

Abstract
BACKGROUND AND PURPOSE
The aim of this study was to evaluate the anti-convulsant effects of magnolol (6, 6′, 7, 12-tetramethoxy-2, 2′-dimethyl-1-β-berbaman, C18H18O2) and the mechanisms involved.
EXPERIMENTAL APPROACH
Mice were treated with magnolol (20, 40 and 80 mg·kg−1) 30 min before injection with pentylenetetrazol (PTZ, 60 mg·kg−1, i.p.). The anti-seizure effects of magnolol were analysed using seizure models of behaviour, EEG and in vitro electrophysiology and c-Fos expression in the hippocampus and cortex.
KEY RESULTS
Magnolol at doses of 40 and 80 mg·kg−1 significantly delayed the onset of myoclonic jerks and generalized clonic seizures, and decreased the seizure stage and mortality compared with those of the vehicle-treated animals. EEG recordings showed that magnolol (40 and 80 mg·kg−1) prolonged the latency of seizure onset and decreased the number of seizure spikes. The anti-epileptic effect of magnolol was reversed by the GABAA/benzodiazepine receptor antagonist flumazenil. Pretreatment with flumazenil decreased the effects of magnolol on prolongation of seizure latency and decline of seizure stage. In a Mg2+-free model of epileptiform activity, using multi-electrode array recordings in mouse hippocampal slices, magnolol decreased spontaneous epileptiform discharges. Magnolol also significantly decreased seizure-induced Fos immunoreactivity in the piriform cortex, dentate gyrus and hippocampal area CA1. These effects were attenuated by pretreatment with flumazenil.
CONCLUSIONS AND IMPLICATIONS
These findings indicate that the inhibitory effects of magnolol on epileptiform activity were mediated by the GABAA/benzodiazepine receptor complex.
Keywords: EEG, epileptiform activity, flumazenil, GABA, magnolol, pentylenetetrazol, seizure
Introduction
Epilepsy is the commonest serious neurological disorder, afflicting 1–2% of the general population worldwide and causing substantial morbidity and mortality (Hirose et al., 2000). Despite rapid progress in the development of antiepileptic medications, one-third of epilepsy patients remain refractory to medication, indicating a need for more and better antiepileptic drugs (Lindekens et al., 2000).
Extracts of Magnolia plants, for instance Magnolia dealbata, M. obovata and M. grandiflora L., are used in Chinese traditional medicine as tranquilizers to treat epilepsy (Mellado et al., 1980; Bastidas Ramirez et al., 1998). An extract of M. grandiflora L. or M. dealbata Zucc. abolishes the seizure activity in maximal electric and pentylenetetrazol (PTZ)-induced seizure models (Bastidas Ramirez et al., 1998; Martinez et al., 2006). Magnolol (Figure 1; 6,6′, 7,12-tetramethoxy-2,2′-dimethyl-1-β-berbaman, C18H18O2;) is the major bioactive constituent of magnolia bark, and the content of magnolol is in the range of 2–11% of the bark's dry weight (el-Feraly and Chan, 1978; Shen et al., 2009). Magnolol exhibits depressant effects on the CNS, to suppress the incidence of spike discharge, to inhibit tonic extensor convulsions and death produced by an intracerebroventricular injection of penicillin G potassium (Watanabe et al., 1975; 1983;) and to increase the threshold of NMDA-induced seizures (Lin et al., 2005). These observations indicate that magnolol might have a potent antiepileptic effect. Therefore, this study was conducted to investigate the anticonvulsant effect of magnolol in the PTZ-induced seizure model in mice.
Figure 1.
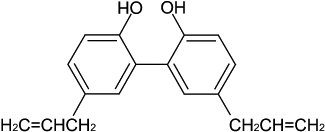
The chemical structure of magnolol.
The GABA receptor complex is the main inhibitory neurotransmitter receptor in the CNS, and the fast-inhibitory activity of GABA is mediated by the GABAA receptor. This receptor is an important target of antiepileptic drugs. Activation of the GABAA receptor causes an anticonvulsive effect, whereas disturbance in GABAA receptor function is often considered as contributing to the pathogenesis of epilepsy (Luddens and Korpi, 1995). The GABAA receptor is an assembly formed by α-, β- and γ-subunits into a pentamer, which can be modulated by allosteric regulators, such as classical benzodiazepines (Pritchett et al., 1989). Benzodiazepines bind to the GABAA/ benzodiazepine receptor subunits α and γ, thereby suppressing epilepsy by increasing GABA-gated chloride ion influx and facilitating GABAergic transmission (Luddens et al., 1995). It has been reported that magnolol increases the binding sites and affinity of the GABAA receptor for GABAA receptor agonist or GABA and then enhances chloride influx (Squires et al., 1999; Ai et al., 2001; Ma et al., 2009). These findings suggest that magnolol might enhance the inhibitory action of GABA on GABAA receptors by binding to sites in the GABA receptor complex. We hypothesized that this GABAA/ benzodiazepine pathway might be responsible for the antiepileptic effects of magnolol.
In the present study, using an in vivo model of behavioural seizures and epileptogenic activity in the EEG and an in vitro model, we showed that magnolol exerted an antiepileptic activity. Magnolol significantly decreased seizure-induced Fos immunoreactivity in the piriform cortex, dentate gyrus and hippocampal area CA1. These effects were attenuated by pretreatment with flumazenil, a benzodiazepine receptor antagonist. Taken together, these findings indicate that magnolol may exert antiepileptiform activity by acting at the GABAA/ benzodiazepine receptor complex.
Methods
Animals
All animal care and experimental procedures were carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and all efforts were made to minimize animal suffering and to use only that number of animals necessary to produce reliable scientific data. Male KunMing strain mice (Experimental Animal Center, Fudan University), 10 weeks old and weighing 18–20 g, were used in the experiments. Six to eight animals were housed in individual cages after they arrived at the lab before surgery in an animal room maintained at a constant temperature (23 ± 1°C) and relative humidity (60 ± 5%) on an automatically controlled 12 h/12 h light/dark cycle (lights on at 07:00 h). Water and food were available ad libitum. All behavioural experiments were carried out between 10:00 and 16:00 h.
EEG recordings and analysis
Under pentobarbitone anaesthesia (50 mg·kg−1, i.p.), mice were chronically implanted with electrodes for polysomnographic EEG recordings. Two stainless steel screws (1 mm in diameter) were inserted through the skull into the cortex (antero–posterior, +1.0 mm; left–right, −1.5 mm from bregma or lambda) according to the atlas of Franklin and Paxinos (1997) and served as EEG electrodes. All electrodes were attached to a microconnector and fixed onto the skull with dental cement. After surgery, animals were singly housed in order to prevent damage of the implanted electrodes. The mice were allowed to recover for 7 days after the implantation of the electrodes. On the day of the experiments, each animal was transferred to a Plexiglas cage (25 × 25 × 40 cm) and habituated for 20 min before EEG recording. The electrode was connected with a slip ring so that the movements of the mice would not be restricted (Nishida et al., 2007).
The baseline of EEG was recorded for 30 min to allow the mice to adapt to the recording leads. The myoclonic jerk, generalized clonus and lethal seizure were identified behaviourally and electrographically by EEG monitoring.
The electrophysiological signals were amplified (1000 times) and filtered (0.3–3 kHz) by using a NeuroLog System (Digitimer Ltd, Hearts, UK) and visualized and stored in a PC computer through an A-D converter, CED 1401 micro (Cambridge Electronic Design, Cambridge, UK). The definition of an EEG spike is based on its amplitude, duration, sharpness and emergence from its background. EEG recordings were analysed by using the peak analysis function of spike2 6.04 software (an analysis programme for CED 1401, Cambridge, UK) (Kong et al., 2010; Qi et al., 2006; Wang et al., 2009). Pharmacologically induced epileptiform activity was monitored for 1 h after the PTZ injection. The latency to seizure onset was defined as the time from injection to the first seizure spike. We recorded, analysed and scored major seizure events and sharply delimited seizure spikes exceeding twice the baseline amplitude (Ziemann et al., 2008).
Seizure behavioural scoring
PTZ (60 mg·kg−1) was intraperitoneally administered 30 min after the injection of magnolol (20, 40 or 80 mg·kg−1), diazepam (2 mg·kg−1) or vehicle. Control animals were given the vehicle. After each injection, the behaviour was monitored, and changes were observed for 60 min. The seizure behaviour was classified as follows (Getova et al., 1998; Chen et al., 2007): stage 0, no response; stage 1, ear and facial twitching; stage 2, myoclonic jerks (MJ); stage 3, clonic forelimb convulsions; stage 4, generalized clonic seizures (GCS), with turning to a side position; and stage 5, generalized clonic–tonic seizures (GTCS) or death within 30 min. Latencies to the onset of MJ and GCS were evaluated during a 30 min period after the PTZ injection (Swinyard and Kupferberg., 1985). In the absence of seizures within 30 min, the latency was taken as 1800 s.
Slice preparation for in vitro electrophysiological study
KunMing mice (21–30 days old) were decapitated under diethyl ether anaesthesia. After dissection, the brain was rapidly removed and placed in oxygenated (95% O2–5% CO2) ice-cold artificial cerebrospinal fluid (ACSF) of the following composition (mM): glycerol, 252; KCl, 5; glucose, 10; CaCl2, 1.6; MgSO4, 1.8; NaHCO3, 27.7; and NaH2PO4, 1.25. Horizontal hippocampal slices (thickness 400–450 µm) were cut by using a Leica VT-1000E vibratome (Leica Microsystems Nussloch GmbH, Nussloch, Germany) and incubated in oxygenated ACSF at temperature 32°C at least 1 h before testing. Extracellular field potential recordings were carried out in a submerged electrophysiological recording chamber perfused with ACSF.
In vitro recordings for determination of field excitatory postsynaptic potentials (fepsps)
Spontaneous fepsps was recorded in the CA3 stratum pyramidale of the hippocamus with 150 mM NaCl-filled glass pipettes (<8 MΩ). ACSF that lacked MgSO4 was used as an in vitro model of interictal and ictal-like epileptiform activity. The details for recording and analysis were described previously (Dzhala et al., 2005). Epileptiform activity was initiated by superfusion with aerated Mg2+-free ACSF. The slices that failed to exhibit seizure-like events in a half hour were discarded. After stable recording of ictal activity for 20 min, magnolol (0.1–100 µM) was supplied. The fepsps were recorded for 80 min by using Clampex software 10.0 (Axon Instrument, Molecular Devices Co., Union City, CA, USA) with a band pass filter of 0.1–5 KHz (×100). Power was calculated by integrating the root mean square value of the signal in frequency bands from 0.1 to 1000 Hz (EEG and fast ripple band) in sequential 10 min time windows proceeding and following drug applications. Slice-to-slice variability and electrode contact variability resulted in substantial variation in signal strength; therefore, subsequent drug-induced changes in burst characteristics were normalized to control bursts before drug application. In all experiments, the recordings were made by using an Axoclamp 200B Amplifier and DigiData 1440A (Axon Instruments).
c-Fos immunohistochemistry
A total of 10 groups of mice were used. The groups were divided into saline- and PTZ (60 mg·kg−1)-treated groups. In the PTZ-treated groups, mice were pretreated with vehicle, magnolol at a dose of 20, 40 or 80 mg·kg−1 or with diazepam at 2 mg·kg−1. To test receptor mechanisms, four groups of mice were used: magnolol 80 mg·kg−1+ flumazenil 5 or 10 mg·kg−1, diazepam 2 mg·kg−1+ flumazenil 10 mg·kg−1 and flumazenil 10 mg·kg−1 groups. Thirty minutes before the magnolol (80 mg·kg−1) or diazepam (2 mg·kg−1) injection, mice were pretreated with flumazenil i.p. at 5 or 10 mg·kg−1. Thirty minutes after the magnolol or diazepam injection, the animals were treated with PTZ. At 45 min after the PTZ administration, the animals were anaesthetized with 10% chloral hydrate and perfused via the heart with saline solution followed by ice-cold 4% paraformaldehyde (PFA) in 0.1 M phosphate buffer (pH 7.4). Their brains were then removed, post-fixed in 4% PFA for 6 h and immersed in 30% sucrose overnight. Thereafter, frozen sections were cut at 30 µm in coronal planes by use of a freezing microtome (Jung Histocut, model 820-II, Leica, Nussloch, Germany). The sections were stored in a cryoprotectant solution at −20°C before histological analysis. Immunohistochemistry was performed in accordance with the free floating method described earlier (Qiu et al., 2003; Qiu et al., 2009). Sections were fixed in 4% PFA for 10 min and incubated with 0.3% H2O2 for 15 min to quench the endogenous peroxidase activity. The sections were next placed for 30 min at 37°C in blocking solution containing 10% normal rabbit serum with 0.3% Triton X-100 in 0.01 M phosphate-buffered saline (PBS, pH 7.2) and then incubated at 4°C for 24 h with a rabbit polyclonal antibody against c-Fos at a 1:1000 dilution in PBS containing 1% normal rabbit serum and 0.3% Triton X-100. On the second day, the sections were incubated with a 1:200 dilution of biotinylated donkey anti-rabbit secondary antibodies for 30 min followed by a 1:200 dilution of avidin–biotin–peroxidase for 45 min at 37°C. The peroxidase reaction was visualized with 0.05% 3,3′-diaminobenzidine tetrahydrochloride in 0.1 M phosphate buffer and 0.01% H2O2. Sections were mounted, dehydrated and coverslipped. As controls, adjacent sections were incubated without the primary antibody to confirm that no non-specific staining had occurred. The sections were examined under bright-field illumination with a Leica DMLB microscope (Leica Microsystems, Wetzlar, Germany). Images were captured with a CoolSNAP-Procf digital camera (RTKE DIAGNOSTIC, SPOT Instruments, Roper Scientific, Marlow, UK).
Study on receptor mechanism involved in antiepileptic effect of magnolol
Dosages of the highly selective antagonist flumazenil that did not affect the seizure threshold were chosen to probe the role of the GABAA/ benzodiazepine receptor complex in the antiepileptic effect of magnolol. Thirty minutes before the magnolol (80 mg·kg−1) or diazepam (2 mg·kg−1) injection, mice were pretreated with flumazenil i.p. at 5 or 10 mg·kg−1. Thirty minutes after the magnolol or diazepam injection, the animals were treated with PTZ. The EEG and behavioural changes were recorded and analysed. In the absence of seizures within 30 min, the latency was taken as 1800 s.
Statistical analysis
All data were expressed as the mean ± SEM (n = 6∼10). Statistical analysis was performed with SPSS 17.0 (SPSS Inc., Chicago, IL, USA). For parametric statistical analysis, Student's t-test was applied to two-group comparisons, and one-factor or multi-factor analysis of variance (anova) was applied to multiple-group comparisons. The anova tests were followed by Tukey's test for seizure latency, seizure spikes and c-Fos expression, or by Dunnett's test for the average power of epileptiform activity. For non-parametric statistical analysis, the Mann–Whitney U-test was applied to seizure stage data. The significance level was set at P < 0.05 for all statistical tests.
Materials
Magnolol was purchased from the National Center for Safety Evaluation of Drugs (NCSED, in China), and was shown to be 98% pure by high-performance liquid chromatography. The drugs PTZ, diazepam and flumazenil, as well as Mg-ATP, were purchased from Sigma-Aldrich (St. Louis, MO, USA), and the other chemicals used were of high-purity analytical grade. All drugs were freshly prepared prior to use, and injection volume (10 mL·kg−1) was kept constant for in vivo experiments. The dosage selections, route of drug administration and injection time of different compounds were based on preliminary experiments and pharmacokinetic considerations. Magnolol and diazepam were suspended in saline containing 0.5% dimethylsulphoxide, and all other drugs were dissolved in the same type of solution.
Results
Antiepileptic effects of Magnolol on PTZ-induced seizure assessed from EEG recordings
To test the antiepileptic effect of magnolol on PTZ-induced seizures, first we examined the seizure onset latency and the number of seizure spikes for 30 min after the PTZ injection. The EEG tracings during seizure showed the time from the PTZ injection to the seizure onset (Figure 2A). Magnolol was given at a dose of 20, 40 or 80 mg·kg−1 30 min before the PTZ injection. In the vehicle-treated mice, the seizure onset latency was 43 ± 3.7 s. As shown in Figure 2B, magnolol at 80 and 40 mg·kg−1 elicited a significant increase in the seizure onset (F(3,20) = 14.1, P < 0.001 overall and post hoc), but not at the low dose of 20 mg·kg−1. Diazepam, used as a positive control, also prolonged the latency to seizure onset (P < 0.01, vs. vehicle). In Figure 2C, the inhibition of epileptic discharges (treatment: F(3,20) = 113.9, P < 0.001; time: F(5,100) = 53.5, P < 0.001; treatment × time interaction: F(15,100) = 2.5, P < 0.01) by magnolol (80 and 40 mg·kg−1) is shown. There were no spikes appearing after 25 min. However, magnolol given at a dose of 20 mg·kg−1 did not suppress seizure spikes after PTZ treatment. Diazepam also suppressed epileptic discharges; the seizure spikes per 5 min interval were 13.5 ± 0.6, 20.3 ± 1.4, 9.7 ± 0.4 and 0.5 ± 0.3 at 5, 10, 15, 20 min, after 20 min, seizure spikes were not observed. As shown in Figure 2D, magnolol (80 and 40 mg·kg−1) and diazepam (2 mg·kg−1) significantly decreased the total number of seizure spikes in 30 min (F(3,20) = 53.9, P < 0.001 overall and post hoc). Magnolol at 20 mg·kg−1 did not significantly affect the number of seizure spikes.
Figure 2.

Magnolol (Mag) prolongs EEG seizure latency and decreases the total number of seizure spikes in PTZ-induced (60 mg·kg−1, i.p.) convulsions. (A) Representative EEG tracings from mice treated with magnolol at 20, 40 and 80 mg·kg−1, diazepam (Dia) at 2 mg·kg−1 and baseline, and quantification of time from the PTZ injection until the first seizure spikes. The open bar with arrow indicates the time of PTZ administration, and the smaller open bars indicate the first seizure spikes. (B) Effects of magnolol on the latency to the onset of the first seizure spikes in the PTZ-induced seizure model. (C) Effects of magnolol at 20, 40 and 80 mg·kg−1 on the total spike number per 5 min in the PTZ-induced seizure model. (D) Effects of magnolol on the total number of seizure spikes in the PTZ-induced seizure model. Each value represents the mean ± SEM (n = 6). *P < 0.05, **P < 0.01, significantly different from the vehicle group.
Antiepileptic effects of magnolol in PTZ-induced seizure assessed by behavioural evaluation
To test the antiepileptic effect of magnolol on behaviour, we examined the behavioural seizure stage score and the latency to onset of MJ and GCS. PTZ caused characteristic behavioural symptoms: first, tremor of the vibrissae and muscles followed by generalized tremor of the body musculature, then GCS or GTCS. Magnolol significantly prolonged the latency of MJ and GCS (MJ: F(3,36) = 40.6, P < 0.001, GCS: F(3,36) = 38 823.4, P < 0.001 overall and post hoc:, Figure 3A and B), at 80 and 40 mg·kg−1 but not at 20 mg·kg−1. Diazepam also significantly increased the MJ latency. There was no GCS or GTCS observed for magnolol at 80 and 40 mg·kg−1 and diazepam at 2 mg·kg−1, 30 min after giving PTZ (Figure 3B). Magnolol (80 and 40 mg·kg−1) also significantly decreased the mean seizure stage (Figure 3C) when given at 80 and 40 mg·kg−1 (P < 0.001 vs. vehicle). The stage distribution analysis (Figure 3D) revealed that all vehicle-treated mice developed GCS (stage 4), with turning to a side position, and 20% of the mice also reached GTCS (stage 5) and died within 30 min. None of the magnolol-treated mice showed GTCS (stage 5) and all mice in the diazepam group showed only tremor of their masticatory muscles (stage 2).
Figure 3.
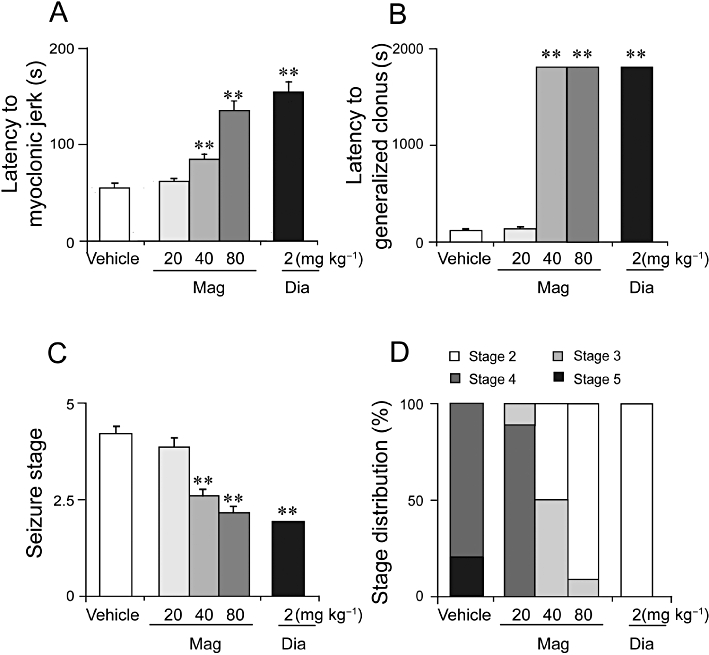
Magnolol (Mag) prolonged behaviour seizure latency and decreased seizure stage in the PTZ-induced (60 mg·kg−1) convulsions. (A) Magnolol prolonged the latency to the onset of MJ and (B) GCS in the PTZ seizure model. (C) Magnolol also decreased the mean seizure stage in the model. (D) Distributions of each stage after magnolol treatment. The divisions within each bar are defined above the bar graph. Each value represents the mean ± SEM (n = 10). **P < 0.01, significantly different from the vehicle group.
Involvement of GABAA/ benzodiazepine pathway in the antiepileptic effect of magnolol in the PTZ-induced seizure model assessed by EEG recording
In order to investigate the probable involvement of the GABAA/ benzodiazepine receptor, we examined the effects of a selective GABAA/ benzodiazepine receptor antagonist, flumazenil, on the anticonvulsant activity of magnolol. As shown in Figure 4A, B, magnolol at 80 mg·kg−1 prolonged the latency to seizure onset, compared with the values for the vehicle group, and pretreatment with flumazenil antagonized the prolonged latency to seizure onset by magnolol (F(2, 25) = 12.7, P < 0.001) or by diazepam (P < 0.01 vs. vehicle).
Figure 4.
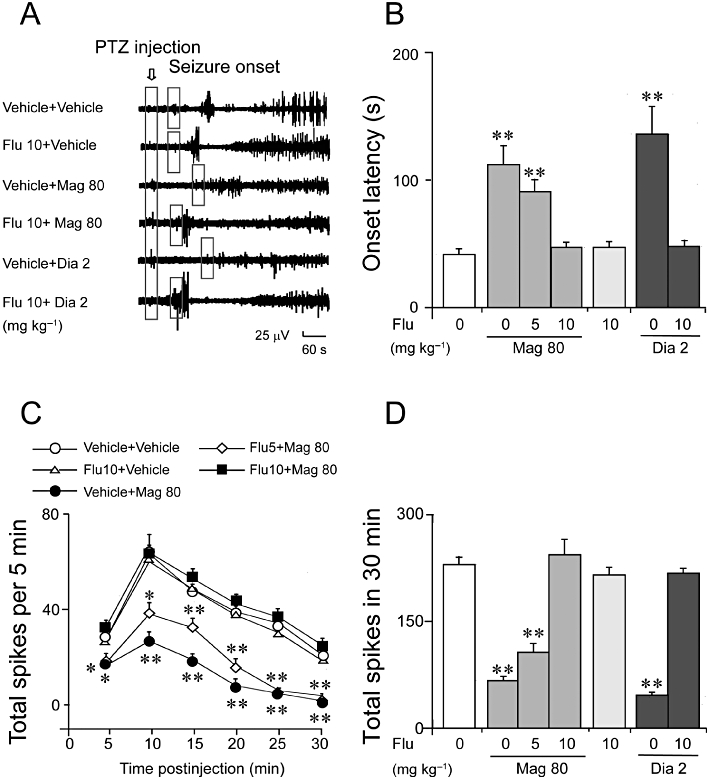
Pretreatment with flumazenil (Flu) blocked the prolonged latency to seizure onset and decreased the total number of seizure spikes of magnolol (Mag) in the PTZ-induced seizure model. (A–B) Flumazenil at 10 mg·kg−1 blocked the prolonged latency to the seizure onset induced by magnolol (80 mg·kg−1) or by diazepam (Dia, 2 mg·kg−1) in the PTZ-induced seizure model. Representative EEG tracings are shown. The open bar with the arrow indicates the time of PTZ administration; the smaller open bars indicate the time of the first seizure spikes. (C-D) Flumazenil at 10 mg·kg−1 antagonized the decrease in the total number of seizure spikes induced by magnolol at 80 mg·kg−1 in the PTZ seizure model. Each value represents the mean ± SEM (n = 6). *P < 0.05, **P < 0.01, significantly different from the vehicle group.
The total number of seizure spikes per 5 min interval was not changed by flumazenil (10 mg·kg−1) given alone, but it did antagonize the decrease in the total number of seizure spikes per 5 min interval induced by magnolol at 80 mg·kg−1 (F(10, 125) = 3.07, P < 0.01). Flumazenil at a lower dose (5 mg·kg−1) was less effective in antagonising the antiepileptic effect of magnolol (80 mg·kg−1) (Figure 4C). Pretreatment with flumazenil (5 or 10 mg·kg−1) also antagonized the reduction in total number of seizure spikes in 30 min, induced by magnolol (80 mg·kg−1) (Figure 4D, F(2, 25) = 53.5, P < 0.001) and diazepam (Figure 4D, P > 0.05 vs. flumazenil 10 mg·kg−1). These results indicate involvement of the GABAA/BZ pathway in the antiepileptic effect of magnolol.
Involvement of GABAA/benzodiazepine pathway in the antiepileptic effect of magnolol in the PTZ-induced behaviour seizure model
As shown in Figure 5, pretreatment with flumazenil (5 or 10 mg·kg−1) dose-dependently antagonized the prolonged latency to onset of MJ (Figure 5A: F(2, 45) = 60.0, P < 0.001) and GCS (F(2, 45) = 16 520.2, P < 0.001) induced by magnolol at 80 mg·kg−1. Flumazenil (10 mg·kg−1) completely antagonized the decrease in stage caused by magnolol at 80 mg·kg−1 (Figure 5C, P > 0.05 vs. flumazenil 10 mg·kg−1). Two mice died in the vehicle-treated group, whereas all mice survived in the magnonol- and diazepam-treated groups, suggesting that flumazenil was unable to reverse the protective effect of magnolol on mortality (Figure 5D). These results suggest that the GABAA/ benzodiazepine pathway was involved in the antiepileptic effect of magnolol in the PTZ-induced behavioural seizure model.
Figure 5.
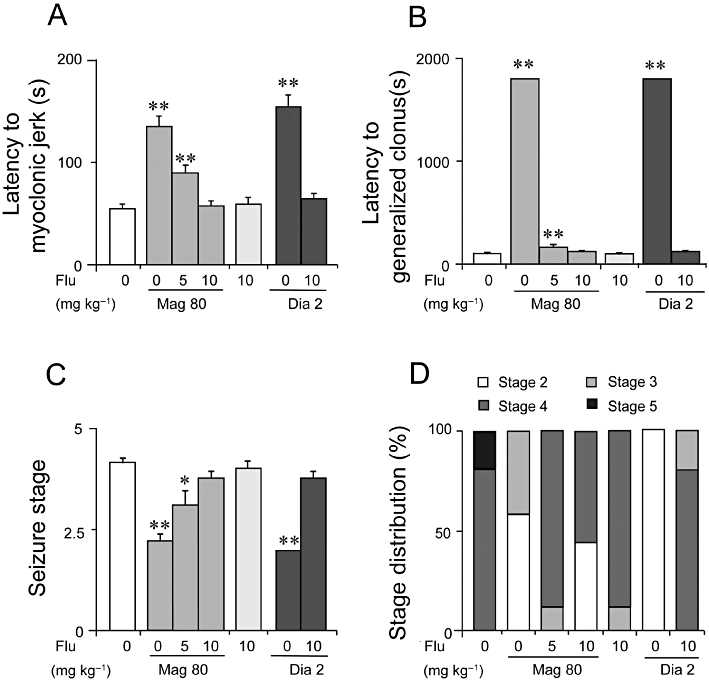
Flumazenil (Flu) antagonized the prolonged latency to MJ (A) and GCS (B) and increased the seizure stage reduced by magnolol in the PTZ seizure model (C). (D) Distributions of each stage after magnolol (Mag) at 80 mg·kg−1 or diazepam (Dia) at 2 mg·kg−1 combined with flumazenil (0, 5, 10 mg·kg−1) treatment. The divisions within each bar are defined above the bar graph. Each value represents the mean ± SEM (n = 10). *P < 0.05, **P < 0.01, significantly different from the vehicle group.
Antiepileptic effects of magnolol in the Mg2+-free model of in vitro epileptiform activity
We used perfusion with Mg2+-free solutions to induce epileptiform activity in hippocampal slices and simultaneously recorded extracellular field potential in the CA3 pyramidal cell layer. Bath application of Mg2+-free ACSF to hippocampal slices resulted in a progressive increase in the neuronal firing that developed to recurrent seizure-like bursts (Figure 6A). Magnolol of different doses (0.1–100 µM) was applied to the bath after the recurrent seizure-like activity had occurred regularly and stably.
Figure 6.
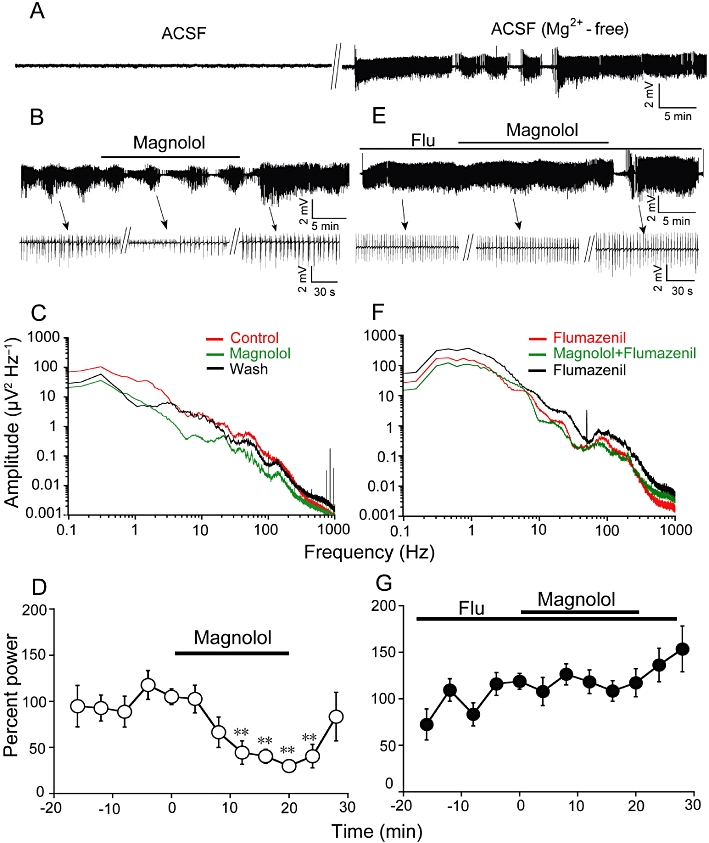
Magnolol attenuated spontaneous epileptiform discharges in mice hippocampus in vitro through GABAA / benzodiazepine receptor. (A) Representative extracellular field potential recording of the tonic–clonic seizure-like events induced by Mg2+-free ACSF in the CA3 pyramidal cell layer of mouse hippocampal slice. (B) Magnolol (100 µM) attenuates seizure-like discharges in CA3 of hippocampal slice in response to Mg2+-free ACSF. Upper traces: example data from CA3. Below: magnified signals of the trace (see arrows for respective locations in recordings) before, during and after magnolol application. (C) Power spectra of epileptiform activity in successive 10 min windows before, during and after magnolol application in frequency range of 0.1–1000 Hz. Magnolol (100 µM) suppressed the power spectra amplitude in hippocampal slice. (D) Averaged power of fEPSP in consecutive 4 min windows from six slices. Magnolol reduced the power of epilesptiform activity by 59 ± 3% (n = 6, **P < 0.01, significantly different from the average power of 4 min before magnolol application, as assessed by anova followed by a Dunnett's test). (E) Flumazenil (1 µM) blocked the anticonvulsant effect of magnolol in hippocampus. Flumazenil was pre-incubated before the magnolol application. Representative trace of tonic–clonic events with magnolol in the presence of flumazenil. (F) Power spectra of epileptiform activity in 10 min windows before, during and after magnolol application in the presence of flumazenil. (G) Averaged power of seizure-like burst is not affected by magnolol in the presence of flumazenil (Flu; n = 6).
Bath application of magnolol at low doses (0.1 or 1 µM) did not suppress epileptiform activity in hippocampal slices (n = 7, data not shown) but at 10 µM attenuated the seizure-like discharges in 2 out of 11 slices. However, the average power of epileptiform activity in the control and after magnolol application was not significantly different with the low doses. High-dose magnolol (100 µM) attenuated the power spectra amplitude of ictal–tonic–clonic discharges (n = 6 from 10 slices, Figure 6B, C). The average power was not significantly changed in control and tended to be decreased by 59 ± 3% after 8 min application of magnolol compared with the average level for the control in 4 min windows before magnolol application (n = 6, P < 0.01, Figure 6D). Flumazenil (1 µM) reversed the anticonvulsive effects of magnolol (n = 6 from eight slices, Figure 6F, G). These data suggest that the GABAA/benzodiazepine receptor was required for the antiepileptic effects of magnolol in vitro.
Magnolol decreased c-fos expression in the hippocampus and piriform cortex
The immunohistochemistry experiments were used to examine the brain regions involved in the antiepileptic effect of magnolol. Figure 7A, J was the composite drawing of representative coronal sections taken at the level of hippocampus and piriform cortex respectively. There were only a few Fos-positive cells in the dentate gyrus (DG), CA1 and piriform cortex of saline-treated group (Figure 7B, K). The mice developed GCS and GTCS after PTZ administration, and a dramatic increase in the number of Fos-positive cells was seen in the DG, CA1 (Figure 7H, P < 0.001 vs. saline) and in the piriform cortex (Figure 7Q, P < 0.001 vs. saline). The animals exhibited mild convulsive events with tremor of the vibrissae and muscles or generalized tremor of the body musculature after magnolol treatment. Magnolol (40 and 80 mg·kg−1) significantly decreased the number of Fos-positive cells in DG and CA1 (DG, F(3,24) = 661.6, P < 0.001; Figure 7H, right panel; CA1, F(3,24) = 50.92, P < 0.001; Figure 7H, left panel) and in the piriform cortex (F(3,24) = 134.2, P < 0.001, overall and post hoc). Diazepam at 2 mg·kg−1 also significantly decreased the number of Fos-positive cells of CA1 and DG (P < 0.001 vs. vehicle) (Figure 7H), and only a few Fos-positive cells were seen in the DG and CA1 in the magnolol-treated (80 mg·kg−1, Figure 7E) and diazepam-treated (2 mg·kg−1, Figure 7F) groups. The enhanced population of Fos-positive cells in the piriform cortex was dose-dependently reduced by magnolol at 40 and 80 mg·kg−1 (Figure 7Q, F(3,24) = 134.2, P < 0.001) or diazepam (2 mg·kg−1). Pretreatment with flumazenil (10 mg·kg−1) completely antagonized the decrease in the number of Fos-positive cells caused by magnolol given at 80 mg·kg−1 (Figure 7I, R, CA1: F(2,30) = 11.5, P < 0.01; DG: F(2,30) = 60.2, P < 0.001; piriform cortex: F(2,30) = 86.9, P < 0.001), and there were densely Fos-positive cells in the CA1 and DG (Figure 7D) and in the piriform cortex (Figure 7M) in the flumazenil-pretreated groups. Magnolol at 80 mg·kg−1 did not show a protective effect when the mice had been pretreated with flumazenil at 10 mg·kg−1, as densely Fos-positive cells were seen in the CA1, DG and piriform cortex (Figure 7G, P), whereas flumazenil at 5 mg·kg−1 only partly blocked the antiepileptic effect of magnolol produced at 80 mg·kg−1 (Figure 7I, R). These results suggest that magnolol inactivated neurons in the hippocampus and piriform cortex to exert its antiepileptic effect.
Figure 7.
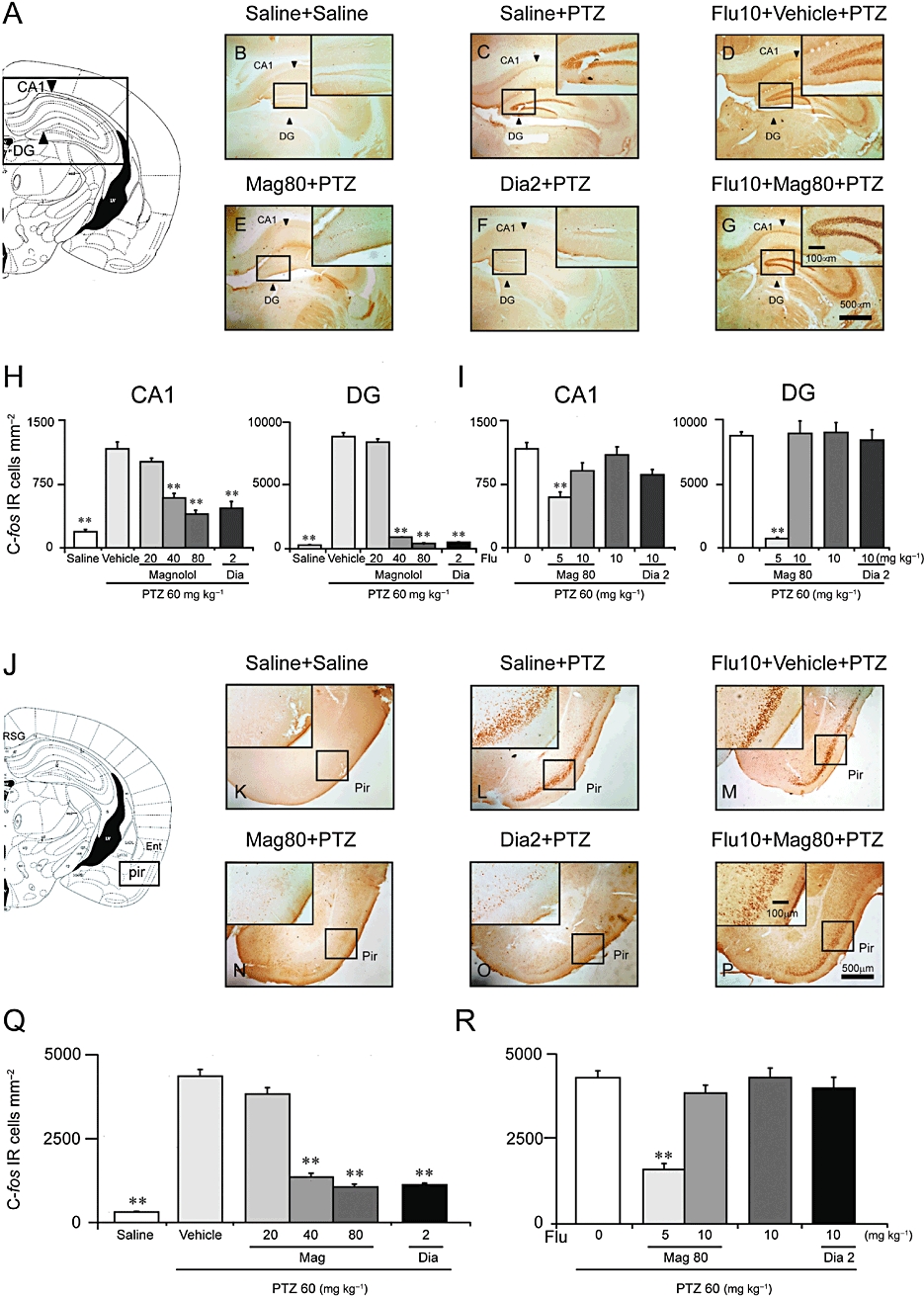
Magnolol (Mag) decreased Fos-positive cells in the hippocampus and piriform cortex (Pir) induced by PTZ 60 mg·kg−1. (A, J) Composite drawing of representative coronal sections was taken at the level of hippocampus and piriform cortex respectively (Paxinos and Watson, 1986). (B, K) Low- and high-power photomicrographs representative of the hippocampus area and piriform cortex, respectively, in which Fos-positive cells were counted. There were only a few Fos-positive cells in the saline group. (C, L) The number of Fos-positive cells in the CA1 and dentate gyrus (DG) parts (arrows) of the hippocampus and in the piriform cortex was high in the vehicle group. (D, M) Pretreatment with flumazenil (Flu) at 10 mg·kg−1 did not influence Fos expression in the CA1 and dentate gyrus (DG) parts (arrows) of the hippocampus or in the piriform cortex induced by PTZ. Magnolol at 80 mg·kg−1 (E, N) and diazepam (Dia) at 2 mg·kg−1 (F, O) significantly decreased the number of Fos-positive cells; positive cell staining was not visible in the DG, and only a few Fos-positive cells were seen in the piriform cortex. (G, P) Flumazenil at 10 mg·kg−1 blocked the decrease in the number of Fos-positive cells in the hippocampus and piriform cortex caused by magnolol at 80 mg·kg−1. (H, Q) Mean number of Fos-positive cells in CA1, DG and in the piriform cortex of saline-, vehicle-, magnolol- and diazepam-treated groups respectively. (I, R) Mean number of Fos-positive cells in CA1, DG and in the piriform cortex of flumazenil (0, 5, 10 mg·kg−1)-pretreated groups. Flumazenil at 5, 10 mg·kg−1 dose-dependently blocked the decrease in the number of Fos-positive cells in the hippocampus and piriform cortex caused by magnolol (Mag; 80 mg·kg−1) or diazepam (2 mg·kg−1). Each value represents the mean ± SEM (n = 6–9). **P < 0.01, significantly different from the vehicle group.
Discussion
This study clearly demonstrated that magnolol exerted a potent antiepileptic effect. The GABAA/ benzodiazepine receptor antagonist flumazenil reversed these effects, indicating that magnolol exerted its anticonvulsant effects by acting through the GABAA/ benzodiazepine receptor complex.
Several in vivo and in vitro studies support our findings that magnolol might elicit anticonvulsant effects by acting on the GABAA/ benzodiazepine receptor complex. Magnolol regulates the release of many neurotransmitters, such as ACh (Tsai et al., 1995b; Hou et al., 2000) and 5-HT (Tsai et al., 1995a; Xu et al., 2008), but 5-HT and cholinergic receptor subtype antagonists do not block the inhibitory effects of magnolol on 5-HT or ACh release (Tsai et al., 1995a; Hou et al., 2000). Recently, some studies have suggested a possible selectivity of magnolol towards the GABAA receptor complex (Ai et al., 2001; Ma et al., 2009). Benzodiazepines such as diazepam, are in wide clinical use as anticonvulsants. It is well known that benzodiazepines facilitate the ability of GABA to activate the GABAA receptor's intrinsic Cl- channel and in turn facilitate inhibitory neurotransmission. The GABAA receptor is an pentameric assembly formed by α-, β- and γ-subunits and can be modulated by allosteric regulators such as the classical benzodiazepines. Magnolol potently enhances either the number of binding sites or the binding affinity of the GABAA agonist [3]H-muscimol for GABAA receptor α-subunit and increases Cl- influx (Squires et al., 1999; Ai et al., 2001). Magnolol also selectively increases the expression of the GABAA receptor α-subunit but does not influence the expression of glutamic acid decarboxylase (Ai et al., 2001; Ma et al., 2009). We have found here that the antiepileptic effects of magnolol were reversed by flumazenil and that magnolol also suppressed the synchronous network activity in hippocampal slices. The low-Mg2+ model of the corticohippocampus represents a well-accepted model of epilepsy in vitro (Walther et al., 1986) and is widely used to study the effects of various antiepileptic drugs (Quilichini et al., 2003; Kilb et al., 2007; Albus et al., 2008). Lowering Mg2+ concentrations has excitatory effects on NMDA receptors by removing the voltage-dependent block by Mg2+. But the GABAA receptors are intact in the Mg2+-free -induced seizure model in vitro. Magnolol attenuated the power of ictal activity in the hippocampus, whereas the interictal-like events were not significantly affected. Facilitation of the GABAA receptor by magnolol leading to the influx of Cl– contributes to an increase in the seizure threshold. Flumazenil slightly enhanced the power of interictal and ictal-like epileptiform activity. A GABAA receptor antagonist causes a prominent increase in the expression of epileptiform events induced by low Mg2+ conditions (Moser et al., 2006). Application of flumazenil in combination with magnolol reversibly blocked the effect of magnolol. Flumazenil at 0.5 µM had a minor potential to inhibit the antiepileptic effects of magnolol in the hippocampus in vitro (in four out of six slices), whereas at 1 µM, it reversed the effect of magnolol (in six out of eight slices). The anticonvulsant effects of magnolol were blocked by flumazenil, indicating that magnolol was acting through the GABAA/ benzodiazepine receptor complex. Cope et al. (2009) reported that enhanced tonic GABAA inhibition is a common feature of typical absence epilepsy, and drugs enhancing GABA transmission may aggravate spike wave discharges. For example, tiagabine, a GABA transporter inhibitor, and vigabatrin, an irreversible inhibitor of GABA transaminase, aggravated non-convulsive spike–wave discharges in both genetic animal models of absence epilepsy and in humans (Coenen and van Luijtelaar, 1989; Coenen et al., 1995; Knake et al., 1999). However, tiagabine and vigabatrin might be anticonvulsive in other forms of idiopathic epilepsy (Akula et al., 2009). Therefore, it seems that drugs that promote GABAergic inhibition have opposite effects in convulsive and non-convulsive epilepsy. Interestingly, benzodiazepines are an exception. They have anti-absence (Coenen and van Luijtelaar, 1989) and anticonvulsive effects. Magnolol could effectively suppress seizure spikes by acting at the GABAA/ benzodiazepine receptor complex. We speculate that magnolol may be effective in generalized absence epilepsy, but further study is needed.
Fos expression is a biological marker of neural activation and is useful for mapping the brain regions involved in seizure generation. As previously reported (Eells et al., 2004; Bastlund et al., 2005), PTZ-induced GTC seizures enhanced Fos expression in many cerebral regions including the cerebral cortex, amygdala and hippocampus, and the highest Fos expression was observed in the hippocampal DG. Magnolol specifically inhibited Fos expression in certain cortico-limbic areas, such as the hippocampus and piriform cortical areas, all of which have been implicated in the generation of convulsive seizures and/or epileptogenesis (Ohno et al., 2009). The hippocampus and piriform cortex are often considered to play major roles in the pathophysiology of temporal lobe seizures (de Guzman et al., 2004; Gavrilovici et al., 2006), and the hallmarks of temporal lobe seizures are hippocampal sclerosis and synaptic reorganization in the DG (Magloczky et al., 2000). Our results suggest that magnolol might be greatly beneficial for suppressing temporal lobe seizures.
In conclusion, the antiepileptic effect of magnolol was antagonized by flumazenil, suggesting that the antiepileptic effect of magnolol might be mediated by the GABAA/ benzodiazepine receptor complex and that magnolol may be a potential therapeutic agent for epilepsy.
Acknowledgments
We are grateful to Ming-Hui Yao for valuable discussion and to Xiao-Jiao Huo for excellent technical support. This study was supported in part by grants-in-aid for scientific research from the National Natural Science Foundation of China (30625021, J0730860, 30770672, 30821002, 30970955, 30901797, 31070957), the Shanghai Committee of Science and Technology (06PJ14008, 09PJ1401800, 09JC1402500, 10XD1400400, 10441901600), National Basic Research Program of China Grants (2009CB5220004, 2011CB711000), Shanghai Leading Academic Discipline Project (B119), Ph.D. Programs Foundation of Ministry of Education of China (20070246186), China National Science and Technology Major Project for Drug Discovery (2009ZX09303-006) and the Program of Basic and Applied Researches for Innovations in Bio-oriented Industry of Japan.
Glossary
Abbreviations
- ACSF
artificial cerebrospinal fluid
- DG
dentate gyrus
- GCS
generalized clonic seizures
- GTCS
generalized clonic-tonic seizures
- MJ
myoclonic jerks
- PFA
paraformaldehyde
- PTZ
pentylenetetrazol
Conflicts of interest
Except as noted in the acknowledgement, all other authors declare that no financial support or compensation has been received from any individual or corporate entity over the past 3 years for research or professional service, and there are no personal financial holdings that could be perceived as constituting a potential conflict of interest.
References
- Ai J, Wang X, Nielsen M. Honokiol and magnolol selectively interact with GABAA receptor subtypes in vitro. Pharmacology. 2001;63:34–41. doi: 10.1159/000056110. [DOI] [PubMed] [Google Scholar]
- Akula KK, Dhir A, Kulkarni SK. Effect of various antiepileptic drugs in a pentylenetetrazol-induced seizure model in mice. Methods Find Exp Clin Pharmacol. 2009;31:423–432. doi: 10.1358/mf.2009.31.7.1393610. [DOI] [PubMed] [Google Scholar]
- Albus K, Wahab A, Heinemann U. Standard antiepileptic drugs fail to block epileptiform activity in rat organotypic hippocampal slice cultures. Br J Pharmacol. 2008;154:709–724. doi: 10.1038/bjp.2008.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastidas Ramirez BE, Navarro Ruiz N, Quezada Arellano JD, Ruiz Madrigal B, Villanueva Michel MT, Garzon P. Anticonvulsant effects of Magnolia grandiflora L. in the rat. J Ethnopharmacol. 1998;61:143–152. doi: 10.1016/s0378-8741(98)00028-2. [DOI] [PubMed] [Google Scholar]
- Bastlund JF, Berry D, Watson WP. Pharmacological and histological characterisation of nicotine-kindled seizures in mice. Neuropharmacology. 2005;48:975–983. doi: 10.1016/j.neuropharm.2005.01.015. [DOI] [PubMed] [Google Scholar]
- Chen CR, Qu WM, Qiu MH, Xu XH, Yao MH, Urade Y, et al. Modafinil exerts a dose-dependent antiepileptic effect mediated by adrenergic alpha1 and histaminergic H1 receptors in mice. Neuropharmacology. 2007;53:534–541. doi: 10.1016/j.neuropharm.2007.06.017. [DOI] [PubMed] [Google Scholar]
- Coenen AM, van Luijtelaar EL. Effects of diazepam and two beta-carbolines on epileptic activity and on EEG and behavior in rats with absence seizures. Pharmacol Biochem Behav. 1989;32:27–35. doi: 10.1016/0091-3057(89)90206-2. [DOI] [PubMed] [Google Scholar]
- Coenen AM, Blezer EH, van Luijtelaar EL. Effects of the GABA-uptake inhibitor tiagabine on electroencephalogram, spike-wave discharges and behaviour of rats. Epilepsy Res. 1995;21:89–94. doi: 10.1016/0920-1211(95)00015-3. [DOI] [PubMed] [Google Scholar]
- Cope DW, Di Giovanni G, Fyson SJ, Orban G, Errington AC, Lorincz ML, et al. Enhanced tonic GABAA inhibition in typical absence epilepsy. Nat Med. 2009;15:1392–1398. doi: 10.1038/nm.2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzhala VI, Talos DM, Sdrulla DA, Brumback AC, Mathews GC, Benke TA, et al. NKCC1 transporter facilitates seizures in the developing brain. Nat Med. 2005;11:1205–1213. doi: 10.1038/nm1301. [DOI] [PubMed] [Google Scholar]
- Eells JB, Clough RW, Browning RA, Jobe PC. Comparative fos immunoreactivity in the brain after forebrain, brainstem, or combined seizures induced by electroshock, pentylenetetrazol, focally induced and audiogenic seizures in rats. Neuroscience. 2004;123:279–292. doi: 10.1016/j.neuroscience.2003.08.015. [DOI] [PubMed] [Google Scholar]
- el-Feraly FS, Chan YM. Isolation and characterization of the sesquiterpene lactones costunolide, parthenolide, costunolide diepoxide, santamarine, and reynosin from Magnolia grandiflora L. J Pharm Sci. 1978;67:347–350. doi: 10.1002/jps.2600670319. [DOI] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. The Mouse Brain in Stereotaxic Coordinates. San Diego: Academic Press; 1997. [Google Scholar]
- Gavrilovici C, D'Alfonso S, Dann M, Poulter MO. Kindling-induced alterations in GABAA receptor-mediated inhibition and neurosteroid activity in the rat piriform cortex. Eur J Neurosci. 2006;24:1373–1384. doi: 10.1111/j.1460-9568.2006.05012.x. [DOI] [PubMed] [Google Scholar]
- Getova D, Froestl W, Bowery NG. Effects of GABAB receptor antagonism on the development of pentylenetetrazol-induced kindling in mice. Brain Res. 1998;809:182–188. doi: 10.1016/s0006-8993(98)00864-6. [DOI] [PubMed] [Google Scholar]
- de Guzman P, D'Antuono M, Avoli M. Initiation of electrographic seizures by neuronal networks in entorhinal and perirhinal cortices in vitro. Neuroscience. 2004;123:875–886. doi: 10.1016/j.neuroscience.2003.11.013. [DOI] [PubMed] [Google Scholar]
- Hirose S, Okada M, Kaneko S, Mitsudome A. Are some idiopathic epilepsies disorders of ion channels?: a working hypothesis. Epilepsy Res. 2000;41:191–204. doi: 10.1016/s0920-1211(00)00141-8. [DOI] [PubMed] [Google Scholar]
- Hou YC, Chao PD, Chen SY. Honokiol and magnolol increased hippocampal acetylcholine release in freely-moving rats. Am J Chin Med. 2000;28:379–384. doi: 10.1142/S0192415X00000441. [DOI] [PubMed] [Google Scholar]
- Kilb W, Sinning A, Luhmann HJ. Model-specific effects of bumetanide on epileptiform activity in the in-vitro intact hippocampus of the newborn mouse. Neuropharmacology. 2007;53:524–533. doi: 10.1016/j.neuropharm.2007.06.015. [DOI] [PubMed] [Google Scholar]
- Knake S, Hamer HM, Schomburg U, Oertel WH, Rosenow F. Tiagabine-induced absence status in idiopathic generalized epilepsy. Seizure. 1999;8:314–317. doi: 10.1053/seiz.1999.0303. [DOI] [PubMed] [Google Scholar]
- Kong S, Qian B, Liu J, Fan M, Chen G, Wang Y. Cyclothiazide induces seizure behavior in freely moving rats. Brain Res. 2010;1355:207–213. doi: 10.1016/j.brainres.2010.07.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YR, Chen HH, Ko CH, Chan MH. Differential inhibitory effects of honokiol and magnolol on excitatory amino acid-evoked cation signals and NMDA-induced seizures. Neuropharmacology. 2005;49:542–550. doi: 10.1016/j.neuropharm.2005.04.009. [DOI] [PubMed] [Google Scholar]
- Lindekens H, Smolders I, Khan GM, Bialer M, Ebinger G, Michotte Y. In vivo study of the effect of valpromide and valnoctamide in the pilocarpine rat model of focal epilepsy. Pharm Res. 2000;17:1408–1413. doi: 10.1023/a:1007559208599. [DOI] [PubMed] [Google Scholar]
- Luddens H, Korpi ER. Biological function of GABAA/benzodiazepine receptor heterogeneity. J Psychiatr Res. 1995;29:77–94. doi: 10.1016/0022-3956(94)00040-x. [DOI] [PubMed] [Google Scholar]
- Luddens H, Korpi ER, Seeburg PH. GABAA/benzodiazepine receptor heterogeneity: neurophysiological implications. Neuropharmacology. 1995;34:245–254. doi: 10.1016/0028-3908(94)00158-o. [DOI] [PubMed] [Google Scholar]
- Ma H, Kim CS, Ma Y, Nam SY, Kim DS, Woo SS, et al. Magnolol enhances pentobarbital-induced sleeping behaviors: possible involvement of GABAergic systems. Phytother Res. 2009;23:1340–1344. doi: 10.1002/ptr.2773. [DOI] [PubMed] [Google Scholar]
- Magloczky Z, Wittner L, Borhegyi Z, Halasz P, Vajda J, Czirjak S, et al. Changes in the distribution and connectivity of interneurons in the epileptic human dentate gyrus. Neuroscience. 2000;96:7–25. doi: 10.1016/s0306-4522(99)00474-1. [DOI] [PubMed] [Google Scholar]
- Martinez AL, Dominguez F, Orozco S, Chavez M, Salgado H, Gonzalez M, et al. Neuropharmacological effects of an ethanol extract of the Magnolia dealbata Zucc. leaves in mice. J Ethnopharmacol. 2006;106:250–255. doi: 10.1016/j.jep.2006.01.003. [DOI] [PubMed] [Google Scholar]
- Mellado V, Chavez Soto MA, Lozoya X. Pharmacological screening of the aqueous extracts of Magnolia grandiflora L. Arch Invest Med (Mex) 1980;11:335–346. [PubMed] [Google Scholar]
- Moser J, Kilb W, Werhahn KJ, Luhmann HJ. Early developmental alterations of low-Mg2+ -induced epileptiform activity in the intact corticohippocampal formation of the newborn mouse in vitro. Brain Res. 2006;1077:170–177. doi: 10.1016/j.brainres.2006.01.024. [DOI] [PubMed] [Google Scholar]
- Nishida N, Huang ZL, Mikuni N, Miura Y, Urade Y, Hashimoto N. Deep brain stimulation of the posterior hypothalamus activates the histaminergic system to exert antiepileptic effect in rat pentylenetetrazol model. Exp Neurol. 2007;205:132–144. doi: 10.1016/j.expneurol.2007.01.021. [DOI] [PubMed] [Google Scholar]
- Ohno Y, Shimizu S, Harada Y, Morishita M, Ishihara S, Kumafuji K, et al. Regional expression of Fos-like immunoreactivity following seizures in Noda epileptic rat (NER) Epilepsy Res. 2009;87:70–76. doi: 10.1016/j.eplepsyres.2009.07.012. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. San Diego: Academic Press; 1986. [Google Scholar]
- Pritchett DB, Sontheimer H, Shivers BD, Ymer S, Kettenmann H, Schofield PR, et al. Importance of a novel GABAA receptor subunit for benzodiazepine pharmacology. Nature. 1989;338:582–585. doi: 10.1038/338582a0. [DOI] [PubMed] [Google Scholar]
- Qi J, Wang Y, Jiang M, Warren P, Chen G. Cyclothiazide induces robust epileptiform activity in rat hippocampal neurons both in vitro and in vivo. J Physiol. 2006;571:605–618. doi: 10.1113/jphysiol.2005.103812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu MH, Qu WM, Xu XH, Yan MM, Urade Y, Huang ZL. D(1)/D(2) receptor-targeting L-stepholidine, an active ingredient of the Chinese herb Stephonia, induces non-rapid eye movement sleep in mice. Pharmacol Biochem Behav. 2009;94:16–23. doi: 10.1016/j.pbb.2009.06.018. [DOI] [PubMed] [Google Scholar]
- Qiu MH, Zhang R, Sun FY. Enhancement of ischemia-induced tyrosine phosphorylation of Kv1.2 by vascular endothelial growth factor via activation of phosphatidylinositol 3-kinase. J Neurochem. 2003;87:1509–1517. doi: 10.1046/j.1471-4159.2003.02110.x. [DOI] [PubMed] [Google Scholar]
- Quilichini PP, Diabira D, Chiron C, Milh M, Ben-Ari Y, Gozlan H. Effects of antiepileptic drugs on refractory seizures in the intact immature corticohippocampal formation in vitro. Epilepsia. 2003;44:1365–1374. doi: 10.1046/j.1528-1157.2003.19503.x. [DOI] [PubMed] [Google Scholar]
- Shen CC, Ni CL, Shen YC, Huang YL, Kuo CH, Wu TS, et al. Phenolic constituents from the stem bark of Magnolia officinalis. J Nat Prod. 2009;72:168–171. doi: 10.1021/np800494e. [DOI] [PubMed] [Google Scholar]
- Squires RF, Ai J, Witt MR, Kahnberg P, Saederup E, Sterner O, et al. Honokiol and magnolol increase the number of [3H] muscimol binding sites three-fold in rat forebrain membranes in vitro using a filtration assay, by allosterically increasing the affinities of low-affinity sites. Neurochem Res. 1999;24:1593–1602. doi: 10.1023/a:1021116502548. [DOI] [PubMed] [Google Scholar]
- Swinyard EA, Kupferberg HJ. Antiepileptic drugs: detection, quantification, and evaluation. Fed Proc. 1985;44:2629–2633. [PubMed] [Google Scholar]
- Tsai TH, Lee TF, Chen CF, Wang LC. Modulatory effects of magnolol on potassium-stimulated 5-hydroxytryptamine release from rat cortical and hippocampal slices. Neurosci Lett. 1995a;186:49–52. doi: 10.1016/0304-3940(95)11279-6. [DOI] [PubMed] [Google Scholar]
- Tsai TH, Westly J, Lee TF, Chen CF, Wang LC. Effects of honokiol and magnolol on acetylcholine release from rat hippocampal slices. Planta Med. 1995b;61:477–479. doi: 10.1055/s-2006-958142. [DOI] [PubMed] [Google Scholar]
- Walther H, Lambert JD, Jones RS, Heinemann U, Hamon B. Epileptiform activity in combined slices of the hippocampus, subiculum and entorhinal cortex during perfusion with low magnesium medium. Neurosci Lett. 1986;69:156–161. doi: 10.1016/0304-3940(86)90595-1. [DOI] [PubMed] [Google Scholar]
- Wang Y, Qi JS, Kong S, Sun Y, Fan J, Jiang M, et al. BDNF-TrkB signaling pathway mediates the induction of epileptiform activity induced by a convulsant drug cyclothiazide. Neuropharmacology. 2009;57:49–59. doi: 10.1016/j.neuropharm.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K, Watanabe HY, Goto Y, Yamamoto N, Yoshizaki M. Studies on the active principles of magnolia bark. Centrally acting muscle relaxant activity of magnolol and honokiol. Jpn J Pharmacol. 1975;25:605–607. doi: 10.1254/jjp.25.605. [DOI] [PubMed] [Google Scholar]
- Watanabe K, Watanabe H, Goto Y, Yamaguchi M, Yamamoto N, Hagino K. Pharmacological properties of magnolol and honokiol extracted from Magnolia officinalis: central depressant effects. Planta Med. 1983;49:103–108. doi: 10.1055/s-2007-969825. [DOI] [PubMed] [Google Scholar]
- Xu Q, Yi LT, Pan Y, Wang X, Li YC, Li JM, et al. Antidepressant-like effects of the mixture of honokiol and magnolol from the barks of Magnolia officinalis in stressed rodents. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32:715–725. doi: 10.1016/j.pnpbp.2007.11.020. [DOI] [PubMed] [Google Scholar]
- Ziemann AE, Schnizler MK, Albert GW, Severson MA, Howard MA, 3rd, Welsh MJ, et al. Seizure termination by acidosis depends on ASIC1a. Nat Neurosci. 2008;11:816–822. doi: 10.1038/nn.2132. [DOI] [PMC free article] [PubMed] [Google Scholar]