

Abstract
Heart mitochondria, which, depending on their location within cardiomyofibers, are classified as either subsarcolemmal or interfibrillar, are the major sources of the high-energy compound, adenosine triphosphate. Physiological differences between these two populations are reflected by differences in the morphology of their cristae, with those of subsarcolemmal mitochondria being mostly lamelliform, and those of interfibrillar mitochondria being mostly tubular. What determines the configuration of cristae, not only in cardiac mitochondria but in mitochondria in general, is unclear. The morphology of cardiac mitochondria, as well as their physiology, is responsive to the exigencies posed by a large variety of pathological situations. Giant cardiac mitochondria make an appearance in certain types of cardiomyopathy and as a result of dietary, pharmacological, and toxicological manipulation; such megamitochondria probably arise by a combination of fusion and true growth. Some of these enlarged organelles occasionally contain a membrane-bound deposit of β-glycogen. Those giant mitochondria induced by experimental treatment usually can be restored to normal dimensions simply by supplying the missing nutrient or by deleting the noxious substance. In some conditions, such as endurance training and ischemia, the mitochondrial matrices become pale. Dense rods or plates are present in the outer compartment of mitochondria under certain conditions. Biochemical alterations in cardiac mitochondria appear to be important in heart failure. In aging, only interfibrillar mitochondria exhibit such changes, with the subsarcolemmal mitochondria unaffected. In certain heart afflictions, biochemical defects are not accompanied by obvious morphological transformations. Mitochondria clearly play a cardinal role in homeostasis of the heart.
1. Introduction
Over the course of an average human life, the heart, beating at a rate of 72 beats per minute, will contract ~2.5 billion times. The energy for this Herculean task is furnished by the cardiac mitochondria, which produce the high energy compound, adenosine triphosphate (ATP). The heart consists principally of cardiac muscle fibers with interspersed blood vessels. Each myofiber is composed of a series of cardiomyocytes that are joined together end-to-end by intercalated disks to form a functional syncitium. The predominant feature of cardiomyocytes is the contractile apparatus, which consists of thin actin filaments and thick myosin filaments. Numerous mitochondria are situated in parallel, longitudinal rows trapped within the contractile apparatus and in monolayers or clusters immediately beneath the sarcolemma. In 1978, Bakeeva et al. described what they termed a mitochondrial reticulum in rat skeletal muscle fibers. Although the mitochondrial constituents of this framework retained their individuality, their close end-to-end juxtaposition [where there are special contact devices (Bakeeva et al., 1983)] gave the impression that the reticulum consisted of a single, continuous organelle. Unfortunately, this misinterpretation has gained widespread currency and has been applied by some workers to the mitochondrial situation in cardiomyocytes. In needs to be emphasized that, irrespective of their propinquity, cardiac mitochondria are not part of a continuous scaffold, but are single entities.
2. Mitochondrial morphology
In general, cardiac mitochondria observed by conventional transmission electron microscopy (TEM) are elliptical, with numerous transverse cristae, which generally are either lamelliform (Fig. 1a) or tubular (Fig 1b). In some skeletal (Luft et al., 1962) or cardiac (Fawcett and McNutt, 1969) muscle mitochondria, the latter may exhibit numerous sharp angulations. Small dense granules that are deposits of divalent cations are present in the mitochondrial matrix.
Fig. 1.

(A) Rat; TEM. A typical cardiac mitochondrion with lamelliform cristae showing its unit membrane substructure to advantage. (B) Human; TEM. A cardiac mitochondrion with apparently tubular cristae. This specimen, from the archives of the Department of Pathology, Case Western Reserve University School of Medicine, was obtained by transjugular bioptome; the tissue sample was from the right side of the interventricular septum.
Because there are great difficulties associated with obtaining samples of human heart in a fresh condition, the forgoing transmission electron microscopy (TEM) description is based mainly on animal hearts, and much of the data presented here are based on experimental animals. Because hearts perform basically the same function with little variation throughout the Mammalia, non-human mammals can be useful in elucidating the dynamics of the human heart.
2.1. High resolution scanning electron microscopy
High resolution scanning electron microscopy (HRSEM) of transected extracted mitochondria yields three-dimensional images of these organelles. Conventional SEM normally yields only surface images of cellular components. Using a method greatly modernized by Riva et al. (1999), the interior of cytoplasmic organelles becomes available for inspection. In brief, tissue samples are lightly fixed, embedded in agar, sectioned at ~100 μm, and subjected to prolonged exposure to osmium tetroxide. Organelles, especially mitochondria, that have been transected during sectioning, are emptied (extracted) of all matrical material, leaving their membranes behind in their original configuration. After critical point-drying and sputter-coating, the membranous structure of the cytoplasmic organelles in a plethora of cells becomes an open book to HRSEM (Fig. 2). Using this methodology, it becomes apparent that the cristae in subsarcolemmal mitochondria (SSM) are lamelliform, whereas those in interfibrillar mitochondria (IFM) are tubular (Riva et al., 2005). These designations– SSM and IFM–are based on the spatial relationship of the mitochondria to the sarcolemma (Figs. 3–6). Those in contact with the sarcolemma are the SSM (Fig. 5); those situated deeper in the cell without obvious contact with the plasma membrane are designated IFM (Fig. 6). This difference in crista morphology is not simply topological, but it appears to have a metabolic underpinning since these two populations possess somewhat different functional properties, i.e., rate of oxidative phosphorylation and enzymatic activity (Palmer et al., 1977). Over and above differences in the structure of cristae per se, immunoelectron microscopic studies by Vogel et al. (2006) have shown a heterogeneous distribution of various mitochondrial proteins, that is, particular proteins may predominate at specific locations—cristae versus inner boundary membranes—reflecting site-related physiological functions.
Fig. 2.

Human; SEM. This micrograph is included to illustrate the advantages of the technique used by us to examine the interior of cardiac mitochondria. Three transected cardiac mitochondria prepared by the osmium-extraction technique are shown. This methodology removes the entire matrix, leaving the internal membranes in their pristine condition. Most of the cristae are tubular although in two organelles such cristae combine to form a limited in extent lamelliform crista (asterisk). This specimen, from the archives of the Department of Cytomorphology, University of Cagliari, was obtained by needle biopsy of the left ventricular myocardium.
Fig. 3.

Dog;TEM. The edges of two longitudinally-sectioned, parallel cardiomyocytes separated by areolar connective tissue that represents the combined epimysia of the respective cells. The high degree of surface scalloping of the two cells is the result of fixative-induced contraction. In each cardiomyocyte, the plasma membrane (sarcolemma) is underlain by a continuous row of mitochondria, the SSM. The more interior mitochondria, which are flanked by myofibrils, are the IFM.
Fig. 6.

Rat; SEM. The edge of a cardiomyocyte illustrating the intimate relationship between SSM and the sarcolemma (arrows).
Fig. 5.

Rat; SEM. A transected cardiomyocyte. The sarcolemma is indicated by the arrows. The lacunae between mitochondria were in life occupied by the contractile apparatus; the myofibrils and myofilaments have been completely effaced by the osmium extraction step. Although at this magnification the mitochondria appear to form a continuous network, the fact that they are each structurally independent entities is shown in Fig. 4.
In addition to the two distinctly different types of cardiac mitochondria in terms of crista structure, there always are in hearts of diverse origins some organelles in both populations that feature both tubular and lamelliform crista, or tubular cristae that form a lattice that often includes a lamelliform segment (Fig. 2); these we designate as mixed.
2.2. High voltage electron microscopy-tomography
Another methodology for determining the internal structure of mitochondria involves combining high voltage electron microscopy of relatively thick sections with computer-based tomography to yield three-dimensional reconstructions of these organelles (Perkins et al., 1997; Manella, 2006). Once completed, such computer reconstructions can be manipulated in space and optically sectioned in any desired plane. A major contribution of this technique is that it has called attention to the previously described (by TEM) but virtually ignored tubular connections of lamelliform cristae to the boundary membrane of mitochondria (Daems and Wisse, 1966). These crista junctions, as they are now known, might play a controlling role in molecular traffic into and out of the cristae. Although this particular methodology is limited in terms of number of mitochondria that are reconstructed for a specific study (in contrast to HRSEM, where literally thousands of organelles can be surveyed) and seems to be restricted to mitochondria of mundane shape, i.e., globular or ovoid, it provides great insight into the internal three-dimensional architecture of individual organelles.
3. Isolated mitochondria
Most of what is known about the functional dynamics of cardiac mitochondria is based on isolated organelles. When cardiac mitochondria are isolated, they retain their essential features although they tend to become globular, but the orientation of their cristae remains largely unaltered (Fig. 7). This circumstance permits the ready distinction of the two populations of cardiac mitochondria, wherein isolated SSM have lamelliform cristae (Fig. 8) and IFM have tubular cristae (Fig. 9) (Riva et al., 2005). The biochemical differences between these two sets of cardiac organelles are quantitative, rather than qualitative. It is true that isolated mitochondria can no longer interact with their cytoplasmic cohorts. However, no data are available to explicate such functional perturbations of isolated cardiac mitochondria.
Fig. 7.

Rat; SEM. A transected pellet of isolated cardiac mitochondria in which the internal structure of a plethora of these organelles is obvious. Such preparations permit quantitation with relative ease of crista configuration.
Fig. 8.
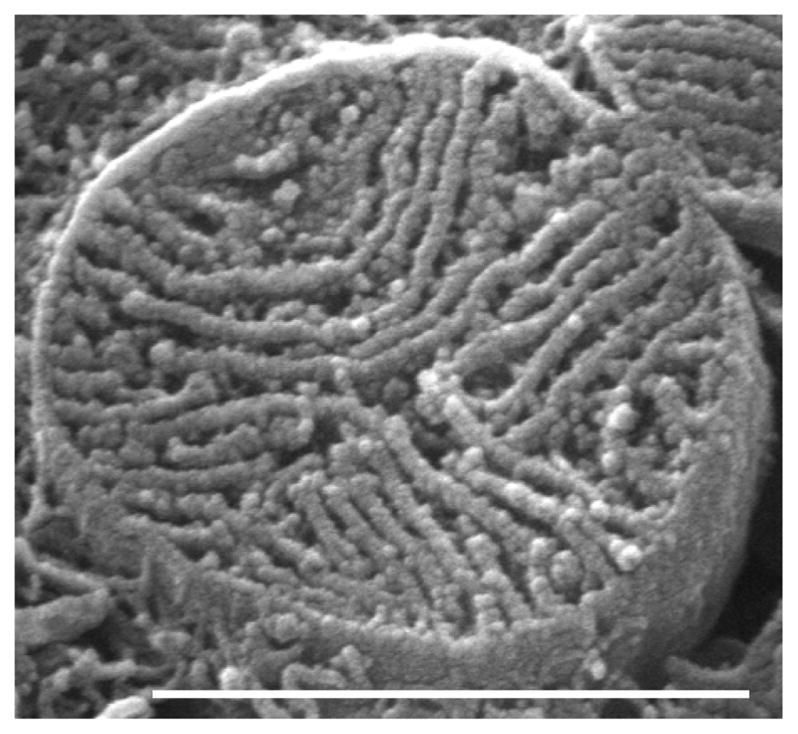
Rat; SEM. An isolated SSM showing its complement of lamelliform cristae.
Fig. 9.
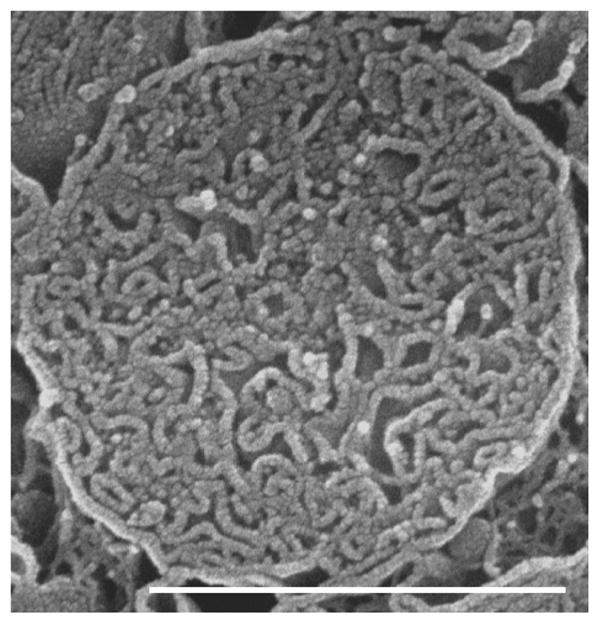
Rat; SEM. An isolated IFM showing its tubular cristae.
In general, isolated mitochondria are the predominant generators of ATP, using fatty acids and pyruvate as their initial substrates. Fatty acids are the preferred fuel for mitochondrial respiration and account for 60–90% of energy production, with glucose oxidation making up 10–40% (Neely and Morgan, 1974). The tricarboxylic acid (TCA) cycle resides in the mitochondrial matrix, where it is anchored to the inner membrane via succinate dehydrogenase. The intermembrane creatine phosphokinase uses ATP and creatine to form ADP and creatine phosphate; the latter is the major storage form of high energy phosphate in the heart; the resultant ADP is available for recycling into ATP. Calcium, a major intracellular metabolic signal, is reversibly sequestered by mitochondria, playing a role in stimulation of the tricarboxylate cycle in the mitochondrial matrix. In heart, the handling of calcium between the two populations of mitochondria is different (Palmer et al., 1986). In SSM, the capacity for calcium uptake is less than that of IFM, leading to release of cytochrome c from only SSM. This release is now known to involve the mitochondrial permeability transition pore, and is a prelude to apoptosis (Kroemer, 2003).
4. Mitochondrial pathology
Mitochondrial structure is highly responsive to changes in the homeostatic status of almost any cell type. Pathological alteration of virtually all non-mitochondrial cellular compartments usually is accompanied by changes in mitochondrial morphology. Such changes are even more apparent when mitochondria are the direct targets of such pathology. This is the case for cardiac mitochondria. These morphological transformations include, but are not limited to: giantism; swelling; distortion of their three-dimensional configuration; loss of cristae; reorientation of cristae; change in shape of cristae; dense rods in their outer compartment; vacuoles or crystalloids in their inner or outer compartment; and marked decreases in the density of their inner compartment. Some of these mitochondrial perturbations as they apply to cardiomyocytes are described in the following section.
4.1. Megamitochondria
Giant mitochondria (megamitochondria) have been described in a number of human cardiomyopathies—in one study (Arbustini et al., 1998), enlarged mitochondria were observed in 85 out of 601 (~14%) cases. Electron microscopy of other cases has revealed megamitochondria of ~30 μm, where a single organelle equals many sarcomeres in length (Kraus and Cain, 1980). Although in many instances their cristae show their usual transverse orientation, in some megamitochondria the cristae are configured in whorls and curlicues, sometimes in a paracrystalline tubular array (Tandler et al., 2002).
Marked increases in mitochondrial size probably are the result of a two-fold process: growth of individual organelles or fusion of abutting mitochondria or some combination of the two. (Of course, an alternative possibility is that superannuated mitochondria are destroyed by autophagy and are then replaced by de novo synthesis of new, very large mitochondria. However, no sign of enhanced autophagy is present in the affected cardiomyocytes.)
In animals, the formation of stacks of cristae in certain megamitochondria is evidence that such organelles are still capable of synthesis of membrane protein, hence growth (Kraus and Cain, 1980). That mitochondria have the ability to fuse with one another even under physiological conditions has long been recognized (Tandler and Hoppel, 1972b). Clearly, pathologically instigated end-to-end fusion of cardiac mitochondria could result in megamitochondria.
With study of mitochondrial fusion in a variety of cell types, a general mechanism for this process has emerged that involves outer membrane proteins, namely, mitofusins. When these proteins on one mitochondrion contact similar proteins on a neighboring organelle, they bind together to tether their host organelles to one another, a prelude to fusion (Chan, 2006). Another protein, OPA1, also is essential for mitochondrial fusion, especially for the step that involves the inner membrane (Chan, 2006). However, the precise mechanism whereby membranes on the conjoined mitochondria meld together remains undetermined in cells of mammalian origin. This is one of the several lacunae in the mammalian mitochondria fusion story, as pointed out in reviews by Chan (2006) and by Detmer and Chan (2007).
In the rat heart, mitochondrial proteins have a half-life of 5.6 (Gross, 1971) to 6.2 days (Aschenbrenner et al., 1970). If these proteins are true indicators of mitochondrial half-life, then these organelles have a high turnover rate, which must involve mitochondrial fission (Tandler and Hoppel, 1972a). Interference with such a division process would of necessity result in enlarged mitochondria as these organelles continue to grow. The morphological steps in mitochondrial division originally were determined by studying the recovery process of giant mitochondria induced by dietary, pharmacological, or toxicological means. In the first such study, hepatic megamitochondria resulting trom ariboflavinosis were restored to normal size within 48 hours of administration of exogenous riboflavin (Tandler et al., 1969). This normalization was brought about by the elongation of a single crista to partition off a portion of the enlarged organelle equivalent to a typical hepatic mitochondrion, followed by ingrowth of the outer membrane in the manner of a closing iris diaphragm. The completion of the outer membrane incursions led to the separation of the small daughter mitochondria from their giant “parent” organelles. This process is repeated many times over until all of the mitochondria are restored to normal dimensions. A similar fission sequence was later demonstrated by Larsen (1970) to take place with great frequency in normal-sized mitochondria in the regenerating fat body of the skipper butterfly, an indication of the ubiquity of this cleavage phenomenon.
Because of the aforementioned relatively short half-life of cardiac mitochondria, these organelles also go through a self-replicative process to maintain their numbers at a requisite level. The fission phenomenon of cardiac mitochondria can be enhanced in frequency by external forces—administration to mice of the copper-chelating agent, cuprizone, results in a significant increase in cardiac mitochondrial number. Copper deficiency in general results in cardiac hypertrophy with a dramatic increase in mitochondria, possibly as a compensatory mechanism for a reduction in cytochrome c oxidase and ATP synthase (Medeiros and Jennings, 2002). With cuprizone, this increase is characterized by the appearance of numerous partitioned mitochondria (Tandler and Hoppel, 1972a). An interesting quirk of these dividing cardiac organelles is that the two sets of cristae on opposite sides of the partition are oriented perpendicular to each other, so that if these internal membranes on one side of the partition are seen in profile, the other set is seen en face.
In addition to its cardiac effects, dietary cuprizone also elicits the formation of hepatic megamitochondria (Tandler and Hoppel, 1973). Deletion of this agent from the diet results in an extraordinarily rapid (7–9 hours) recovery (in terms of size) of these organelles. Partitions are not involved in this recovery. Instead, the giant mitochondria undergo medial attenuation, becoming thinner and thinner until membranes from opposite sides meet and fuse, liberating two smaller daughter mitochondria. As is the case with partitioned megamitochondria, repetition of this bisection process quickly results in size normalization of the hepatocyte mitochondrial population. Thus, there appears to exist two different morphological pathways for mitochondrial division. The selection of which pathway will be operative is under the control of an unknown factor. The medial attenuation pathway of mitochondrial division fits the molecular model based on yeast studies, as proposed by Chan (2006). This model supposes that a necklace of a dynamin-related protein (Drp1) encircles a mitochondrion and by constricting ultimately separates the organelle into two smaller versions of itself. Clearly, much remains to be elucidated concerning the molecular mechanisms of mitochondrial division.
4.2. Mitochondrial glycogen
A rather consistent occurrence in cardiac megamitochondria is inclusions of β-glycogen (monoparticulate). According to Kraus and Cain (1980), such inclusions are due to the accidental entrapment of cytosolic glycogen during the enlargement process (obviously meaning by fusion—ed. note). If this were the case, then the deposits of glycogen should be enclosed in a double membrane representing the two limiting membranes of an involved mitochondrion—instead, they are delimited by just one membrane, indicating that they are ensconced in the mitochondrial outer compartment. Buja et al. (1972) encountered intramitochondrial β-glycogen deposits in cardiomyocytes of dogs exposed to 2–4 weeks of normothermic anoxic cardiac arrest. They propose several mechanisms for the intramitochondrial formation of glycogen, including the notion that solubilized enzymes of glycogen synthesis diffuse into the mitochondria, ultimately leading to glycogen formation. It should be noted that intramitochondrial glycogen is not the exclusive province of cardiac mitochondria -- such inclusions were first reported to occur in mitochondria of the oncocytes of Warthin’s tumor (Tandler and Shipkey, 1964) and of oncocytomas [where they may be composed of α-glycogen (clustered particles) as well as β-glycogen (monoparticulate)] (Tandler et al., 1970) and in hepatic mitochondria of mice subjected to dietary (Tandler et al., 1968) or pharmacological (Walter et al., 1980) manipulation.
4.3. Endurance training
In the course of enlargement brought about by endurance training of rats, cristae in giant mitochondria are disorganized and reduced in number, with a concomitant loss of matrix density (Coleman et al., 1987). Although there are no direct measurements of mitochondrial function in hearts from endurance-trained human beings, certain physiological parameters dealing with this organ have been examined. Such training leads to hearts that have an enhanced stroke volume and cardiac output, indicating an enlargement of the heart itself. As is the case with copper deficiency, this enlargement may have been brought about by an increase in mitochondrial number, or, alternatively, by a marked increase in megamitochondria. Administration of cobalt to dogs leads to extreme swelling of cardiac mitochondria with attendant matrix rarefaction accompanied by loss of cristae (Sandusky et al., 1981). A similar reduction of matrix density is brought about in rat normal-sized cardiac mitochondria by ischemia (Lesnefsky et al., 2004).
4.4. Ischemic mitochondrial damage
Human hearts respond to ischemia with a series of degenerative events, some involving mitochondria, ultimately resulting in cell death via apoptosis and necrosis. Although it was originally thought that it was during reperfusion that mitochondria suffered their major damage, recent studies in rats have shown that it actually is during ischemia that injury to both SSM and IFM takes place. In marked contrast, ischemia of rabbit heart does not result in a reduction of matrix density of cardiac mitochondria and the induced mitochondrial injury (decreased oxidative phosphorylation through cytochrome oxidase) affects only the SSM, not the IFM (Lesnefsky et al., 1997); with prolongation of the ischemic period, the biochemical damage (cardiolipin and cytochrome c reduction) incurred by these organelles progressively increases.
In rabbits, this ischemic progression occurs only in SSM -- neither these nor IFM exhibit any reduction in matrix density. Irrespective of species or type of cardiac mitochondria, there is early in ischemia a reversible reduction in ANT and ATPase (complex V), followed by a loss of activity of complexes III and IV – the net result is that oxidative phosphorylation is significantly impaired (Lesnefsky et al., 2001). The loss of cytochrome c signals a point of no return from ischemia – once past, these biochemical impairments are irreversible. If reperfusion is initiated before this juncture, the opening of the mitochondrial permeability transition pore in cardiac mitochondria exacerbates the mitochondrial damage (Halestrap, 2006; Honda and Ping, 2006).
4.5. Intramitochondrial rods and crystalloids
Dense rods (more probably narrow plates) originally were reported to occur in beef heart mitochondria (Hall and Crane, 1970, 1971). These were described as occurring within cristae, but subsequent studies of heart ultrastructure in many other species revealed that such rods occurred not only within cristae, but in the outer compartment in general. Because these structures have not to our knowledge been observed in hearts that were fixed by vascular perfusion, the rods may be an artifact engendered by the delay (and resultant hypoxia) inherent in immersion fixation. Their number increases if the ablated heart is cooled on ice for one hour (Saito et al., 1974). The increased presence of such rods in isolated heart mitochondria probably is due to the lack of oxygenation during the extended time required for their preparation. Because rods were not present in the mitochondria of freeze-dried myocardial cells but appeared after conventional fixation and dehydration, Heine and Schaeg (1979) concluded that they were artifacts of processing. It should be noted, however, that their freeze-drying technique completely effaced the outer compartment and its intracristal spaces, which could have affected rods that were there in the first place. The composition of these rods is completely unknown but they appear to have little or no influence on mitochondrial performance (Saito et al., 1974).
Paracrystalline plate-like inclusions, which in profile appear as rods, are engendered in giant mitochondria of creatine-free cultured cardiomyocytes (Eppenberger-Eberhardt et al., 1991). Similar intramitochondrial inclusions have been experimentally induced in rat skeletal muscle (O’Gorman et al., 1997) where octamers of creatine kinase appear to be their principal, if not sole, constituent (Stadhouders et al., 1994). Compared to controls, the mitochondria in such experimental muscles had slower rates of state 4 and of maximal state 3 respiration (O’Gorman et al., 1997). The three-dimensional structure of intramitochondrial crystalloids in human mitochondrial myopathies has been examined by conventional electron microscopy by high angle tilting of thin sections and by using freeze-fracture techniques (Mukherjee et al., 1986). The crystalloids consist of planar arrays of 8 nm particles. Like the case of rods, the pathophysiological significance of these structures remains undetermined.
4.6 Cardiomyopathy
Strong evidence for the differences between the two populations of mitochondria in the heart is provided by a strain of hamsters (BIO14.6) with congenital dystrophic myopathy that evolves into cardiomyopathy. Approximately four months postnatally, these hamsters develop a mitochondrial defect in the heart (Hoppel et al., 1982). This defect is restricted to the IFM, in which oxidative phosphorylation falls 40–50%. In contrast, oxidative phosphorylation in SSM is unaffected.
Heart failure is the inability of the heart to carry out its canonical pumping function. In this section, we will deal with only the energetic components of the pumping process, leaving the contractile apparatus to more qualified investigators. A Focus Article in the New England Journal of Medicine (Neubauer, 2007)) placed the defect in heart failure squarely in the province of mitochondrial dysfunction, a sentiment echoed by Marin-Garcia and Goldenthal (2008). Although studies of human heart are relatively few in number, most publications strongly support this supposition.
Sixty-six endocardial biopsies obtained from 48 patients with cardiomyopathy (a form of heart failure) showed variable cellular architecture that often involved mitochondria (Baandrup et al., 1981). In some cells, mitochondrial alterations included a striking increase in number, rarefaction of their matrices, and occasional giant mitochondria. Initial biochemical studies relied entirely on tissue homogenates of explanted hearts, but did not make use of isolated mitochondria. Such homogenates revealed a significant decrease in creatine and creatine phosphate, suggesting a depletion of energetic reserves (Kalsi et al., 1999). Coupled with a decrease in citrate synthase (a mitochondrial marker enzyme) content, the foregoing observations point to a loss of mitochondria as the basis for the bioenergetic decrements. Further studies by Jarreta et al. (2000) of homogenates of human hearts from patients with idiopathic and ischemic dilated cardiomyopathy found a decrease in mitochondrial electron transport chain complex III activity. These workers concluded that the mitochondrial respiratory chain dysfunction is secondary to the heart failure and not a primary mitochondrial disease. Using similar heart preparations, Quigley et al. (2000) -- in contrast to Jarreta et al. (2000) -- found decreased tissue citrate synthase activity, indicating decreased mitochondrial content, a finding consonant with that of Kalsi et al. (1999). In addition, Quigley et al. (2000) employed isolated mitochondria, which were found to have a decreased ratio of cytochrome c oxidase/citrate synthase activity. They concluded that, in humans, the slowing of ATP production by mitochondria might be a key factor in cardiac ventricular dysfunction.
Dörner et al. (2006) subsequently studied the kinetics of ANT isoforms in dilated cardiomyopathy and found a switch in the two isoforms present in this pathological condition. This switch limits entry of ADP into mitochondria so that ATP formation is slowed. This finding applied exclusively to dilated cardiomyopathy and not to other forms of the disease.
Sharov et al. (2000), using permeabilized fibers from myocardium of patients in heart failure, found that oxidative phosphorylation was, on the average, approximately halved. Both idiopathic and ischemic heart failure gave the same results.
In order to determine the precise role of mitochondria in heart failure, Sabbah et al. (1991) developed a canine model of heart failure. The model is based on repeated intracoronary injection of microspheres, producing microembolization of the ventricles, and leading to chronic heart failure (Sabbah, 1991, 1992). In this model, the number of mitochondria per unit volume increased, but these organelles were smaller than conventional ones. Mitochondrial abnormalities included loss of matrix density and the appearance of intraorganelle myelin figures. Using skinned fibers from dog heart, Sharov et al. (1998) obtained results similar to those from skinned fibers of human origin supporting the usefulness of the dog model as a surrogate for human heart.
A further advance in elucidating the role of mitochondria in heart failure was achieved by the isolation of these organelles from the aforementioned dog heart model (Rosca et al, 2008). Both SSM and IFM had a 50% reduction in oxidative phosphorylation without a decrease in the activity of the ETC complexes. This seemingly contradictory finding is explainable by a decrease in the proposed functional supercomplexes, namely, respirasomes. The latter are assemblages of complex I, the dimer of complex III, and from one to four copies of complex IV (Schägger, 2001). This structural unit is believed to facilitate and coordinate electron transfer in the most efficient way possible. When all is said and done, it appears that mitochondria are pivotal structures in heart failure and that further detailed information on their structure and function will provide greater insight into the human scourge of heart failure.
5. Aging
Although strictly speaking aging is not a disease, it does have profound effects on cardiac mitochondrial performance. All things being equal, older hearts are less amenable to therapy than are hearts in younger individuals. Although the source(s) of this phenomenon is(are) not immediately apparent, the mitochondrial theory of aging (reviewed by Lesnefsky and Hoppel, 2006), which is steadily gaining acceptance, suggests that mitochondria in older hearts could be the origin of waning cardiac capacity. Here again, studies in this regard require laboratory animals. There is very little evidence that mitochondria increase to giant size in cells of aging mammals and when it does occur, only in hepatocytes. A study by Sohal (1970) showed enlarged mitochondria in cardiomyocytes in Drosophila, but this study is based on a short-lived invertebrate. What relationship such a finding has to mammalian heart cells is unclear.
Experimental studies in both adult and aged rats highlighted the significance of the two populations of mitochondria in cardiomyocytes (Fannin et al., 1999). Although SSM remain at adult levels of activity, the IFM of hearts from aged rats show a marked decrease both in number and in certain functional activities. The latter include state 3 respiration (oxidative phosphorylation) and complexes III and IV activity. These functional changes take place in the absence of structural alterations in the affected organelles.
Another arena in which aging may have cardiac effects is in ischemia (Lesnefsky and Hoppel, 2003). We previously described the effects of ischemia on cardiac mitochondrial function. These effects are exaggerated in older individuals. Here again, the SSM sustain a degree of damage comparable to those in adults, whereas in IFM ischemia elicits a degree of damage over and above that produced by aging alone. We postulate that this additive phenomenon is responsible for the increased cardiac damage following ischemia in elderly patients compared to adults.
These mitochondrial findings relative to ischemia and aging have practical implications for human patients. If the changes in IFM that accompany aging can be outweighed by induction of new, pristine components of the affected complexes, then the ischemia that accompanies heart attacks would have less dire consequences. This has been accomplished in rats through the prophylactic use of acetylcarnitine (Lesnefsky et al., 2006). If this therapeutic regimen (with appropriate modifications) has similar effects in human patients, it will be a boon to mankind.
6. Conclusions
Mitochondria are the engines that drive the pumping action of the heart. Many afflictions of the heart originate in mitochondria or have a mitochondrial component. Much of what we know concerning myocardial pathology is based on study of laboratory animals-- this information often is extrapolated to the human condition. What currently is lacking and is urgently needed is direct information concerning the human heart. Biochemists need to make use of human disposable cardiac tissue obtainable at surgery, following ethical guidelines. Examination of such specimens will help to determine whether human cardiac mitochondria are true simulacra of those in laboratory animals. If these human mitochondria turn out to be identical to those in animals, especially in terms of the two populations, it would increase confidence in the use of animals to investigate the most basic mechanisms of cardiac mitochondrial pathology and to facilitate the design of rational therapies.
A major problem hardly touched upon by recent studies concerns the significance of crista configuration. Why do SSM have lamelliform cristae, whereas IFM have predominately tubular cristae? Is this disparate morphology related to mitochondrial function? Is it connected in some fashion to respirasomes, which are components of the cristae membranes? To take this question one step farther, how are respirasomes per se assembled and what directs them to their final location? With questions such as these in mind, it can be concluded that mitochondria once again have returned to the central position in heart research that they formerly occupied.
Fig. 4.

Rat; SEM. Several rows of in situ IFM have mainly lamelliform cristae.
Acknowledgments
This work was supported in part by NIH grants PO1 AG015885 and P01 HL074237 and by grants from the Fondazione Banco di Sardegna and MIUR. The expert technical assistance of Dr. Gabriele Conti and of Kiet Luc is gratefully acknowledged.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
EXPLANATION OF FIGURES
Note: In each micrograph (with the exception of Fig 1B), the scale line equals 1 μm; in Fig. 1A, the scale line equals 0.5 μm.
References
- Arbustini E, Diegoli M, Fasani R, Grasso M, Morbini P, Banchieri N, Bellini O, Dal Bello B, Pilotto A, Magrini G, Campana C, Fortini P, Gavazzi A, Narula J, Viganò M. Mitochondrial DNA mutations and mitochondrial abnormalities in dilated cardiomyopathy. Am J Pathol. 1998;153:1501–10. doi: 10.1016/S0002-9440(10)65738-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aschenbrenner V, Druyan R, Rabinowitz M. Haem o, cytochrome c and total protein turnover in mitochondria from rat heart and liver. Biochem J. 1970;119:157–60. doi: 10.1042/bj1190157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakeeva LE, Yu S, Chentsov, Skulachev VP. Mitochondrial framework (Reticulum mitochondriale) in rat diaphragm muscle. Biochim Biophys Acta. 1978;501:349–69. doi: 10.1016/0005-2728(78)90104-4. [DOI] [PubMed] [Google Scholar]
- Bakeeva LE, Yu S, Chentsov, Skulachev VP. Intermitochondria contacts in myocardiocytes. J Mol Cell Cardiol. 1983;15:413–20. doi: 10.1016/0022-2828(83)90261-4. [DOI] [PubMed] [Google Scholar]
- Baandrup U, Florio RA, Roters F, Olsen EG. Electron microscopic investigation of endomyocardial biopsy samples in hypertrophy and cardiomyopathy. A semiquantitative study in 48 patients. Circulation. 1981;63:1289–98. doi: 10.1161/01.cir.63.6.1289. [DOI] [PubMed] [Google Scholar]
- Bhatnagar MK, Yamashiro S. Ultrastructural alterations of the myocardium of rats fed rapeseed oil. Res Vet Sci. 1979;26:183–8. [PubMed] [Google Scholar]
- Buja LM, Ferrans VJ, Levitsky S. Occurrence of intramitochondrial glycogen in canine myocardium after prolonged anoxic cardiac arrest. Mol Cell Cardiol. 1972;4:237–54. doi: 10.1016/0022-2828(72)90061-2. [DOI] [PubMed] [Google Scholar]
- Chan DC. Mitochondrial fusion and fission in mammals. Annu Rev Cell Dev Biol. 2006;22:79–99. doi: 10.1146/annurev.cellbio.22.010305.104638. [DOI] [PubMed] [Google Scholar]
- Coleman R, Silberstein M, Gershon D, Reznick AZ. Giant mitochondria in the myocardium of aging and endurance-trained mice. Gerontology. 1987;33:34–9. doi: 10.1159/000212851. [DOI] [PubMed] [Google Scholar]
- Daems WT, Wisse E. Shape and attachment of the cristae mitochondriales in mouse hepatic cell mitochondria. J Ultrastruct Res. 1966;16:123–40. doi: 10.1016/s0022-5320(66)80027-8. [DOI] [PubMed] [Google Scholar]
- Detmer SA, Chan DC. Functions and dysfunctions of mitochondrial dynamics. Nature Rev Mol Cell Biol. 2007;8:870–9. doi: 10.1038/nrm2275. [DOI] [PubMed] [Google Scholar]
- Dörner A, Giessen S, Gaub R, Grosse Siestrup H, Schwimmbeck PL, Hetzer R, Poller W, Schultheiss HP. An isoform shift in the cardiac adenine nucleotide translocase expression alters the kinetic properties of the carrier in dilated cardiomyopathy. Eur J Heart Fail. 2006;8:81–9. doi: 10.1016/j.ejheart.2005.05.003. [DOI] [PubMed] [Google Scholar]
- Eppenberger-Eberhardt M, Riesinger I, Messerli M, Schwarb P, Müller M, Eppenberger HM, Wallimann T. Adult rat cardiomyocytes cultured in creatine-deficient medium display large mitochondria with paracrystalline inclusions, enriched for creatine kinase. J Cell Biol. 1991;113:289–302. doi: 10.1083/jcb.113.2.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fannin SW, Lesnefsky EJ, Slabe TJ, Hassan HO, Hoppel CL. Aging selectively decreases oxidative capacity in rat heart interfibrillar mitochondria. Arch Biochem Biophys. 1999;372:399–407. doi: 10.1006/abbi.1999.1508. [DOI] [PubMed] [Google Scholar]
- Fawcett DW, McNutt NS. The ultrastructure of the cat myocardium. I. Ventricular papillary muscle. J Cell Biol. 1969;42:1–45. doi: 10.1083/jcb.42.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross NJ. Control of mitochondrial turnover under the influence of thyroid hormone. J Cell Biol. 1971;48:29–40. doi: 10.1083/jcb.48.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halestrap AP. Calcium, mitochondria and reperfusion injury: a pore way to die. Biochem Soc Trans. 2006;34:232–7. doi: 10.1042/BST20060232. [DOI] [PubMed] [Google Scholar]
- Hall JD, Crane FL. An intracristal structure in beef heart mitochondria. Exp Cell Res. 1970;62:480–3. doi: 10.1016/0014-4827(70)90583-5. [DOI] [PubMed] [Google Scholar]
- Hall JD, Crane FL. A new structure in beef heart mitochondria. J Cell Biol. 1971;48:420–5. doi: 10.1083/jcb.48.2.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heine H, Schaeg G. Origin and function of ‘rod-like structures’ in mitochondria. Cells Tissues Organs. 1979;103:1–10. doi: 10.1159/000144992. [DOI] [PubMed] [Google Scholar]
- Honda HM, Ping P. Mitochondrial permeability transition in cardiac cell injury and death. Cardiovasc Drugs Ther. 2006;20:425–32. doi: 10.1007/s10557-006-0642-0. [DOI] [PubMed] [Google Scholar]
- Hoppel CL, Tandler B, Parland W, Turkaly JS, Albers LD. Hamster cardiomyopathy—A defect in oxidative phosphorylation in the cardiac interfibrillar mitochondria. J Biol Chem. 1982;257:1540–8. [PubMed] [Google Scholar]
- Jarreta D, Orús J, Barrientos A, Miró O, Roig E, Heras M, Moraes CT, Cardellach F, Casademont J. Mitochondrial function in heart muscle from patients with idiopathic dilated cardiomyopathy. Cardiovasc Res. 2000;45:860–5. doi: 10.1016/s0008-6363(99)00388-0. [DOI] [PubMed] [Google Scholar]
- Kalsi KK, Smolenski RT, Protchard RD, Khaghani A, Seymour AM, Yacoub MH. Energetics and function of the failing human heart with dilated or hypertrophic cardiomyopathy. Eur J Clin Invest. 1999;29:469–77. doi: 10.1046/j.1365-2362.1999.00468.x. [DOI] [PubMed] [Google Scholar]
- Kraus B, Cain H. Giant mitochondria in the human myocardium—morphogenesis and fate. Virchows Arch B Cell Pathol. 1980;33:77–89. doi: 10.1007/BF02899172. [DOI] [PubMed] [Google Scholar]
- Kroemer G. mitochondrial control of apoptosis: an introduction. Biochem Biophys Res Commun. 2003;304:433–5. doi: 10.1016/s0006-291x(03)00614-4. [DOI] [PubMed] [Google Scholar]
- Larsen WJ. Genesis of mitochondria in insect fat body. J Cell Biol. 1970;47:373–83. [PMC free article] [PubMed] [Google Scholar]
- Lesnefsky EJ, Chen Q, Slabe TJ, Stoll MSK, Minkler PE, Hassan M, Tandler B, Hoppel CL. Ischemia, rather than perfusion, inhibits respiration through cytochrome oxidase in the isolated, perfused rabbit heart: role of cardiolipin. Am J Physiol. 2004;287:H258–67. doi: 10.1152/ajpheart.00348.2003. [DOI] [PubMed] [Google Scholar]
- Lesnefsky EJ, He DC, Moghaddas S, Hoppel CL. Reversal of mitochondrial defects before ischemia protects the aged heart. FASEB J. 2006;20:1543–5. doi: 10.1096/fj.05-4535fje. [DOI] [PubMed] [Google Scholar]
- Lesnefsky EJ, Hoppel CL. Ischemia-reperfusion injury in the aged heart: Role of mitochondria—Minireview. Arch Biochem Biophys. 2003;420:287–97. doi: 10.1016/j.abb.2003.09.046. [DOI] [PubMed] [Google Scholar]
- Lesnefsky EJ, Hoppel CL. Oxidative phosphorylation and aging. Ageing Res Rev. 2006;5:402–33. doi: 10.1016/j.arr.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Lesnefsky EJ, Moghaddas S, Tandler B, Kerner J, Hoppel CL. Mitochondrial dysfunction in cardiac disease: ischemia-reperfusion, aging, and heart failure. J Mol Cell Cardiol. 2001;33:1065–89. doi: 10.1006/jmcc.2001.1378. [DOI] [PubMed] [Google Scholar]
- Lesnefsky EJ, Tandler B, Ye J, Slabe TJ, Turkaly J, Hoppel Cl. Myocardial ischemia decreases oxidative phosphorylation through cytochrome oxidase in subsarcolemmal mitochondria. Am J Physiol. 1997;273:H1544–54. doi: 10.1152/ajpheart.1997.273.3.H1544. [DOI] [PubMed] [Google Scholar]
- Luft R, Ikkos D, Palmieri G, Ernster L, Afzelius B. A case of severe hypermetabolism of nonthyroid origin with a defect in the maintenance of mitochondrial respiratory control: a correlated clinical, biochemical, and morphological study. J Clin Invest. 1962;41:1776–804. doi: 10.1172/JCI104637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manella C. The relevance of mitochondrial membrane topology to mitochondrial function. Biochim Biophys Acta. 2006;1762:140–7. doi: 10.1016/j.bbadis.2005.07.001. [DOI] [PubMed] [Google Scholar]
- Marin-Garcia J, Goldenthal MJ. Mitochondrial centrality in heart failure. Heart Fail Rev. 2008;13:137–50. doi: 10.1007/s10741-007-9079-1. [DOI] [PubMed] [Google Scholar]
- Medeiros DM, Jennings D. Role of copper in mitochondrial biogenesis via interaction with ATP synthase and cytochrome c synthase. J Bioenerg Biomembr. 2002;34:389–95. doi: 10.1023/a:1021206220851. [DOI] [PubMed] [Google Scholar]
- Mukherjee TM, Dixon BR, Blumbergs PC, Swift JG, Hallpike JF. The fine structure of the intramitochondrial crystalloids in mitochondrial myopathy. J Submicrosc Cytol. 1986;18:595–604. [PubMed] [Google Scholar]
- Neely JR, Morgan HW. Relationship between carbohydrate and lipid metabolism and the energy balance of heart muscle. Annu Rev Biochem. 1974;36:413–59. doi: 10.1146/annurev.ph.36.030174.002213. [DOI] [PubMed] [Google Scholar]
- Neubauer S. The failing heart—an engine out of fuel. N Engl J Med. 2007;356:1140–51. doi: 10.1056/NEJMra063052. [DOI] [PubMed] [Google Scholar]
- O’Gorman E, Fuchs KH, Tittman P, Wallimann T. Crystalline mitochondrial inclusion bodies isolated from creatine depleted soleus muscle. J Cell Sci. 1997;110:1403–11. doi: 10.1242/jcs.110.12.1403. [DOI] [PubMed] [Google Scholar]
- Palmer JW, Tandler B, Hoppel CL. Biochemical properties of subsarcolemmal and interfibrillar mitochondria isolated from rat cardiac muscle. J Biol Chem. 1977;252:8731–9. [PubMed] [Google Scholar]
- Palmer JW, Tandler B, Hoppel CL. Heterogeneous response of subsarcolemmal heart mitochondria to calcium. Am J Physiol. 1986;250:H741–8. doi: 10.1152/ajpheart.1986.250.5.H741. [DOI] [PubMed] [Google Scholar]
- Perkins G, Renken C, Martone ME, Young SJ, Ellisman M, Frey T. Electron tomography of neuron mitochondria. Three-dimensional structure and organization of cristae and membrane contacts. J Struct Biol. 1997;119:260–72. doi: 10.1006/jsbi.1997.3885. [DOI] [PubMed] [Google Scholar]
- Quigley AF, Kapsa RM, Esmore D, Hale G, Byrne E. Mitochondrial respiratory chain activity in idiopathic dilated cardiomyopathy. J Card Fail. 2000;6:47–55. doi: 10.1016/s1071-9164(00)00011-7. [DOI] [PubMed] [Google Scholar]
- Riva A, Faa G, Loffredo F, Piludu M, Testa Riva F. An improved OsO4 maceration method by high resolution scanning electron microscopy. Scanning Microsc. 1999;13:111–22. [Google Scholar]
- Riva A, Tandler B, Lesnefsky EJ, Conti G, Loffredo F, Vazquez E, Hoppel CL. Structure of cristae in cardiac mitochondria of aged rat. Mech Age Develop. 2006;127:917–921. doi: 10.1016/j.mad.2006.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riva A, Tandler B, Loffredo F, Vazquez E, Hoppel CL. Structural differences in two biochemically defined populations of cardiac mitochondria. Am J Physiol. 2005;289:H868–72. doi: 10.1152/ajpheart.00866.2004. [DOI] [PubMed] [Google Scholar]
- Rosca MG, Vazquez EJ, Kerner J, Parland W, Chandler MP, Stanley W, Sabbah HN, Hoppel CL. Cardiac mitochondria in heart failure: Decrease in respirasomes and oxidative phosphorylation. Cardiovasc Res. 2008;80:30–9. doi: 10.1093/cvr/cvn184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabbah HN, Sharov V, Riddle JM, Kono T, Lesch M, Goldstein S. Mitochondrial abnormalities in myocardium of dogs with chronic heart failure. J Mol Cell Cardiol. 1992;24:1333–47. doi: 10.1016/0022-2828(92)93098-5. [DOI] [PubMed] [Google Scholar]
- Sabbah HN, Stein PD, Kono T, Gheorghiade M, Levine TB, Jafri S, Hawkins ET, Goldstein S. A canine model of chronic heart failure produced by multiple sequential microembolizations. Am J Physiol. 1991;260:H1379–84. doi: 10.1152/ajpheart.1991.260.4.H1379. [DOI] [PubMed] [Google Scholar]
- Saito A, Smigel M, Fleischer S. Membrane junctions in the intermembrane space of mitochondria from mammalian tissues. J Cell Biol. 1974;60:653–63. doi: 10.1083/jcb.60.3.653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandusky GE, Henk WG, Roberts ED. Histochemistry and ultrastructure of the heart in experimental cobalt cardiomyopathy in the dog. Toxicol Appl Pharmacol. 1981;61:89–98. doi: 10.1016/0041-008x(81)90010-7. [DOI] [PubMed] [Google Scholar]
- Schägger H. Respiratory chain supercomplexes. IUBMB Life. 2001;52:119–28. doi: 10.1080/15216540152845911. [DOI] [PubMed] [Google Scholar]
- Sharov VG, Goussey A, Lesch M, Goldstein S, Sabbah HN. Abnormal mitochondrial function in myocardium of dogs with chronic heart failure. J Mol Cell Cardiol. 1998;30:1757–62. doi: 10.1006/jmcc.1998.0739. [DOI] [PubMed] [Google Scholar]
- Sharov VG, Todor AV, Silverman N, Goldstein S, Sabbah HN. Abnormal mitochondrial respiration in failed human myocardium. J Mol Cell Cardiol. 2000;32:2361–7. doi: 10.1006/jmcc.2000.1266. [DOI] [PubMed] [Google Scholar]
- Sohal RS. Mitochondrial changes in the heart of Drosophila replete Wollaston with age. Exp Gerontol. 1970;5:213–6. doi: 10.1016/0531-5565(70)90040-9. [DOI] [PubMed] [Google Scholar]
- Stadhouders AM, Jap PH, Winkler HP, Eppenberger HM, Wallimann T. Mitochondrial creatine kinase: a major constituent of pathological inclusions seen in mitochondrial myopathies. Proc Natl Acad Sci USA. 1994;91:5089–93. doi: 10.1073/pnas.91.11.5089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tandler B, Dunlap M, Hoppel CL, Hassan M. Giant mitochondria in a cardiomyopathic heart. Ultrastruct Pathol. 2002;26:177–83. doi: 10.1080/01913120290076847. [DOI] [PubMed] [Google Scholar]
- Tandler B, Erlandson RA, Smith AL, Wynder EL. Riboflavin and mouse hepatic cell structure and function. II. Division of mitochondria during recovery from simple deficiency. J Cell Biol. 1969;41:477–93. doi: 10.1083/jcb.41.2.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tandler B, Erlandson RA, Wynder EL. Riboflavin and mouse hepatic cell structure and function. I. Ultrastructural alterations in simple deficiency. Am J Pathol. 1968;52:69–95. [PMC free article] [PubMed] [Google Scholar]
- Tandler B, Hoppel CL. Possible division of cardiac mitochondria. Anat Rec. 1972a;173:309–24. doi: 10.1002/ar.1091730306. [DOI] [PubMed] [Google Scholar]
- Tandler B, Hoppel CL. Mitochondria. Academic Press; New York: 1972b. [Google Scholar]
- Tandler B, Hoppel CL. Division of giant mitochondria during recovery from cuprizone intoxication. J Cell Biol. 1973;56:266–72. doi: 10.1083/jcb.56.1.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tandler B, Hutter RVP, Erlandson RA. Ultrastructure of oncocytoma of the parotid gland. Lab Invest. 1970;23:567–80. [PubMed] [Google Scholar]
- Tandler B, Shipkey FH. Ultrastructure of Warthin’s tumor. I. Mitochondria. J Ultrastruct Res. 1964;11:292–305. doi: 10.1016/s0022-5320(64)90034-6. [DOI] [PubMed] [Google Scholar]
- Vogel F, Bornhövd C, Neupert W, Reichert AS. Dynamic subcompartmentalization of the mitochondrial inner membrane. J Cell Biol. 2006;175:237–47. doi: 10.1083/jcb.200605138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter RJ, Tandler B, Hoppel CL. Ultrastructural and biochemical effects of D-penicillamine on mouse hepatocytes. Anat Rec. 1980;197:289–95. doi: 10.1002/ar.1091970303. [DOI] [PubMed] [Google Scholar]