

Abstract
The unexpectedly high reactivity of a (2-benzoxazolyl) per-O-benzoyl-β-D-thioglucoside and related donors in reactions promoted by copper(II) trifluoromethanesulfonate is revealed, by comparison with the unreactive α-anomer, to be the result of neighboring group participation. Revision of the armed-disarmed concept for glycosyl donors is not required.
According to the armed-disarmed concept of Fraser-Reid glycosyl donors protected with ether protecting groups are armed and can be activated in the presence of disarmed donors carrying ester protecting groups.1 This seminal concept builds on the observations of earlier workers in the field,2 is widely applied in modern oligosaccharide synthesis,3 and is at the heart of the one pot oligosaccharide protocols developed by the Ley and Wong groups.4 Mechanistically, the effect is readily understood in terms of the destabilization of positive charge, whether full or partial, at the anomeric center in the course of most glycosylation reactions by electron-withdrawing protecting groups. Recently, it was observed that while the perbenzyl donor 1 in the series of benzoxazolyl thioglycosides 1–5 was the most reactive as expected on the basis of the armed-disarmed rationale, the 2-O-benzyl S-benzoxazolyl glycosides 2 and 3 were less reactive than the corresponding 2-O-acyl donors 4 and 5 on activation with copper(II) triflate.5
Similarly, it was found inter alia that the disaccharide 6 could be activated with copper(II) triflate in the presence of the acceptor 7 giving a 70% yield of trisaccahride 8 (Scheme 1) in apparent contravention of the armed-disarmed concept.
Scheme 1.

Trisaccharide synthesis
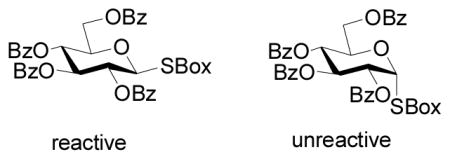
A rationale was advanced based on the differential stabilization of a fully developed positive charge (oxacarbenium ion) at the anomeric center by the protecting groups at O2 and/or O6, and on the changing ability of the “O5 lone pair” to eject the leaving group from the anomeric center according to the nature of the O6 protecting group.5 It occurred to us that the at first sight unusual reactivity patterns of Figure 1 and Scheme 1 are better explained on the basis of classical anchimeric assistance, and, if so, that they would be dependent on the anomeric configuration. We report here on the results of our investigation and confirm that neighboring group participation is the underlying cause of the observed reactivity sequence.
Figure 1.

Donor reactivity sequence with Cu(OTf)2
The α-analog (11) of 2 was synthesized from the known bromide 96 by hydrolysis to the pyranose and subsequent Mitsunobu reaction with benzoxazolethiol (Scheme 2).
Scheme 2.

Synthesis of the α-SBox donor 8
In a similar manner 2,3,4,6-tetra-O-benzoyl-D-glucopyranose 127 was transformed into a separable mixture of the α- and β-SBox glycosides 13 and 4, and the α-NBox glycoside 15 (Scheme 3). The β-SBox donors 1 and 2 were prepared according to the literature protocols.8
Scheme 3.

Synthesis of the α-SBox donor 13
The identity of donors 11 and 13 as the α-SBox derivatives, as opposed to the corresponding NBox derivatives (e.g., 14) was determined by inspection of the 13C-NMR spectra in which C2 of the benzoxazole is found around δ160 as compared to the more downfield shift of this carbon resonance in the N-glycosides (δ ~178), consistent with the thiocarbonyl nature of the latter system. These assignments were confirmed by the UV spectra, which showed the anticipated differences between 2-alkylthiobenzoxazoles and N-alkylbenzoxazol-2-thiones.9
A series of reactions were then conducted in which donors 1, 2, 4, 11, and 13 were activated with Cu(OTf)2 in dichloromethane at room temperature in the presence of 5A molecular sieves10 and 1,2;3,4-di-O-isopropylidene-α-D-galactopyranose, essentially according to the original conditions. The results obtained with donors 1, 2, and 4 (Table 1) conform to the pattern reported previously (Fig. 1). Thus, the perbenzyl donor 1 was the most reactive and was consumed within 2 h (Table 1, entry 1), whereas the perbenzoyl system 4 required 14 h (Table 1, entry 2). The 2-O-benzyl-3,4,6-tri-O-benzoyl-β-SBox derivative 2 was recovered in 85% yield after 14 h in a reaction that afforded only 14% of the anticipated glycoside (Table 1, entry 3). Turning to the α-SBox donors 11 and 13, the 2-O-benzyl-3,4,6-tri-O-benzoyl-α-SBox system 11 showed comparably poor reactivity to its β-anomer (Table 1, entry 4), whereas the per-O-benzoyl α-SBox donor 13 was completely unreactive under these conditions (Table 1, entry 5).
Table 1.
Coupling Reactions with 1,2;3,4-Di-O-isopropylidene-α-D-galactopyranosea
| entry | donor | time (h) | product | yield | α:β ratio | recovered donor |
|---|---|---|---|---|---|---|
| 1 |
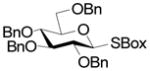 1 |
2 |
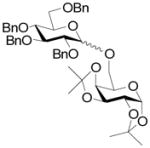 15 |
93% | 1:1.4 | 0% |
| 2 |
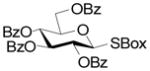 4 |
14 |
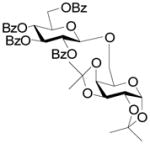 16 |
95% | β-only | 0% |
| 3 |
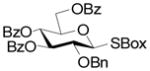 2 |
14 |
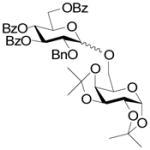 17 |
14% | α-only | 85% |
| 4 |
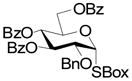 11 |
14 |
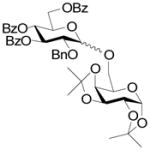 17 |
13% | α-only | 88% |
| 5 |
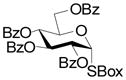 13 |
14 | - | No reaction | - | 95% |
all reactions were conducted in dichloromethane at room temperature with stoichiometric amounts of donor and acceptor, 2.4 equivalents of Cu(OTf)2, and in the presence of 5 A molecular sieves
The complete contrast in reactivity of the α- and β-anomers of the perbenzoyl donors 4 and 13 (Table 1, entries 2 and 5) is informative, especially when viewed alongside the very similar reactivity of the two anomers of the 2-O-benzyl-3,4,6-O-benzyl system 2 and 11 (Table 1, entris 3 and 4). We conclude that the enhanced reactivity of donor 4 with respect to its α-isomer 13 is simply a manifestation of the weak promoting system (copper(II) triflate) being assisted by participation of the 2-O-benzoate ester. Thus, unlike the case of the fully armed per-O-benzyl ether, the promoter is unable to cause departure of the anomeric leaving group from the formally more disarmed donor 4 without participation by the antiperiplanar ester group. When participation is stereoelectronically prevented, as in the case of 13, no reaction occurs at all. The 2-O-benzyl-3,4,6-O-benzoyl systems, both of which are moderately disarmed, but which can not benefit from neighboring group participation from the 2-position, show the anticipated reactivity between 1 and 13. It is anticipated that with stronger Lewis acids capable of promoting ionization without the need for neighboring group participation the reactivity order of the equatorial donors 1–5, and of donors 6 and 7, will revert to that predicted by the armed/disarmed hypothesis.
Comparable effects have been observed previously in carbohydrate chemistry. For example, Paulsen and Herold reported that β-glucosyl pentacetate 18 underwent reaction with antimony pentachloride to give a dioxolenium ion 19 and subsequent rearrangement products, under conditions in which the corresponding α-anomer 20 was unreactive (Fig. 2).11 Under the same conditions, the more reactive chlorides 21 and 22 were consummed irrespective of anomeric configuration. Konradsson studied the relative rates of reaction of a series of pentenyl glycosides and reported, inter alia, that the per-O-benzyl systems 23 and 24 showed only a 1.7 fold difference in reactivity favoring the β-isomer. In contrast, in the peracetylated systems 25 and 26 the β-anomer was found to be 5.2 times more reactive than its α-isomer (Fig. 2).12 Both groups attributed the enhanced reactivity of the 2-O-acetate protected β-configured donors to neighboring group participation by the ester.
Figure 2.

Literature examples of the phenomenon
We conclude that the unusual reactivity sequence reported for donors 1–5 on promotion with copper(II) trifluoromethanesulfonate is the result of a weakly promoted system being facilitated by neighboring group assistance from an antiperiplanar ester. The acceleration of solvolysis reactions by antiperiplanar vicinal ester groups is one of the classical concepts of physical organic chemistry from which carbohydrates are not immune.13 Revision of the seminal armed-disarmed concept is not required.
Supplementary Material
Acknowledgments
We thank the NIH (GM62160) for support of our work on the mechanisms of glycosylation reactions, and Professor A. V. Demchenko (University of Missouri St. Louis) for helpful discussions.
Footnotes
Supporting Information Available. Full experimental details and characterization data for new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Mootoo DR, Konradsson P, Udodong U, Fraser-Reid B. J Am Chem Soc. 1988;110:5583–5584. [Google Scholar]
- 2.(a) Glaudemans CPJ, Fletcher HG. J Am Chem Soc. 1965;87:4636–4641. [Google Scholar]; (b) Green LG, Ley SV. In: Carbohydrates in Chemistry and Biology. Ernst B, Hart GW, Sinaÿ P, editors. Vol. 1. Wiley-VCH; Weinheim: 2000. pp. 427–448. [Google Scholar]; (c) Horenstein NA. Adv Phys Org Chem. 2006;41:275–314. [Google Scholar]
- 3.(a) Garegg PJ. Adv Carbohydr Chem Biochem. 2004;59:69–134. doi: 10.1016/S0065-2318(04)59003-0. [DOI] [PubMed] [Google Scholar]; (b) Fraser-Reid B, Tatsuta K, Thiem J, editors. Glycoscience: Chemistry and Biology. Springer; Berlin: 2001. [Google Scholar]; (c) Toshima K, Tatsuta K. Chem Rev. 1993;93:1503–1531. [Google Scholar]
- 4.(a) Douglas NL, Ley SV, Lucking U, Warriner SL. J Chem Soc, Perkin Trans 1. 1998:51–65. [Google Scholar]; (b) Zhang Z, Ollmann IR, Ye X-S, Wischnat R, Baasov T, Wong C-H. J Am Chem Soc. 1999;121:734–753. [Google Scholar]
- 5.Kamat MN, Demchenko AV. Org Lett. 2005;7:3215–3218. doi: 10.1021/ol050969y. [DOI] [PubMed] [Google Scholar]
- 6.Lichtenthaler FW, Köhler B. Carbohydr Res. 1994;258:77–85. [Google Scholar]
- 7.Sebesan S, Neira S. Carbohydr Res. 1992;223:169–185. [Google Scholar]
- 8.Demchenko AV, Kamat MN, De Meo C. Synlett. 2003:1287–1290. [Google Scholar]
- 9.Zinner H, Peseke K. Chem Ber. 1965;98:3515–3519. [Google Scholar]
- 10.Without commenting on the reason, we note that reactions conducted in the presence of 4 rather than 5A molecular sieves were much slower with donors 1 and 4, and prohibitively so with donors 2, 11, and 13.
- 11.Paulsen H, Herold CP. Chem Ber. 1970;103:2450–2462. [Google Scholar]
- 12.Konradsson P. In: Glycoscience: Chemistry and Biolog. Fraser-Reid B, Tatsuta K, Thiem J, editors. Vol. 1. Springer; Berlin: 2001. pp. 535–549. [Google Scholar]
- 13.(a) Winstein S, Grunwald E, Ingraham LL. J Am Chem Soc. 1948;70:821–828. [Google Scholar]; (b) Capon B, McManus SP. Neighboring Group Participation. Plenum Press; New York: 1976. [Google Scholar]; (c) Anslyn EV, Dougherty DA. Modern Physical Organic Chemistry. University Science Books; Sausalito: 2006. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.