

Abstract
FoxO1 integrates multiple metabolic pathways. Nutrient levels modulate FoxO1 acetylation, but the functional consequences of this posttranslational modification are unclear. To answer this question, we generated mice bearing alleles that encode constitutively acetylated and acetylation-defective FoxO1 proteins. Homozygosity for an allele mimicking constitutive acetylation (Foxo1KQ/KQ) results in embryonic lethality, due to cardiac and angiogenesis defects. In contrast, mice homozygous for a constitutively deacetylated Foxo1 allele (Foxo1KR/KR) display a unique metabolic phenotype of impaired insulin action on hepatic glucose metabolism, but decreased plasma lipid levels and low respiratory quotient, consistent with a state of preferential lipid usage. Moreover, Foxo1KR/KR mice show a dissociation between weight gain and insulin resistance in predisposing conditions (high fat diet, diabetes and Insulin receptor mutations), possibly due to decreased cytokine production in adipose tissue. Thus acetylation inactivates FoxO1 during nutrient excess whereas deacetylation selectively potentiates FoxO1 activity, protecting against excessive catabolism during nutrient deprivation.
INTRODUCTION
The discovery of FoxO transcription factors as distal effectors of insulin signaling marks a watershed in metabolic research (Accili and Arden, 2004; Kimura et al., 1997). A burgeoning consensus indicates that FoxOs (especially FoxO1) regulate liver glucose production (Dong et al., 2008; Matsumoto et al., 2007; Nakae et al., 2002; Nakae et al., 2001a), hypothalamic neuropeptide synthesis (Kitamura et al., 2006) and processing (Plum et al., 2009), adipocyte and myoblast differentiation (Hribal et al., 2003; Nakae et al., 2003), and pancreatic β cell turnover and function (Kitamura et al., 2002; Kitamura et al., 2005; Nakae et al., 2002; Okamoto et al., 2006).
FoxO proteins link the metabolic state of the organism to cellular gene expression, allowing target tissues to adopt a posture that reflects nutrient availability (FoxO off) or deprivation (FoxO on). As with other nutrient and stress sensors (Allen-Jennings et al., 2001), FoxOs are regulated via post-translational modifications. Akt-dependent phosphorylation inactivates FoxOs by causing their nuclear exclusion. In addition, FoxOs are acetylated by Cbp, and deacetylated by Class I-II and Class III (NAD-dependent) deacetylases (Brunet et al., 2004; Daitoku et al., 2004; van der Horst et al., 2004).
SirT1, a Class III deacetylase, affects lifespan in lower eukaryotes and requires the FoxO ortholog daf-16 for this effect (Imai et al., 2000; Tissenbaum and Guarente, 2001). As Sirt1 activation has beneficial effects on mammalian metabolism (Banks et al., 2008; Pfluger et al., 2008), including improvements in glucose tolerance and decreased hepatic glucose production, we proposed that it acts by promoting FoxO1 deacetylation (Banks et al., 2008). There has been considerable interest in understanding the effects of FoxO acetylation. At the cellular level, it can be shown that acetylation affects FoxO trafficking (Brunet et al., 2004; Frescas et al., 2005; Kitamura et al., 2005) and DNA binding (Daitoku et al., 2004; Kitamura et al., 2005). But the phenotypic consequences of this post-translational modification remain unclear (Daitoku et al., 2004; Kitamura et al., 2005; Motta et al., 2004), owing partly to the difficulty of conducting rigorous quantitative assessments of in vivo acetylation, partly to the interplay between acetylation, phosphorylation and protein stability (Qiang et al., 2010), and partly to the intrinsic shortcomings of assaying FoxO function in cultured cells. To answer this question, we generated two mouse models by introducing targeted mutations in the Foxo1 locus that yield alleles encoding either constitutively acetylated or acetylation-defective mutant FoxO1. In this study, we report that appropriate regulation of FoxO1 by acetylation is essential for survival. We show that acetylation of FoxO1 controls development, energy balance and nutrient homeostasis. We link these phenotypes with a selective regulation of gene expression that possibly reflects metabolic changes in response to prolonged fasting, or calorie restriction.
RESULTS
Generation of Foxo1 knock-in alleles
FoxO1 acetylation is physiologically regulated by nutritional status: it decreases when animals are fasted, and it increases after re-feeding (Figure 1A). We have previously identified seven lysine acetylation site at amino acid residues 219, 242, 245, 259, 262, 271, and 291 (Kitamura et al., 2005; Qiang et al., 2010). To understand the physiologic function of FoxO1 acetylation, we used homologous recombination in embryonic stem cells to generate mice bearing recombinant Foxo1 alleles in which the endogenous exon 2 has been replaced with a modified exon 2 encoding either a constitutively acetylated allele or a constitutively deacetylated allele (Figure S1). We refer to the former as Foxo1KQ allele (in which lysine has been replaced by glutamine) and to the latter as Foxo1KR allele (in which lysine has been replaced by arginine).
Figure 1. FoxO1 acetylation and localization.

(A) Immunoblot analysis of FoxO1 acetyl-lysine content in epididymal adipose tissue extracts from mice subjected to fasting and re-refeeding. (B) Fluorescence microscopy of primary mouse hepatocytes transduced with FoxO1-GFP-WT, -KR or -KQ adenoviruses and maintained in serum-free medium (upper panel) or incubated with insulin (lower panel). We show representative images. (C–D) Immunoblotting analysis of FoxO1 content in nuclear and cytosolic fractions isolated from epididymal adipose tissue extracts of fasted (C) or refed (D) wild type (WT) or Foxo1KR/KR (KR) animals. We used nucleophosmin (NPM) or tubulin as markers of cellular fractions in (C) and Hdac1 or βactin in (D).
We analyzed the subcellular distribution of KQ and KR FoxO1 by transduction of adenoviruses encoding the two mutants as GFP fusion proteins in primary mouse hepatocytes. In response to insulin, wild type FoxO1 redistributes from the nucleus to the cytoplasm. We observed that the KQ mutant was predominantly cytoplasmic, whereas the KR mutant was predominantly nuclear (Figure 1B). Consistent with these data, we observed increased FoxO1 immunoreactivity in nuclear fractions isolated from adipocytes of Foxo1KR/KR mice compared to wild type littermates, and decreased immunoreactivity in cytoplasmic fractions (Figure 1C). This pattern persisted in re-fed animals, consistent with impaired nuclear exclusion of the KR mutant (Fig. 1D). These findings indicate that the Foxo1KR allele confers gain-of-function, whereas the Foxo1KQ allele confers loss-of-function.
Embryonic lethality in Foxo1KQ/KQ mice due to defective vasculogenesis
Foxo1KQ/+ mice are viable and fertile. But no viable homozygous offspring resulted from the intercrossing of Foxo1KQ/+ mice, determined by genotyping of litters on postnatal day (P) 12 (Table S1). Between embryonic (E) day 9.5 and E11.5, we observed the expected Mendelian frequency of genotypes (χ2 = 4.10); however, all homozygous mutants were abnormal. At E11.5, all Foxo1KQ/KQ embryos (n = 3) lacked a heartbeat and were deteriorating (Figure 2A). At E10.5, the single Foxo1KQ/KQ embryo recovered had a heartbeat, but showed significant growth retardation and an enlarged pericardium (Figure 2B). At E9.5, Foxo1KQ/KQ embryos had a hypoplastic first branchial arch (I) and the second branchial arch (II) was either absent or hypoplastic; they also displayed a distended heart and expanded atrio-ventricular canal (n=8/10 assessed) (Figures 2C and 2D). Histological sections from a stage-matched Foxo1KQ/KQ and Foxo1+/+ pair further demonstrated the expanded atrio-ventricular canal and revealed disorganized trabeculi within the ventricle (Figures 2E and 2F).
Figure 2. Embryonic lethality in mice homozygous for the Foxo1KQ/KQ mutation.

(A–B) Microphotographs of Foxo1+/+ and Foxo1KQ/KQ embryos at E10.5 and E11.5. (C–D) Microphotographs of E9.5 embryos showing first (I) and second branchial arch (II), as well as atrioventricular canal (AVC) and ventricle (V). Arrow in D indicates the distended AVC in the mutant. (E–F) Histological sections of E9.5 embryos across the AVC (arrows) and ventricle. (G–L) Whole-mount PECAM-1 immunostaining with details of the aortic arch arteries in the head (arrowheads in I, missing in J) and the dorsal aorta (arrows) in the tail region (K–L) of a Foxo1+/+ (G, I, and K) and Foxo1KQ/KQ embryo (H, J, and L). Asterisks in k and l mark somites which are smaller in the mutant. (M–N) Histological sections through the first branchial arch showing dorsal aortae (arrows in M) and the first branchial arch artery (arrowheads in M). (M′-N′) Sections through more posterior regions showing paired dorsal aortae (arrows that are distended in the mutant (N′) compared with wild type (M′). (O–P) Vascular plexus in the yolk sac in Foxo1KQ/KQ and wild type embryos shown by immunostaining for PECAM.
Based on the defective angiogenesis and vascular remodeling in Foxo1−/− mice (Furuyama et al., 2004), we determined the extent of vascularization in Foxo1KQ/KQ mutants using whole-mount immunostaining with the endothelial-specific marker, PECAM-1 (Figures 2G through 2L). The vasculature in the head was significantly reduced in the Foxo1KQ/KQ compared to wild type, and the aortic arch arteries could not be resolved in the mutant (Figures 2I and 2J). Moreover, while the dorsal aorta appeared to be present in both wild type and mutant, the Foxo1KQ/KQ embryos displayed aortic PECAM-1 staining pattern that was more evident in the posterior of the embryo (Figures 2G through 2L). Confirmation of the underdeveloped or missing aortic arch arteries was apparent with histological analyses of transverse section (Figures 2M and 2N). Sections also showed minimal development of the dorsal aortae in the head (Figures 2M, 2N, 2E and 2F) but large, distended dorsal aortae in the posterior (Figures 2M′ and 2N′). Not noted in Foxo1 null embryos (Furuyama et al., 2004; Hosaka et al., 2004) was a defect in somite development that is apparent in Foxo1KQ/KQ embryos as small and irregular somites (Figures 2K and 2L). Finally, the vascular plexus in the yolk sac appears to have formed in the Foxo1KQ/KQ mutant (Figures 2O and 2P), but further development is compromised (data not shown).
Metabolic characterization of Foxo1KR/KR mutants
Unlike Foxo1KQ/KQ, Foxo1KR/KR mutants were born at term in Mendelian ratios and grew normally. Immunoblotting analyses of FoxO1 immunoprecipitated from liver extracts of 12-wk-old mice indicated that its acetyl-lysine content was substantially decreased, consistent with the notion that the seven mutated amino acid residues account for the bulk of FoxO1 acetylation (Figure S1).
Foxo1KR/KR mice showed normal glucose and insulin levels, as well as glucose tolerance and insulin tolerance, following 4-hr fast (data not shown). However, glucose and insulin levels were significantly increased after 18-hr fast or 4-hr re-feeding (Table S2). Accordingly, under these conditions Foxo1KR/KR mice showed a 50% increase of their HOMA-IR index, compared to littermate controls (Figure 3A). Given FoxO1’s role in hepatic glucose production, we subjected Foxo1KR/KR mice to pyruvate tolerance tests (Matsumoto et al., 2007). They showed increased glucose levels, consistent with increased hepatic glucose production (Figure 3B). We next performed hyperinsulinemic euglycemic clamp studies. Consistent with the slight increases of glucose and insulin, we found decreased glucose infusion rates (Figure 3C). In addition, rates of hepatic glucose production in the presence of maximal insulin concentrations (60 μU/ml) were 2.5-fold higher in Foxo1KR/KR mice, consistent with insulin resistance of hepatic glucose metabolism–even though the difference did not achieve statistical significance, owing to elevated individual variations among mice tested (Figure 3D). Whole body glucose uptake was unchanged, and glycolysis showed a non-significant trend toward reduction (Figure 3E). Several additional parameters measured during the clamps were normal (Figure S2).
Figure 3. Metabolic data in Foxo1KR/KR mice.

(A) HOMA-IR values calculated from the data in Table S2. (B) Pyruvate tolerance tests in WT and Foxo1KR/KR mice. (C) Glucose infusion rates, (D) Endogenous glucose production under hyperinsulinemic conditions, and (E) Glycolysis during euglycemic hyperinsulinemic clamps. (F) Determination of TG content in lipoprotein fractions isolated by FPLC analysis. (G) Basal and isoproterenol-stimulated glycerol release from primary cultures of epididymal fat pads. n= ≥ 6 mice per group and each experiment. *= P<0.05; ** =P<0.01 by 2-way ANOVA. Data are presented as means ± SEM.
In contrast with the data on glucose and insulin, we observed a significant decrease of plasma free fatty acids (FFA), triglyceride (TG) and total cholesterol levels, as well as liver TG content in the re-fed state (Table S2). Analysis of lipoprotein composition indicated that the reduction in TG levels was due primarily to decreased very-low-density lipoprotein (VLDL) TG (Figure 3F). However, the main apoprotein component of VLDL, ApoB, was expressed at normal levels (data not shown), as was the LDL receptor (Figure S2), and TG secretion was normal (data not shown), indicating that the reduction in TG levels is not secondary to decreased secretion or accelerated clearance through hepatic LDL receptors. Similar to TG, cholesterol levels were reduced in all lipoprotein fractions (Figure S2). De novo lipogenesis, measured using incorporation of 3H2O into fatty acids, decreased by ~20% in Foxo1KR/KR mice (21.86 vs. 27.03 to μmol/g protein/hr).
To determine the contribution of adipose tissue lipolysis to the observed changes in TG and FFA levels, we measured FFA suppression during glucose clamps and glycerol release from isolated epididymal fat pads under basal and isoproterenol-stimulated conditions. The former was identical between the two strains (Figure S2), while the latter was slightly increased in Foxo1KR/KR mice in the presence of isoproterenol (Figure 3G), indicating that the lower plasma TG and FFA in mutant mice are not secondary to decreased adipocyte lipolysis.
Selective regulation of FoxO1-dependent gene expression in Foxo1KR/KR mice
To understand the pathophysiology of the dissociation between impaired glucose and improved lipid metabolism, we analyzed gene expression in fasted and re-fed Foxo1KR/KR mice. In both conditions, we found 1.5 to 2-fold increases in the expression of hepatic gluconeogenetic genes, including G6pc, Pck1, and Pgc1α (Matsumoto et al., 2007; Puigserver et al., 2003) (Figure 4A). Conversely, we found 50–70% decrease of Gck and 30% decrease of Pkrl, two key regulators of glycolysis. These changes dovetail with the decreased sensitivity of HGP to insulin suppression, and with the decreased glycolysis observed in the clamps. This concerted shift to increase glucose production and inhibit glucose breakdown was specific, as several additional FoxO1 targets, including Hnf4α, InsR, and Irs2, were unaffected (data not shown).
Figure 4. Gene expression analysis.

(A)Hepatic expression of genes involved in glucose and (B, C) lipid metabolism in 2-month-old male mice. (D) Adipose expression of fatty acid and TG synthetic genes in the same cohort. n = ≥ 6 for each genotype. *=P<0.05, **=P<0.01 by Student’s t-test. Data are presented as means ± SEM.
With regard to lipid metabolism, we found decreases of Scd1 and Elovl3, genes required for unsaturated fatty acid synthesis and fatty acid elongation, of the fatty acid transporter Cd36, of the liver hormone Fgf21, and of Pparγ (Figure 4B). TG synthetic genes Srebf1, Chrebp and their targets Fasn and Acc1 were normal, whereas Pdk4 was increased, suggesting that carbons are being diverted to lipid oxidation (Figure 4C). These data are consistent with the observed decrease of fatty acid synthesis, and with increased lipid oxidation, and might explain the decreases in plasma TG and FFA levels. To understand whether these changes resulted from direct hepatic effects of the Foxo1KR/KR mutation, we performed analyses of basal and hormone-regulated gene expression in isolated primary hepatocytes. The results were largely consistent with those described above, supporting the conclusion that the KR mutation affects hepatic gene expression in a cell-autonomous manner (Figure S3).
We next analyzed adipose tissue. Unlike in liver, we detected substantial increases of Chrebp and its targets Fasn and Acc1, as well as Elovl3 and Dgat2. Similar to the liver, expression of Scd1 was decreased (Figure 4D). These findings suggest that Foxo1KR/KR adipocytes are poised to synthesize TG from saturated FFA (Guillou et al., 2010), and might thus explain the reduction of plasma TG through increased deposition in adipocytes, consistent with the increased weight gain of Foxo1KR/KR mice when exposed to nutrient excess (see below). We didn’t find significant gene expression changes in skeletal muscle, consistent with the fact that the latter is not a site of FoxO1 expression (not shown) (Kitamura et al., 2007).
The KR allele fails to exacerbate insulin resistance on predisposed backgrounds
To explore in more detail the phenotype of Foxo1KR/KR mice, we analyzed their metabolic profiles in response to nutritional and genetic forms of insulin resistance. The prediction of these studies was that, if the KR allele acted as a bona fide gain-of-function allele, it ought to exacerbate insulin resistance and promote the transition to frank diabetes. In the first experiment, we backcrossed Foxo1KR/KR mice on the C57BL6 background and subjected them to high-fat diet (HFD). Foxo1KR/KR mice consistently gained more weight than littermate controls (Figure 5A), due to increased fat mass (Figure 5B). Moreover, adipose tissue of HFD-fed Foxo1KR/KR mice revealed substantial increases of Elovl3 and Fasn (Figure S4), while liver TG and cholesterol content remained lower than wild type littermates (Figure S4). Contrary to our predictions, glycemia and intra-peritoneal glucose tolerance were identical between Foxo1KR/KR and controls (Figure 5C). There were no differences in plasma leptin, adiponectin, and resistin levels (Table S3), while TNF-α values were decreased, and adipocyte Mcp1 expression was lower (Figure S4), in the absence of histological changes to adipose tissue between two genotypes (data not shown). These findings are consistent with the possibility that adipose tissue of Foxo1KR/KR mice is less proinflammatory, and might thus protect them against worsening glucose tolerance despite the increased body weight.
Figure 5. Characterization of Foxo1KR/KR mice in dietary and genetic models of insulin resistance.

(A) Body weight curves, (B) body composition, and (C) intraperitoneal glucose tolerance tests in WT and Foxo1KR/KR mice during high fat feeding. (D) Body weight, (E) 16-hr fasting glucose levels, (F) insulin levels in random fed, 16-hr fasted, and 4-hr re-fed, (G) glucose tolerance tests in male and (H) female db/db and Foxo1KR/KR;db/db mice. n=6–12 for each genotype and each experiment. *=P<0.05, **=P<0.01 by 2-way ANOVA. Data are presented as means ± SEM.
Likewise, when Foxo1KR/KR mice were intercrossed with db/db mice, they tended to have greater body weights than db/db controls (Figure 5D), but had similar glucose (Figure 5E) and insulin levels (Figure 5F) in the random-fed and 4-hr re-fed states. Interestingly, fasting insulin levels were significantly higher in Foxo1KR/KR:db/db than in db/db mice (Figure 5F). Moreover, both genders of Foxo1KR/KR:db/db mice displayed worsening glucose tolerance (Figures 5G and 5H).
The results in these two models indicate that the KR substitutions predispose to weight gain, but hardly exacerbate insulin resistance. To test the potential synergistic effects of the Foxo1KR/KR mutation with insulin resistance more directly, we introduced the KR allele on a background of non-obese insulin resistance, Insulin Receptor haploinsufficient mice (InsR+/−) (Kido et al., 2000). Foxo1KR/KRInsR+/− double mutant mice showed a trend toward higher fasting glucose levels (Figure 5I), associated with increased fasting and fed insulin levels (Figure 5J). However, glucose levels following an intra-peritoneal glucose challenge were identical (Figure 5K), and the hypoglycemic response to insulin was slightly impaired only at late time points (≥30 min) during intraperitoneal insulin tolerance tests (data not shown).
In sum, these experiments indicate that the KR mutations predispose to weight gain, but fail to cause overt diabetes in strains predisposed to it by underlying insulin resistance.
Energy balance in Foxo1KR/KR mice
The findings above suggest a selective effect of the KR allele on energy balance. We therefore analyzed Foxo1KR/KR mice in indirect calorimetry experiments. Food intake and body weight in Foxo1KR/KR were similar to controls (Figures 6A and 6B). Continuous monitoring of respiratory exchanges and locomotor activity in metabolic cages showed normal VO2 and locomotion in Foxo1KR/KR mice, but decreased CO2 generation and resting energy expenditure (Figures 6C through 6E). These data indicate increased reliance on lipids as energy source, and can possibly explain the lower FFA and TG levels. To investigate differences in basal metabolic rate, we compared the energy expenditure required for locomotor activity in Foxo1KR/KR and control mice (Banks et al., 2008; Ravussin et al., 1986). Foxo1KR/KR mice showed lower resting energy expenditure in the face of unchanged activity (Figure 6F), possibly reflecting altered substrate preference.
Figure 6. Energy homeostasis studies.
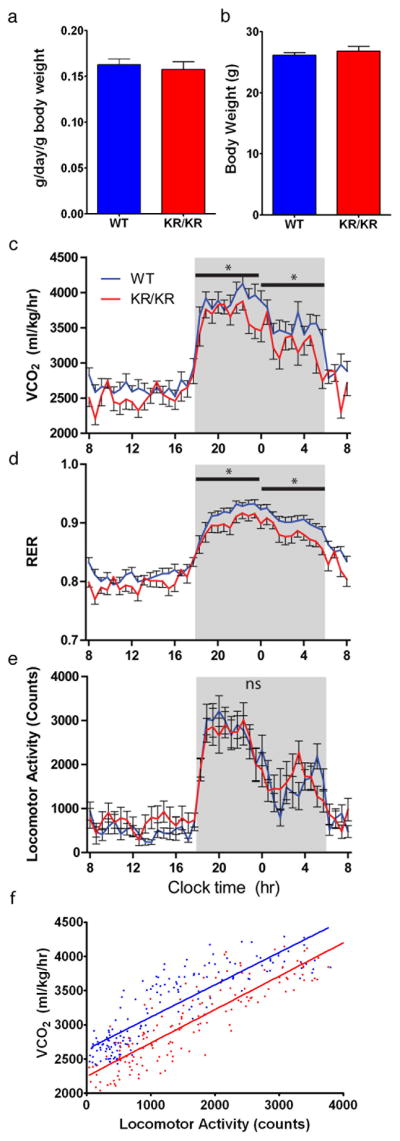
(A) Food intake, (B) body weight, (C) energy expenditure as measured by CO2 produced (VCO2), (D) respiratory exchange ratio (CO2 produced/O2 consumed) (RER), (E) locomotor activity, and (F) metabolic efficiency plots in 2 month old WT and Foxo1KR/KR mice maintained on standard chow diet. n=8 for each genotype and each experiment. *=P<0.05 by 2-way ANOVA. Data are presented as means ± SEM.
Discussion
Functional role of acetylation in FoxO1 function
There has been controversy on the role of FoxO1 acetylation. Most (Daitoku et al., 2004; Kitamura et al., 2005; van der Heide and Smidt, 2005; van der Horst et al., 2004), but not all reports (Fukuoka et al., 2003; Motta et al., 2004; Yang et al., 2005) conclude that it decreases FoxO1 activity. Our data demonstrate conclusively that acetylation is an “off” signal, as Foxo1KQ/KQ embryos are phenotypically undistinguishable from Foxo1−/− mutants (Furuyama et al., 2004; Hosaka et al., 2004).
The Foxo1KQ/KQ mutant as a model of nutritional embryopathy
FoxO1 acetylation occurs in response to nutrient excess, for example hyperglycemia (Cheng et al., 2009; Kitamura et al., 2005). Thus, the KQ allele can be construed to mimic the effects of nutrient excess on embryonic development. In this regard, the embryonic lethality of Foxo1KQ/KQ mice provides intriguing similarities with elusive aspects of diabetic embryopathy (Zabihi and Loeken, 2010). While we found no evidence of the characteristic defects in neural tube formation of this condition in Foxo1KQ/KQ mice, the latter display abnormalities in branchial arch and cardiovascular development reminiscent of those found in diabetic embryos (Morgan et al., 2008). Moreover, some of the genes implicated in the pathogenesis of diabetic embryopathy have also been shown to be FoxO1 targets, for example Gpx1 (Wentzel et al., 2008).
Mixed sensitivity and resistance to insulin in Foxo1KR/KR mutants
The Foxo1KR/KR mutants display three striking metabolic sub-phenotypes: (i) Altered hepatic glucose metabolism. The decreased sensitivity to insulin-mediated suppression of glucose production likely results from abnormal regulation of FoxO1-dependent gene expression and impaired regulation of FoxO1 localization. Together with decreased rates of glycolysis, this phenotype mirrors the defects seen in the diabetic liver (Bouche et al., 2004). (ii) Decreased plasma lipid levels and hepatic lipid content. The decreased expression of lipogenic genes and increased lipid oxidation genes—together with a lower respiratory quotient are consistent with a state of preferential lipid usage. (iii) Increased weight gain under predisposing conditions without further worsening of glucose metabolism. This finding is likely explained by increased TG synthesis in adipose tissue, leading to preferential fat storage in adipocytes vs. liver or other tissues.
To help interpret these seemingly disparate observations, it behooves us to compare the sub-phenotypes linked to the gain-of-function KR allele with those of mice expressing another gain-of-function FoxO1 mutant: the phosphorylation-deficient, but acetylation-competent S253A (Table S3) (Nakae et al., 2002). This mutation prevents insulin-dependent phosphorylation, and results in a constitutively nuclear FoxO1 (Nakae et al., 2001b). The S253A mutant increases expression of FoxO1 target genes in glucose production, as does the KR allele (Nakae et al., 2002; Okamoto et al., 2006). But the similarities end there, for the S253A mutant increases TG and cholesterol levels (Altomonte et al., 2004), as well as VLDL secretion (Kamagate et al., 2008), while the KR mutant has opposite effects. Another phosphorylation-defective FoxO1 mutant, bearing mutations of the three main phosphorylation sites (ADA) (Nakae et al., 2001b), also promotes lipogenesis and hepatic lipid deposition, while inhibiting lipid oxidation. But the ADA mutant, unlike the KR mutant, increases Akt phosphorylation. Thus, the increase in lipid synthesis in that instance is probably secondary to Akt activation (Matsumoto et al., 2006). While comparisons between knock-in mice and transgenics or adenovirus-mediated overexpression experiments have to be interpreted circumspectly, this body of work indicates that the KR allele has a distinctive gene expression signature. Without oversimplifications, it can be said that it acts like a pure gain-of-function mutant on hepatic glucogenesis, and as dominant negative on lipid metabolism, lipoprotein turnover and food intake.
The decrease in circulating plasma lipid levels is likely multifactorial, owing to increased expression of TG synthetic genes in adipose tissue, associated with changes in hepatic lipid metabolism genes: the decreases in Scd1 and Pparγ would be expected to decrease hepatic lipid content (Gavrilova et al., 2003; Gutierrez-Juarez et al., 2006), while that of Elovl3 can be associated with low VLDL (Zadravec et al., 2010). Interestingly, FoxO1 has been shown to inhibit PPARγ (Dowell et al., 2003; Nakae et al., 2003) potentially accounting for aspects of the gene expression profile. Indeed, mice lacking either InsR or its main substrates Irs1 and Irs2 in liver–two situations that can also be construed as FoxO1 gain of function–phenocopy key aspects of the KR allele (Biddinger et al., 2008; Dong et al., 2008). In sum, FoxO1 deacetylation promotes TG deposition in adipose tissue and lipid catabolism in liver.
Mechanism and physiologic significance of the KR phenotype
These findings raise two interrelated questions: what is the mechanism by which the KR allele alters the FoxO1-dependent gene expression signature, and what physiologic situation does it recapitulate?
With regard to the first question, the gene expression data can be explained by recalling the dual mechanism of action of FoxO1: as a transcription factor, and as a co-regulator of gene expression (Kitamura et al., 2007; Matsumoto et al., 2006; Ramaswamy et al., 2002). Pure transcriptional targets of FoxO1 (e.g., G6pc, Igfbp1, Pgc1α) are equally induced by the deacetylated vs. dephosphorylated mutant, whereas genes in which FoxO1 has the ability to act as a coregulator (e.g., Srebf1, Pck1, Gck) (Matsumoto et al., 2006) appear to be regulated in a diverging manner by acetylation- vs. phosphorylation-defective mutants. We speculate that this is due to different coactivator and corepressor complexes recruited by the two mutants (Kitamura et al., 2006).
The physiologic significance of these observations is best understood by considering that metabolic pathways are highly regulated by acetylation and that protein deacetylation occurs in response to prolonged fasting or calorie restriction (Schwer and Verdin, 2008). Under these conditions, dampening the FoxO1-dependent catabolic response can facilitate the transition from a physiologic fast (4–6 hr in a mouse) to a more prolonged state of nutrient deprivation. Thus, changes in gene expression seen in Foxo1KR/KR mice could be construed as mimicking the shift from a pure glucogenic state (short fast) to a lipolytic state (prolonged fast), as demonstrated by the increased glycerol release from isoproterenol-stimulated fat pads (Figure 3F). In other words, acetylation becomes a failsafe mechanism to prevent excessive FoxO1 activity.
Relationship between FoxO1 deacetylation and SirT1
We have reported that gain-of-function of the FoxO1 deacetylase SirT1 mimics some effects of calorie restriction, promotes insulin sensitivity, and causes FoxO1 deacetylation in mice (SirBACO mice) (Banks et al., 2008). We had inferred that the effects of SirT1 were mediated in large part through FoxO1. The present analysis prompts a critical reevaluation of that inference. Foxo1KR/KR and SirBACO mice share limited metabolic features. Thus, we conclude that the effects of SirT1 are largely FoxO1-independent in mice. Conversely, it’s likely that deacetylases other than SirT1 play a role in modulating FoxO1 function (Mihaylova et al., 2011; Wang et al., 2011). The identification of these proteins has important therapeutic implications.
MATERIALS AND METHODS
Generation of knock-in mice
We amplified the Foxo1 locus around exon 2 from 129/SvEv DNA using Phusion polymerase (Finnzymes), and replaced the EcoR I/Hind III fragment with sequences encoding either FoxO1-KR or FoxO1-KQ (Kitamura et al., 2005). We selected homologous recombinants using a floxed pGK-neomycin cassette inserted 1 kb upstream of exon 2, and a diphtheria toxin-A cassette and detection by Southern blotting of ES cell DNA digested with Kpn I with a probe external to the targeting vector. ES cell manipulations were as described (Accili et al., 1996), and were followed by intercrosses with ROSA26-cre transgenics to delete the neomycin cassette prior to breeding to homozygosity. PCR genotyping primers are: 5′-GCACCTTCAGTCGCCGTCAA and 5′-CCACAGGAGAATACAAGAGGAAGGC. The wild-type allele is 394-bp and the knock-in allele is 448 bp. Animals were backcrossed onto C57BL/6Jdb/+mice [B6.BKS(D)-Leprdb/LeprJ, JAX stock 000697] for ten generations prior to diet studies and indirect calorimetry.
Adenovirus Generation and Cell Culture
We generated adenoviruses encoding WT-, KR- or KQ-FoxO1-GFP using AdEasy (Stratagene) and obtained primary hepatocytes as described (Banks et al., 2008). We carried out adenovirus transduction 4 hr after isolation.
Mice and Embryo collection
We collected embryos from timed matings between Foxo1KQ/+ mice, considering 12 PM on the day of appearance of a vaginal plug embryonic day (E) 0.5. We assessed litters at E9.5, E10.5, E11.5, and postnatal day (P) 12, and carried out embryo dissections in cold PBS, using a dissecting microscope (Leica MZ8). We fixed embryos in 4% paraformaldehyde (PFA), washed them three times in PBS, 1:1 PBS/MeOH, and 100% MeOH, and performed immunostaining. We genotyped embryos using yolk sac specimens.
Wholemount immunostaining
We carried out wholemount immunohistochemistry with rat anti-mouse CD31 and platelet endothelial cell adhesion molecule-1 (PECAM-1) antibodies (BD Biosciences). Briefly, after bleaching with MeOH:DMSO:H2O2 (4:1:1) for 5-hr, we rehydrated embryos with 50% MeOH in PBT (PBS, 0.2% BSA, 0.5% Triton-X), followed by one wash in PBT and five washes in PBSMT (PBS, 2% non-fat milk powder, 0.5% Triton-X). We incubated embryos with primary antibody in PBSMT, washed and incubated them with horseradish peroxidase-conjugated antibody (sc-2005, Santa Cruz). We performed DAB staining in PBT containing 0.08% NaCl, 0.25 mg/ml DAB, and 0.03% H2O2 for 20–60 min, followed by fixation in 4% PFA, or fluorescent staining with Cy3-donkey anti-rat secondary antibody (Jackson Immunoresearch). We imaged embryos using a Nikon SMZ1500 microscope or a Zeiss LSM5 Exciter confocal microscope.
Histology
For H&E histological analysis, we embedded embryos in OCT, after fixation with 4% PFA and cryopreservation in 30% sucrose. We obtained transverse frozen sections (8 μm) using a cryostat (Microm HM525, Thermo Scientific), and imaged them using a Leica CTR6500 microscope.
Metabolic characterization and energy balance studies
We measured blood or plasma glucose using a glucose monitor (One Touch Ultra, Lifescan); insulin, leptin, resistin, and adiponectin (Linco/Millipore) and TNF–α (BD Biosciences) by ELISA, plasma triglyceride (Infinity), cholesterol and non-esterified fatty acids by colorimetric assays (Cholesterol E and NEFA C, Wako Pure Chemicals). Glucose, insulin, and pyruvate tolerance tests have been described (Matsumoto et al., 2007; Nakae et al., 2002). We collected blood between 10:00 AM and 12:00 PM to reduce variability. For lipoprotein separation, we pooled serum samples of six mice per genotype and applied them to FPLC gel filtration on two Superose 6 columns in series (Amersham Bioscience). We collected eluates in 0.5ml fractions at a flow rate of 0.7 ml/min. We assayed TG and cholesterol from column eluates and from liver extracts as described (Haeusler et al., 2010). To measure hepatic glycogen content, we homogenized frozen liver in 6% perchloric acid, adjusted to pH 6–7 with KOH followed by incubation with 1 mg/ml amyloglucosidase (Sigma) in 0.2 M acetate (pH 4.8) and quantification of glucose released (glycogen breakdown value – PCA value). We carried out hyperinsulinemic-euglycemic clamp studies as described (Lin et al., 2010). We performed indirect calorimetry using the Oxymax Comprehensive Lab Animal Monitoring System (Columbus Instruments) and measured body composition with either Piximus DEXA scanner (GE Healthcare) or NMR (Bruker Optics) (Banks et al., 2008).
RNA and protein analysis
We used standard RNA extraction and real-time RT-PCR and western blotting procedures (Matsumoto et al., 2006). Primer sequences are available on request. Anti-acetylated-FoxO1 antibody was from Santa Cruz Biotechnology (#sc-49437).
Statistical analysis
All results are presented as mean ± SEM. We calculate P values by unpaired Student’s t – tests or 2-way ANOVA.
Supplementary Material
Hghlights.
FoxO1 acetylation results in its loss-of-function
Homozygosity for acetylated Foxo1 alleles phenocopies Foxo1 knockout
Homozygosity for deacetylated Foxo1 alleles impairs glucose metabolism, lowers TG
Deacetylation regulates FoxO1 target gene selection during nutrient deprivation
Acknowledgments
Supported by NIH grants DK079496 (to ASB), DK057539 and HL087123 (to DA), DK63608 (Columbia University Diabetes & Endocrinology Research Center) and DK76169 (Yale Mouse Metabolic Phenotyping Center). We thank members of the Accili laboratory for helpful discussions of the data and critical reading of the manuscript, Dr. Thomas Ludwig for the gift of the DT-A plasmid, Dr. Lori Sussel for making her laboratory available for embryo analysis, and Dr. Bruce Spiegelman for guidance and support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Accili D, Arden KC. FoxOs at the crossroads of cellular metabolism, differentiation, and transformation. Cell. 2004;117:421–426. doi: 10.1016/s0092-8674(04)00452-0. [DOI] [PubMed] [Google Scholar]
- Accili D, Drago J, Lee EJ, Johnson MD, Cool MH, Salvatore P, Asico LD, Jose PA, Taylor SI, Westphal H. Early neonatal death in mice homozygous for a null allele of the insulin receptor gene. Nat Genet. 1996;12:106–109. doi: 10.1038/ng0196-106. [DOI] [PubMed] [Google Scholar]
- Allen-Jennings AE, Hartman MG, Kociba GJ, Hai T. The roles of atf3 in glucose homeostasis. a transgenic mouse model with liver dysfunction and defects in endocrine pancreas. J Biol Chem. 2001;276:29507–29514. doi: 10.1074/jbc.M100986200. [DOI] [PubMed] [Google Scholar]
- Altomonte J, Cong L, Harbaran S, Richter A, Xu J, Meseck M, Dong HH. Foxo1 mediates insulin action on apoC-III and triglyceride metabolism. J Clin Invest. 2004;114:1493–1503. doi: 10.1172/JCI19992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks AS, Kon N, Knight C, Matsumoto M, Gutierrez-Juarez R, Rossetti L, Gu W, Accili D. SirT1 gain of function increases energy efficiency and prevents diabetes in mice. Cell Metab. 2008;8:333–341. doi: 10.1016/j.cmet.2008.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biddinger SB, Hernandez-Ono A, Rask-Madsen C, Haas JT, Aleman JO, Suzuki R, Scapa EF, Agarwal C, Carey MC, Stephanopoulos G, et al. Hepatic insulin resistance is sufficient to produce dyslipidemia and susceptibility to atherosclerosis. Cell Metab. 2008;7:125–134. doi: 10.1016/j.cmet.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouche C, Serdy S, Kahn CR, Goldfine AB. The cellular fate of glucose and its relevance in type 2 diabetes. Endocr Rev. 2004;25:807–830. doi: 10.1210/er.2003-0026. [DOI] [PubMed] [Google Scholar]
- Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- Cheng Z, Guo S, Copps K, Dong X, Kollipara R, Rodgers JT, Depinho RA, Puigserver P, White MF. Foxo1 integrates insulin signaling with mitochondrial function in the liver. Nat Med. 2009;15:1307–1311. doi: 10.1038/nm.2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daitoku H, Hatta M, Matsuzaki H, Aratani S, Ohshima T, Miyagishi M, Nakajima T, Fukamizu A. Silent information regulator 2 potentiates Foxo1-mediated transcription through its deacetylase activity. Proc Natl Acad Sci U S A. 2004;101:10042–10047. doi: 10.1073/pnas.0400593101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong XC, Copps KD, Guo S, Li Y, Kollipara R, DePinho RA, White MF. Inactivation of hepatic Foxo1 by insulin signaling is required for adaptive nutrient homeostasis and endocrine growth regulation. Cell Metab. 2008;8:65–76. doi: 10.1016/j.cmet.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowell P, Otto TC, Adi S, Lane MD. Convergence of peroxisome proliferator-activated receptor gamma and Foxo1 signaling pathways. J Biol Chem. 2003;278:45485–45491. doi: 10.1074/jbc.M309069200. [DOI] [PubMed] [Google Scholar]
- Frescas D, Valenti L, Accili D. Nuclear trapping of the forkhead transcription factor FoxO1 via Sirt-dependent deacetylation promotes expression of glucogenetic genes. J Biol Chem. 2005;280:20589–20595. doi: 10.1074/jbc.M412357200. [DOI] [PubMed] [Google Scholar]
- Fukuoka M, Daitoku H, Hatta M, Matsuzaki H, Umemura S, Fukamizu A. Negative regulation of forkhead transcription factor AFX (Foxo4) by CBP-induced acetylation. Int J Mol Med. 2003;12:503–508. [PubMed] [Google Scholar]
- Furuyama T, Kitayama K, Shimoda Y, Ogawa M, Sone K, Yoshida-Araki K, Hisatsune H, Nishikawa S, Nakayama K, Nakayama K, et al. Abnormal angiogenesis in Foxo1 (Fkhr)-deficient mice. J Biol Chem. 2004;279:34741–34749. doi: 10.1074/jbc.M314214200. [DOI] [PubMed] [Google Scholar]
- Gavrilova O, Haluzik M, Matsusue K, Cutson JJ, Johnson L, Dietz KR, Nicol CJ, Vinson C, Gonzalez FJ, Reitman ML. Liver peroxisome proliferator-activated receptor gamma contributes to hepatic steatosis, triglyceride clearance, and regulation of body fat mass. J Biol Chem. 2003;278:34268–34276. doi: 10.1074/jbc.M300043200. [DOI] [PubMed] [Google Scholar]
- Guillou H, Zadravec D, Martin PG, Jacobsson A. The key roles of elongases and desaturases in mammalian fatty acid metabolism: Insights from transgenic mice. Prog Lipid Res. 2010;49:186–199. doi: 10.1016/j.plipres.2009.12.002. [DOI] [PubMed] [Google Scholar]
- Gutierrez-Juarez R, Pocai A, Mulas C, Ono H, Bhanot S, Monia BP, Rossetti L. Critical role of stearoyl-CoA desaturase-1 (SCD1) in the onset of diet-induced hepatic insulin resistance. J Clin Invest. 2006;116:1686–1695. doi: 10.1172/JCI26991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeusler RA, Han S, Accili D. Hepatic FoxO1 ablation exacerbates lipid abnormalities during hyperglycemia. J Biol Chem. 2010;285:26861–26868. doi: 10.1074/jbc.M110.134023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosaka T, Biggs WH, 3rd, Tieu D, Boyer AD, Varki NM, Cavenee WK, Arden KC. Disruption of forkhead transcription factor (FOXO) family members in mice reveals their functional diversification. Proc Natl Acad Sci U S A. 2004;101:2975–2980. doi: 10.1073/pnas.0400093101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hribal ML, Nakae J, Kitamura T, Shutter JR, Accili D. Regulation of insulin-like growth factor-dependent myoblast differentiation by Foxo forkhead transcription factors. J Cell Biol. 2003;162:535–541. doi: 10.1083/jcb.200212107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- Kamagate A, Qu S, Perdomo G, Su D, Kim DH, Slusher S, Meseck M, Dong HH. FoxO1 mediates insulin-dependent regulation of hepatic VLDL production in mice. J Clin Invest. 2008;118:2347–2364. doi: 10.1172/JCI32914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kido Y, Burks DJ, Withers D, Bruning JC, Kahn CR, White MF, Accili D. Tissue-specific insulin resistance in mice with mutations in the insulin receptor, IRS-1, and IRS-2. J Clin Invest. 2000;105:199–205. doi: 10.1172/JCI7917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura KD, Tissenbaum HA, Liu Y, Ruvkun G. daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science. 1997;277:942–946. doi: 10.1126/science.277.5328.942. [DOI] [PubMed] [Google Scholar]
- Kitamura T, Feng Y, Kitamura YI, Chua SC, Jr, Xu AW, Barsh GS, Rossetti L, Accili D. Forkhead protein FoxO1 mediates Agrp-dependent effects of leptin on food intake. Nat Med. 2006;12:534–540. doi: 10.1038/nm1392. [DOI] [PubMed] [Google Scholar]
- Kitamura T, Kitamura YI, Funahashi Y, Shawber CJ, Castrillon DH, Kollipara R, DePinho RA, Kitajewski J, Accili D. A Foxo/Notch pathway controls myogenic differentiation and fiber type specification. J Clin Invest. 2007;117:2477–2485. doi: 10.1172/JCI32054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura T, Nakae J, Kitamura Y, Kido Y, Biggs WH, 3rd, Wright CV, White MF, Arden KC, Accili D. The forkhead transcription factor Foxo1 links insulin signaling to Pdx1 regulation of pancreatic beta cell growth. J Clin Invest. 2002;110:1839–1847. doi: 10.1172/JCI200216857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura YI, Kitamura T, Kruse JP, Raum JC, Stein R, Gu W, Accili D. FoxO1 protects against pancreatic beta cell failure through NeuroD and MafA induction. Cell Metab. 2005;2:153–163. doi: 10.1016/j.cmet.2005.08.004. [DOI] [PubMed] [Google Scholar]
- Lin HV, Plum L, Ono H, Gutierrez-Juarez R, Shanabrough M, Borok E, Horvath TL, Rossetti L, Accili D. Divergent regulation of energy expenditure and hepatic glucose production by insulin receptor in agouti-related protein and POMC neurons. Diabetes. 2010;59:337–346. doi: 10.2337/db09-1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto M, Han S, Kitamura T, Accili D. Dual role of transcription factor FoxO1 in controlling hepatic insulin sensitivity and lipid metabolism. J Clin Invest. 2006;116:2464–2472. doi: 10.1172/JCI27047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto M, Pocai A, Rossetti L, Depinho RA, Accili D. Impaired regulation of hepatic glucose production in mice lacking the forkhead transcription factor Foxo1 in liver. Cell Metab. 2007;6:208–216. doi: 10.1016/j.cmet.2007.08.006. [DOI] [PubMed] [Google Scholar]
- Mihaylova MM, Vasquez DS, Ravnskjaer K, Denechaud PD, Yu RT, Alvarez JG, Downes M, Evans RM, Montminy M, Shaw RJ. Class IIa histone deacetylases are hormone-activated regulators of FOXO and mammalian glucose homeostasis. Cell. 2011;145:607–621. doi: 10.1016/j.cell.2011.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan SC, Relaix F, Sandell LL, Loeken MR. Oxidative stress during diabetic pregnancy disrupts cardiac neural crest migration and causes outflow tract defects. Birth Defects Res A Clin Mol Teratol. 2008;82:453–463. doi: 10.1002/bdra.20457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motta MC, Divecha N, Lemieux M, Kamel C, Chen D, Gu W, Bultsma Y, McBurney M, Guarente L. Mammalian SIRT1 represses forkhead transcription factors. Cell. 2004;116:551–563. doi: 10.1016/s0092-8674(04)00126-6. [DOI] [PubMed] [Google Scholar]
- Nakae J, Biggs WH, 3rd, Kitamura T, Cavenee WK, Wright CV, Arden KC, Accili D. Regulation of insulin action and pancreatic beta-cell function by mutated alleles of the gene encoding forkhead transcription factor Foxo1. Nat Genet. 2002;32:245–253. doi: 10.1038/ng890. [DOI] [PubMed] [Google Scholar]
- Nakae J, Kitamura T, Kitamura Y, Biggs WH, 3rd, Arden KC, Accili D. The forkhead transcription factor Foxo1 regulates adipocyte differentiation. Dev Cell. 2003;4:119–129. doi: 10.1016/s1534-5807(02)00401-x. [DOI] [PubMed] [Google Scholar]
- Nakae J, Kitamura T, Ogawa W, Kasuga M, Accili D. Insulin regulation of gene expression through the forkhead transcription factor Foxo1 (Fkhr) requires kinases distinct from Akt. Biochemistry. 2001a;40:11768–11776. doi: 10.1021/bi015532m. [DOI] [PubMed] [Google Scholar]
- Nakae J, Kitamura T, Silver DL, Accili D. The forkhead transcription factor Foxo1 (Fkhr) confers insulin sensitivity onto glucose-6-phosphatase expression. J Clin Invest. 2001b;108:1359–1367. doi: 10.1172/JCI12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto H, Hribal ML, Lin HV, Bennett WR, Ward A, Accili D. Role of the forkhead protein FoxO1 in beta cell compensation to insulin resistance. J Clin Invest. 2006;116:775–782. doi: 10.1172/JCI24967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfluger PT, Herranz D, Velasco-Miguel S, Serrano M, Tschop MH. Sirt1 protects against high-fat diet-induced metabolic damage. Proc Natl Acad Sci U S A. 2008;105:9793–9798. doi: 10.1073/pnas.0802917105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plum L, Lin HV, Dutia R, Tanaka J, Aizawa KS, Matsumoto M, Kim AJ, Cawley NX, Paik JH, Loh YP, et al. The obesity susceptibility gene Cpe links FoxO1 signaling in hypothalamic pro-opiomelanocortin neurons with regulation of food intake. Nat Med. 2009;15:1195–1201. doi: 10.1038/nm.2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puigserver P, Rhee J, Donovan J, Walkey CJ, Yoon JC, Oriente F, Kitamura Y, Altomonte J, Dong H, Accili D, et al. Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1alpha interaction. Nature. 2003;423:550–555. doi: 10.1038/nature01667. [DOI] [PubMed] [Google Scholar]
- Qiang L, Banks AS, Accili D. Uncoupling of acetylation from phosphorylation regulates FoxO1 function independent of its subcellular localization. The Journal of biological chemistry. 2010;285:27396–27401. doi: 10.1074/jbc.M110.140228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswamy S, Nakamura N, Sansal I, Bergeron L, Sellers WR. A novel mechanism of gene regulation and tumor suppression by the transcription factor FKHR. Cancer Cell. 2002;2:81–91. doi: 10.1016/s1535-6108(02)00086-7. [DOI] [PubMed] [Google Scholar]
- Ravussin E, Lillioja S, Anderson TE, Christin L, Bogardus C. Determinants of 24-hour energy expenditure in man. Methods and results using a respiratory chamber. The Journal of clinical investigation. 1986;78:1568–1578. doi: 10.1172/JCI112749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwer B, Verdin E. Conserved metabolic regulatory functions of sirtuins. Cell Metab. 2008;7:104–112. doi: 10.1016/j.cmet.2007.11.006. [DOI] [PubMed] [Google Scholar]
- Tissenbaum HA, Guarente L. Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature. 2001;410:227–230. doi: 10.1038/35065638. [DOI] [PubMed] [Google Scholar]
- van der Heide LP, Smidt MP. Regulation of FoxO activity by CBP/p300-mediated acetylation. Trends Biochem Sci. 2005;30:81–86. doi: 10.1016/j.tibs.2004.12.002. [DOI] [PubMed] [Google Scholar]
- van der Horst A, Tertoolen LG, de Vries-Smits LM, Frye RA, Medema RH, Burgering BM. FOXO4 is acetylated upon peroxide stress and deacetylated by the longevity protein hSir2(SIRT1) J Biol Chem. 2004;279:28873–28879. doi: 10.1074/jbc.M401138200. [DOI] [PubMed] [Google Scholar]
- Wang B, Moya N, Niessen S, Hoover H, Mihaylova MM, Shaw RJ, Yates JR, 3rd, Fischer WH, Thomas JB, Montminy M. A hormone-dependent module regulating energy balance. Cell. 2011;145:596–606. doi: 10.1016/j.cell.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wentzel P, Gareskog M, Eriksson UJ. Decreased cardiac glutathione peroxidase levels and enhanced mandibular apoptosis in malformed embryos of diabetic rats. Diabetes. 2008;57:3344–3352. doi: 10.2337/db08-0830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Hou H, Haller EM, Nicosia SV, Bai W. Suppression of FOXO1 activity by FHL2 through SIRT1-mediated deacetylation. EMBO J. 2005;24:1021–1032. doi: 10.1038/sj.emboj.7600570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabihi S, Loeken MR. Understanding diabetic teratogenesis: where are we now and where are we going? Birth Defects Res A Clin Mol Teratol. 2010;88:779–790. doi: 10.1002/bdra.20704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zadravec D, Brolinson A, Fisher RM, Carneheim C, Csikasz RI, Bertrand-Michel J, Boren J, Guillou H, Rudling M, Jacobsson A. Ablation of the very-long-chain fatty acid elongase ELOVL3 in mice leads to constrained lipid storage and resistance to diet-induced obesity. Faseb J. 2010;24:4366–4377. doi: 10.1096/fj.09-152298. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.