; Lechner et al., 2004). This was corroborated in the present study, as ADP caused inhibition of electrically-evoked noradrenaline release from SCG neurons, which was reversed to facilitation by the P2Y12 antagonist cangrelor (Jacobson and Boeynaems, 2010). Likewise, in SCG neurons treated with pertussis toxin to inactivate the signalling cascades of inhibitory P2Y12 and P2Y13 receptors, ADP also enhanced electrically-evoked noradrenaline release. Moreover, this effect was observed when release was triggered by depolarizing K+ concentrations in the presence of TTX in order to block action potential propagation. Thus, the site of action for the facilitation of transmitter release by ADP must be in close proximity to the sites of vesicle exocytosis (i.e. must be a bona fide presynaptic receptor).
The presynaptic receptor mediating the facilitation of noradrenaline release was a P2Y receptor, more precisely a P2Y1 receptor, as indicated by the following results: (i) 2-MesATP, an agonist at rat P2Y1 receptors (Dixon, 2000) enhanced stimulation-evoked noradrenaline release from SCG neurons treated with pertussis toxin; (ii) 2-MesATP was more potent than ADP as previously demonstrated for rat P2Y1 receptors (Vohringer et al., 2000); (iii) the facilitation by ADP was abolished by suramin and reactive blue 2, which are both known to be P2Y1 antagonists (von Kugelgen, 2006); (iv) the selective P2Y1 antagonist MRS 2179 (Boyer et al., 1998) also abolished the facilitatory affect of ADP; (v) finally, the specific P2Y1 agonist MRS 2365 (Chhatriwala et al., 2004) enhanced stimulation-evoked noradrenaline release even when the signalling cascades of inhibitory P2Y receptors had not been impaired by pertussis toxin.
The ability of P2Y1 receptors to mediate facilitation of transmitter release was also confirmed using recombinant receptors. In PC12 cells, the activation of heterologously expressed rat P2Y1 receptors led to an increase in K+-evoked noradrenaline release when signalling via inhibitory GPCRs was prevented by pertussis toxin; this effect was not observed in the absence of P2Y1 receptors. In PC12 cells expressing P2Y1 receptors but not treated with the bacterial toxin, ADP caused a reduction of depolarization-evoked noradrenaline release as it did in non-transfected PC12 cells. However, this inhibitory effect of ADP was much more pronounced in non-transfected PC12 cells than in cells expressing P2Y1 receptors. This indicates that ADP simultaneously activates the facilitatory P2Y1 receptors and inhibitory P2Y12 or possibly P2Y13 receptors.
P2Y1 receptors are most commonly linked to phospholipase C via proteins of the Gq family (Abbracchio et al., 2006). In accordance with this notion, the PLC inhibitor U73122 abolished the facilitation by ADP, but removal of Gs proteins by a cholera toxin treatment had no such effect. Activated PLC employs membrane phosphatidylinositol 4,5-bisphosphates to generate inositol trisphosphate and diacylglycerol, which then mediate increases in intracellular Ca2+ and activation of PKC, respectively (Suh and Hille, 2007). However, none of these effects was involved in the facilitation of noradrenaline release via presynaptic P2Y1 receptors, as neither the depletion of intracellular Ca2+ stores nor the inhibition of a set of protein kinases including PKC were sufficient to prevent this facilitation.
Heterologously expressed P2Y1 receptors mediate inhibition of Kv7 channels in SCG neurons (Brown et al., 2000) as well as in PC12 cells (Moskvina et al., 2003). Moreover, ADP has been found to inhibit Kv7 channels in SCG neurons, but it was concluded that this effect is mediated by endogenously expressed P2Y6 receptors (Boehm, 1998). Rat SCG neurons are known to express endogenous P2Y1, P2Y2, P2Y4, P2Y12 and P2Y13 in addition to P2Y6 receptors (Moskvina et al., 2003; Lechner et al., 2004). The present results clearly show that endogenous P2Y1 receptors contribute to the regulation of Kv7 channels in SCG neurons by nucleotides, as the inhibition by ADP was abolished by the selective P2Y1 antagonist MRS 2179 (Boyer et al., 1998) and mimicked by the specific P2Y1 agonist MRS 2365 (Chhatriwala et al., 2004). The inhibition of IM by UDP, in contrast, was not altered by MRS 2179, thus indicating that P2Y1 and P2Y6 receptors can control Kv7 channels independently of each other. While this work was in progress, an inhibition of Kv7 channels in rat SCG neurons by MRS 2365 via endogenous P2Y1 receptors has been reported by others (Filippov et al., 2010).
Activation of presynaptic Kv7 channels leads to a decrease in transmitter release from cerebrocortical nerve terminals, while inhibition causes the opposite effect (Luisi et al., 2009), and the same holds true for SCG neurons (Hernandez et al., 2008). Moreover, presynaptic muscarinic receptors have been shown to facilitate transmitter release through inhibition of Kv7 channels (Martire et al., 2007). By analogy, the present results suggest that the inhibition of Kv7 channels via P2Y1 receptors is the basis for the facilitation of noradrenaline release in both PC12 cells and SCG neurons.
In hippocampal neurons, activation of P2Y1 receptors was found to lead to inhibition of transmitter release (Rodrigues et al., 2005; Heinrich et al., 2008). Although the underlying mechanisms are still obscure, endogenous as well as recombinant P2Y1 receptors are known to mediate inhibition of neuronal voltage-gated Ca2+ channels (Hussl and Boehm, 2006), and this is a prime mechanism of presynaptic inhibition (Brown and Sihra, 2008). Recently, P2Y1 receptors were also reported to mediate inhibition of Ca2+ currents in SCG neurons (Filippov et al., 2010). In the preparation used for this study, ADP also elicits inhibition of voltage-activated Ca2+ channels, but this latter effect excludes P2Y1 and is mediated only by P2Y12 receptors (Kulick and von Kugelgen, 2002; Lechner et al., 2004). The reasons for this discrepancy remain enigmatic, but it is obvious that inhibition of voltage-activated Ca2+ channels cannot be the basis for the presynaptic facilitation described here, but rather underlies presynaptic inhibition.
It appears puzzling that one GPCR type like P2Y1 can mediate presynaptic facilitation or presynaptic inhibition depending on the neuron being investigated. However, this has also been found with other presynaptic Gq-linked receptors, for instance with M1 muscarinic cholinoceptors: on the one hand, M1 receptors mediate an enhancement of noradrenaline release from sympathetic nerve terminals through activation of PKC (Costa et al., 1993; Somogyi et al., 1996); on the other hand, these receptors mediate presynaptic inhibition through the depletion of membrane phosphatidylinositol 4,5-bisphosphate via PLC and the resulting closure of voltage-activated Ca2+ channels (Kubista et al., 2009). As mentioned above, P2Y1 receptors have been reported to inhibit Kv7 channels as well as voltage-activated Ca2+ channels in SCG neurons, and the latter effect is determined by the presence or absence of a scaffold protein (Filippov et al., 2010). Hence, depending on the scaffold proteins nerve terminals are endowed with, one type of GPCR might mediate presynaptic facilitation or inhibition.
In conclusion, the present results demonstrate that ADP controls sympathetic transmitter release not only via inhibitory presynaptic P2Y12 (and maybe P2Y13) receptors but also via facilitatory presynaptic P2Y1 receptors. The principle that two separate GPCRs for one transmitter family, such as adenine nucleotides, mediate opposing effects at sympathetic nerve terminals is not unknown: noradrenaline and adrenaline activate presynaptic α2- and β2-adrenoceptors and thereby cause inhibition and facilitation of noradrenaline release, respectively (Boehm and Kubista, 2002). Since noradrenaline and adenine nucleotides are co-transmitters in postganglionic sympathetic neurons, the pair of inhibitory presynaptic P2Y12 and facilitatory presynaptic P2Y1 receptors can be viewed as a novel counterpart of the well-established presynaptic adrenoceptors.
Acknowledgments
This study was supported by grant P17611 from the Austrian Science Funds (FWF); GKC and IS are members of the doctoral programme CCHD supported by the FWF, the Medical University of Vienna and the Austrian Academy of Sciences. The perfect technical assistance of Gabi Gaupmann is gratefully acknowledged.
Glossary
Abbreviations
- 2-MeSATP
2-methylthio-ATP
- DMSO
dimethyl sulphoxide
- H-7
1-(5-isoquinolinylsulphonyl)-2-methylpiperazine
- IM
M-type K+ current
- MRS
2179, 2′-deoxy-N6-methyladenosine 3′,5′-bisphosphate tetrasodium salt
- MRS
2365, [[(1R,2R,3S,4R,5S)-4-[6-amino-2-(methylthio)-9H-purin-9 -yl]-2,3-dihydroxybicyclo[3.1.0]hex-1-yl]methyl] diphosphoric acid mono ester trisodium salt
- PIP2
phosphatidylinositol 4,5-bisphosphate
- PTX
pertussis toxin
- SCG
superior cervical ganglion
- TTX
tetrodotoxin
- U73122
1-[6-[((17β)-3-methoxyestra-1,3,5[10]-trien-17-yl)amino]hexyl]-1H-pyrrole-2,5-dione
Conflicts of interest
None.
References
- Abbracchio MP, Burnstock G, Boeynaems JM, Barnard EA, Boyer JL, Kennedy C, et al. International Union of Pharmacology LVIII: update on the P2Y G protein-coupled nucleotide receptors: from molecular mechanisms and pathophysiology to therapy. Pharmacol Rev. 2006;58:281–341. doi: 10.1124/pr.58.3.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm S. Selective inhibition of M-type potassium channels in rat sympathetic neurons by uridine nucleotide preferring receptors. Br J Pharmacol. 1998;124:1261–1269. doi: 10.1038/sj.bjp.0701956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm S. ATP stimulates sympathetic transmitter release via presynaptic P2X purinoceptors. J Neurosci. 1999;19:737–746. doi: 10.1523/JNEUROSCI.19-02-00737.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm S, Kubista H. Fine tuning of sympathetic transmitter release via ionotropic and metabotropic presynaptic receptors. Pharmacol Rev. 2002;54:43–99. doi: 10.1124/pr.54.1.43. [DOI] [PubMed] [Google Scholar]
- Boehm S, Huck S, Motejlek A, Drobny H, Singer EA, Freissmuth M. Cholera toxin induces cyclic AMP-independent down-regulation of Gs alpha and sensitization of alpha 2-autoreceptors in chick sympathetic neurons. J Neurochem. 1996;66:1019–1026. doi: 10.1046/j.1471-4159.1996.66031019.x. [DOI] [PubMed] [Google Scholar]
- Bofill-Cardona E, Vartian N, Nanoff C, Freissmuth M, Boehm S. Two different signaling mechanisms involved in the excitation of rat sympathetic neurons by uridine nucleotides. Mol Pharmacol. 2000;57:1165–1172. [PubMed] [Google Scholar]
- Boyer JL, Mohanram A, Camaioni E, Jacobson KA, Harden TK. Competitive and selective antagonism of P2Y1 receptors by N6-methyl 2′-deoxyadenosine 3′,5′-bisphosphate. Br J Pharmacol. 1998;124:1–3. doi: 10.1038/sj.bjp.0701837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DA, Sihra TS. Presynaptic signaling by heterotrimeric G-proteins. Handb Exp Pharmacol. 2008;184:207–260. doi: 10.1007/978-3-540-74805-2_8. [DOI] [PubMed] [Google Scholar]
- Brown DA, Filippov AK, Barnard EA. Inhibition of potassium and calcium currents in neurones by molecularly-defined P2Y receptors. J Auton Nerv Syst. 2000;81:31–36. doi: 10.1016/s0165-1838(00)00150-8. [DOI] [PubMed] [Google Scholar]
- Burnstock G. Pathophysiology and therapeutic potential of purinergic signaling. Pharmacol Rev. 2006;58:58–86. doi: 10.1124/pr.58.1.5. [DOI] [PubMed] [Google Scholar]
- Chhatriwala M, Ravi RG, Patel RI, Boyer JL, Jacobson KA, Harden TK. Induction of novel agonist selectivity for the ADP-activated P2Y1 receptor versus the ADP-activated P2Y12 and P2Y13 receptors by conformational constraint of an ADP analog. J Pharmacol Exp Ther. 2004;311:1038–1043. doi: 10.1124/jpet.104.068650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa M, Barrington M, Majewski H. Evidence that M1 muscarinic receptors enhance noradrenaline release in mouse atria by activating protein kinase C. Br J Pharmacol. 1993;110:910–916. doi: 10.1111/j.1476-5381.1993.tb13899.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csolle C, Heinrich A, Kittel A, Sperlagh B. P2Y receptor mediated inhibitory modulation of noradrenaline release in response to electrical field stimulation and ischemic conditions in superfused rat hippocampus slices. J Neurochem. 2008;106:347–360. doi: 10.1111/j.1471-4159.2008.05391.x. [DOI] [PubMed] [Google Scholar]
- Dixon CJ. Evidence that 2-methylthioATP and 2-methylthioADP are both agonists at the rat hepatocyte P2Y(1) receptor. Br J Pharmacol. 2000;130:664–668. doi: 10.1038/sj.bjp.0703350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorostkar MM, Boehm S. Presynaptic lonotropic receptors. Handb Exp Pharmacol. 2008;184:479–527. doi: 10.1007/978-3-540-74805-2_15. [DOI] [PubMed] [Google Scholar]
- Filippov AK, Simon J, Barnard EA, Brown DA. The scaffold protein NHERF2 determines the coupling of P2Y1 nucleotide and mGluR5 glutamate receptor to different ion channels in neurons. J Neurosci. 2010;30:11068–11072. doi: 10.1523/JNEUROSCI.2597-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerevich Z, Borvendeg SJ, Schroder W, Franke H, Wirkner K, Norenberg W, et al. Inhibition of N-type voltage-activated calcium channels in rat dorsal root ganglion neurons by P2Y receptors is a possible mechanism of ADP-induced analgesia. J Neurosci. 2004;24:797–807. doi: 10.1523/JNEUROSCI.4019-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncalves J, Queiroz G. Presynaptic adenosine and P2Y receptors. Handb Exp Pharmacol. 2008;184:339–372. doi: 10.1007/978-3-540-74805-2_11. [DOI] [PubMed] [Google Scholar]
- Heinrich A, Kittel A, Csolle C, Sylvester Vizi E, Sperlagh B. Modulation of neurotransmitter release by P2X and P2Y receptors in the rat spinal cord. Neuropharmacology. 2008;54:375–386. doi: 10.1016/j.neuropharm.2007.10.013. [DOI] [PubMed] [Google Scholar]
- Hernandez CC, Zaika O, Tolstykh GP, Shapiro MS. Regulation of neural KCNQ channels: signalling pathways, structural motifs and functional implications. J Physiol. 2008;586:1811–1821. doi: 10.1113/jphysiol.2007.148304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidaka H, Inagaki M, Kawamoto S, Sasaki Y. Isoquinolinesulfonamides, novel and potent inhibitors of cyclic nucleotide dependent protein kinase and protein kinase C. Biochemistry. 1984;23:5036–5041. doi: 10.1021/bi00316a032. [DOI] [PubMed] [Google Scholar]
- Hussl S, Boehm S. Functions of neuronal P2Y receptors. Pflugers Arch. 2006;452:538–551. doi: 10.1007/s00424-006-0063-8. [DOI] [PubMed] [Google Scholar]
- Jacobson KA, Boeynaems JM. P2Y nucleotide receptors: promise of therapeutic applications. Drug Discov Today. 2010;15:570–578. doi: 10.1016/j.drudis.2010.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubista H, Boehm S. Molecular mechanisms underlying the modulation of exocytotic noradrenaline release via presynaptic receptors. Pharmacol Ther. 2006;112:213–242. doi: 10.1016/j.pharmthera.2006.04.005. [DOI] [PubMed] [Google Scholar]
- Kubista H, Kosenburger K, Mahlknecht P, Drobny H, Boehm S. Inhibition of transmitter release from rat sympathetic neurons via presynaptic M(1) muscarinic acetylcholine receptors. Br J Pharmacol. 2009;156:1342–1352. doi: 10.1111/j.1476-5381.2009.00136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Kugelgen I. Pharmacological profiles of cloned mammalian P2Y-receptor subtypes. Pharmacol Ther. 2006;110:415–432. doi: 10.1016/j.pharmthera.2005.08.014. [DOI] [PubMed] [Google Scholar]
- von Kugelgen I, Starke K. Noradrenaline-ATP co-transmission in the sympathetic nervous system. Trends Pharmacol Sci. 1991;12:319–324. doi: 10.1016/0165-6147(91)90587-i. [DOI] [PubMed] [Google Scholar]
- Kulick MB, von Kugelgen I. P2Y-receptors mediating an inhibition of the evoked entry of calcium through N-type calcium channels at neuronal processes. J Pharmacol Exp Ther. 2002;303:520–526. doi: 10.1124/jpet.102.037960. [DOI] [PubMed] [Google Scholar]
- Lechner SG, Mayer M, Boehm S. Activation of M1 muscarinic receptors triggers transmitter release from rat sympathetic neurons through an inhibition of M-type K+ channels. J Physiol. 2003;553:789–802. doi: 10.1113/jphysiol.2003.052449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechner SG, Dorostkar MM, Mayer M, Edelbauer H, Pankevych H, Boehm S. Autoinhibition of transmitter release from PC12 cells and sympathetic neurons through a P2Y receptor-mediated inhibition of voltage-gated Ca2+ channels. Eur J Neurosci. 2004;20:2917–2928. doi: 10.1111/j.1460-9568.2004.03760.x. [DOI] [PubMed] [Google Scholar]
- Luisi R, Panza E, Barrese V, Iannotti FA, Viggiano D, Secondo A, et al. Activation of pre-synaptic M-type K+ channels inhibits [3H]d-aspartate release by reducing Ca2+ entry through P/Q-type voltage-gated Ca2+ channels. J Neurochem. 2009;109:168–181. doi: 10.1111/j.1471-4159.2009.05945.x. [DOI] [PubMed] [Google Scholar]
- Martire M, D'Amico M, Panza E, Miceli F, Viggiano D, Lavergata F, et al. Involvement of KCNQ2 subunits in [3H]dopamine release triggered by depolarization and pre-synaptic muscarinic receptor activation from rat striatal synaptosomes. J Neurochem. 2007;102:179–193. doi: 10.1111/j.1471-4159.2007.04562.x. [DOI] [PubMed] [Google Scholar]
- Moskvina E, Unterberger U, Boehm S. Activity-dependent autocrine-paracrine activation of neuronal P2Y receptors. J Neurosci. 2003;23:7479–7488. doi: 10.1523/JNEUROSCI.23-20-07479.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Queiroz G, Talaia C, Goncalves J. ATP modulates noradrenaline release by activation of inhibitory P2Y receptors and facilitatory P2X receptors in the rat vas deferens. J Pharmacol Exp Ther. 2003;307:809–815. doi: 10.1124/jpet.103.054809. [DOI] [PubMed] [Google Scholar]
- Quintas C, Fraga S, Goncalves J, Queiroz G. The P2Y(1) and P2Y(12) receptors mediate autoinhibition of transmitter release in sympathetic innervated tissues. Neurochem Int. 2009;55:505–513. doi: 10.1016/j.neuint.2009.05.002. [DOI] [PubMed] [Google Scholar]
- Rodrigues RJ, Almeida T, Richardson PJ, Oliveira CR, Cunha RA. Dual presynaptic control by ATP of glutamate release via facilitatory P2X1, P2X2/3, and P2X3 and inhibitory P2Y1, P2Y2, and/or P2Y4 receptors in the rat hippocampus. J Neurosci. 2005;25:6286–6295. doi: 10.1523/JNEUROSCI.0628-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholze T, Moskvina E, Mayer M, Just H, Kubista H, Boehm S. Sympathoexcitation by bradykinin involves Ca2+-independent protein kinase C. J Neurosci. 2002;22:5823–5832. doi: 10.1523/JNEUROSCI.22-14-05823.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz DD, Malik KU. Cyclic AMP modulates but does not mediate the inhibition of [3H]norepinephrine release by activation of alpha-2 adrenergic receptors in cultured rat ganglion cells. Neuroscience. 1993;52:107–113. doi: 10.1016/0306-4522(93)90186-j. [DOI] [PubMed] [Google Scholar]
- Somogyi GT, Tanowitz M, Zernova G, De Groat WC. M1 muscarinic receptor-induced facilitation of ACh and noradrenaline release in the rat bladder is mediated by protein kinase C. J Physiol. 1996;496:245–254. doi: 10.1113/jphysiol.1996.sp021681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperlagh B, Heinrich A, Csolle C. P2 receptor-mediated modulation of neurotransmitter release-an update. Purinergic Signal. 2007;3:269–284. doi: 10.1007/s11302-007-9080-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh BC, Hille B. Regulation of KCNQ channels by manipulation of phosphoinositides. J Physiol. 2007;582:911–916. doi: 10.1113/jphysiol.2007.132647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vohringer C, Schafer R, Reiser G. A chimeric rat brain P2Y1 receptor tagged with green-fluorescent protein: high-affinity ligand recognition of adenosine diphosphates and triphosphates and selectivity identical to that of the wild-type receptor. Biochem Pharmacol. 2000;59:791–800. doi: 10.1016/s0006-2952(99)00390-1. [DOI] [PubMed] [Google Scholar]