

Abstract
Genetic disorders of homocysteine (Hcy) or folate metabolism or high-methionine diets elevate plasma Hcy and its atherogenic metabolite Hcy-thiolactone. In humans, severe hyperhomocysteinemia due to genetic alterations in cystathionine β-synthase (Cbs) or methylenetetrahydrofolate reductase (Mthfr) results in neurological abnormalities and premature death from vascular complications. In mouse models, dietary or genetic hyperhomocysteinemia results in liver or brain pathological changes and accelerates atherosclerosis. Hcy-thiolactone has the ability to form isopeptide bonds with protein lysine residues, which generates modified proteins (N-Hcy-protein) with autoimmunogenic and prothrombotic properties. Our aim was to determine how N-Hcy-protein levels are affected by genetic or nutritional disorders in Hcy or folate metabolism in mice. We found that plasma N-Hcy-protein was elevated 10-fold in mice fed a high-methionine diet compared with the animals fed a normal commercial diet. We also found that inactivation of Cbs, Mthfr, or the proton-coupled folate transporter (Pcft) gene resulted in a 10- to 30-fold increase in plasma or serum N-Hcy-protein levels. Liver N-Hcy-protein was elevated 3.4-fold in severely and 11-fold in extremely hyperhomocysteinemic Cbs-deficient mice, 3.6-fold in severely hyperhomocysteinemic Pcft mice, but was not elevated in mildly hyperhomocysteinemic Mthfr-deficient animals, suggesting that mice have a capacity to prevent accumulation of N-Hcy-protein in their organs. These findings provide evidence that N-Hcy-protein is an important metabolite associated with Hcy pathophysiology in the mouse.—Jakubowski, H., Perła-Kaján, J., Finnell, R. H., Cabrera, R. M., Wang, H., Gupta, S., Kruger, W. D., Kraus, J. P., and Shih, D. M. Genetic or nutritional disorders in homocysteine or folate metabolism increase protein N-homocysteinylation in mice.
Keywords: methionine, thiolactone, liver damage, atherothrombosis
In mammals such as mice and humans, homocysteine (Hcy) is formed from the dietary methionine (Met) (1). Levels of Hcy are regulated by remethylation to Met, catalyzed by Met synthase and betaine-Hcy methyltransferase, and transsulfuration to cysteine, the first step of which is catalyzed by cystathionine β-synthase (CBS). The remethylation requires betaine or vitamin B12 and 5-methyltetrahydrofolate, generated by 5,10-methylenetetrahydrofolate reductase (MTHFR) (which requires riboflavin), whereas the transsulfuration requires vitamin B6. The requirements for cofactors are met entirely from the absorption of dietary B vitamins and folates in the small intestine.
Hcy is also metabolized to the thioester Hcy-thiolactone in an error-editing reaction in protein biosynthesis when Hcy is mistakenly selected in place of Met by methionyl-tRNA synthetase (2,3,4,5). The flow through the Hcy-thiolactone pathway increases when the remethylation or transsulfuration reaction is impaired by genetic alterations of enzymes, such as CBS (6, 7) or MTHFR (7, 8), by an inadequate supply of cofactors, e.g., 5-methyltetrahydrofolate (6, 9, 10), or by a high-Met diet (7).
Although there is a preponderance of data showing that total Hcy (tHcy) is a risk factor for cardiovascular disease in the general population, the question of causality remains unanswered, and the results of Hcy-lowering clinical trials have fallen short of expectations (11). In contrast, there is less debate regarding the toxicity of Hcy or its metabolite in patients with hyperhomocysteinemia caused by the inborn errors of metabolism (1). Furthermore, studies of genetic and nutritional hyperhomocysteinemia in mouse models provide additional support for a causal role of Hcy or its metabolite in atherosclerosis (12).
Among the pathological manifestations of human CBS deficiency, which include mental retardation, ectopia lentis, and osteoporosis, vascular complications remain the major cause of morbidity and mortality in untreated CBS-deficient patients (1); in these patients cerebrovascular incidents are 8 times more frequent than myocardial infarctions (13). Hcy lowering by cofactor supplementation improves vascular outcomes in CBS-deficient patients. Specifically, untreated CBS-deficient patients have 1 vascular event per 25 patient-years (13), whereas treated CBS-deficient patients have only 1 vascular event per 263 patient-years (relative risk 0.091, P<0.001) (14). Hcy-lowering therapy started early in life also prevents neurological abnormalities from human MTHFR deficiency (15, 16). These findings suggest that Hcy or its metabolite plays a causal role in atherothrombosis and brain disease.
However, mechanisms by which CBS or MTHFR deficiency leads to specific clinical manifestations are not clear. Because of parallel changes in the concentrations of Hcy metabolites under most clinical and experimental circumstances, it has not been possible to assign the observed toxicity to a specific Hcy metabolite. Hcy itself, S-adenosyl-Hcy, and Hcy-thiolactone remain potential candidates, and possible mechanisms have been assigned to each (12, 17,18,19).
One hypothesis suggests that the metabolic conversion to Hcy-thiolactone contributes to Hcy-associated pathological changes (6). Hcy-thiolactone could be detrimental because of its ability to form N-Hcy-protein in which Hcy is N-linked by an isopeptide bond to a protein lysine residue (4, 19). In humans, N-Hcy-protein accumulation has been linked to an autoimmune response and atherothrombosis (8, 17, 18). Plasma Hcy-thiolactone and N-Hcy-protein, originally discovered in human fibroblasts and endothelial cells (6, 9, 10, 20), occur in the human body (7, 10, 21,22,23) and are greatly elevated under conditions predisposing to atherothrombosis, such as hyperhomocysteinemia caused by mutations in the CBS or MTHFR gene in humans (7, 23). Recent human clinical studies show that plasma N-Hcy-protein levels are associated with a risk of coronary heart disease (24), whereas plasma Hcy-thiolactone levels are associated with the development and progression of diabetic macrovasculopathy (25). In ApoE-deficient mice, N-Hcy-protein accumulates in atherosclerotic lesions, and the accumulation increases in the animals fed a high-Met diet (26).
The purpose of the current research was to extent the hypothesis that Hcy-thiolactone is a toxic metabolite and that one of its potential mechanisms of toxicity is the formation of N-Hcy-protein. With this in mind, in the present study we examined effects of dietary (high-Met diet) or genetic modifications in Cbs(27), Mthfr(28), and proton-coupled folate transporter (Pcft) (29) genes on N-Hcy-protein levels in mice and their relationships to Hcy.
MATERIALS AND METHODS
Mice
Wild-type mice (6–9 mo old, strain C57BL/6J; The Jackson Laboratory, Bar Harbor, ME) were fed a control diet containing 0.5% Met or a high-Met isocaloric diet containing 1.5% Met (TD.04352 or TD.04353, respectively; Harlan Teklad, Madison, WI, USA). One group of mice (n=8) was fed the 1.5% Met diet for 2 wk followed by a control 0.5% Met diet for 4 wk. Another group of mice (n=8) was fed the 1.5% Met diet for 8 and 18 wk.
Cbs-deficient (27) and Mthfr-deficient mice (28) were created as described previously. Cbs-deficient mice are on a C57BL6 background, whereas Mthfr-deficient mice are on a mixed BALB/cAnNCrlBR background. The mice were fed a standard chow diet. Animals between 15 and 20 wk (Cbs-deficient) or 6 and 14 mo Mthfr-deficient) of age were used in experiments.
Transgenes harboring wild-type human CBS (Tg-hCBS), S466L (Tg-S466L), or I278T (Tg-I278T) variant were used to rescue the neonatal lethality associated with homozygosity for Cbs−(30, 31). In these animals, the human CBS is under control of the zinc-inducible metallothionein promoter, which allows one to rescue the neonatal lethality phenotype of Cbs−/− in mice by supplementing the drinking water of pregnant dams with 25 mM zinc. Zinc water is replaced by plain water after weaning. All transgenic mice are on a C57BL/6J strain background. The transgenic mice were fed a standard chow diet (Teklad 2018SX; Harlan Teklad).
The murine Pcft gene was disrupted in embryonic stem (ES) cells by homologous recombination. A replacement vector was designed to delete a 2.9-kb fragment of Pcft gene containing exons 1-3 of the wild-type locus. The deleted sequence was replaced by pKO Scrambler targeting vector carrying LacZ/Neo. The targeting vector was electroporated into 129SvEvBrd-derived ES cells for homologous recombination. The ES medium was replaced with M15 medium containing genticin G418 (350 μg/ml) 24 h after electroporation. ES cells carrying the disrupted gene were identified by Southern blot analysis using 5′ internal and 3′ external probes. Briefly, 7–8 days after electroporation and selection in G418 media, individual colonies were picked and expanded, and genomic DNA was isolated. Five positive clones containing 5′ 9.3-kb ApaL1 and 3′ 12.3-kb EcoRI inserts were identified by Southern blot. ES cell clones that were characterized by correct homologous recombination of the targeting vector and a single integration event were selected for blastocyst injections. The chimeric mice were obtained and bred to C57BL/6J albino mice to generate F1 heterozygous animals. DNA samples were isolated from the tails of ES cell-derived animals and were analyzed for the presence of a disrupted Pcft by Southern blot analysis. Germline transmission by chimeras of disrupted Pcft was identified from the Pcft-derived clone 1D1. These progeny were intercrossed to generate F2 wild-type, heterozygous, and homozygous mutant progeny. For genotyping, DNA was isolated and subjected to PCR analysis using primers specific to exon 3 (28-mer), intron 3 (29-mer), neo (neo3a), and intron 3 (rev). Primers sequences were 5′-CCCAACTCCAAAGCGCAGGTTCAT-3′ (28-mer) and 5′-TCCAGATGGGAAAGAAGAGGTCTA-3′ (29-mer) for wild-type Pcft and 5′-GCAGCGCATCGCCTTCTATC-3′ (neo3a) and 5′-CTTGACCACAACTGTCCATGTGC-3′ (rev) for the mutant allele carrying neo. The Pcft-deficient mice were fed a standard chow diet; 6- to 8-wk-old animals were used in the experiments.
Determination of N-Hcy-protein, Hcy-thiolactone, and tHcy
Plasma N-Hcy-protein (32), Hcy-thiolactone (21), and tHcy (22) were assayed as described previously. Serum and tissue tHcy for Tg-S466L, Tg-I278T, and Tg-hCBS animals were measured as described previously (30, 31). Tissue N-Hcy-protein and tHcy for other mice were assayed by similar procedures. In brief, 100–150 mg of frozen mouse liver or heart tissue was pulverized with dry ice in a mortar prechilled to −80°C, suspended in 10 vol of 20 mM sodium phosphate buffer (pH 7.4), 0.1 M NaCl, 0.5 mM EDTA, and 2 mM DTT, and further disintegrated by sonication (two strokes of 15 s, 70 W power, on wet ice). Crude extracts were clarified by centrifugation, and aliquots containing 0.5 mg of protein were used for N-Hcy-protein (32) and tHcy assays (21).
Statistics
Each variable was calculated as mean ± sd. The two-sample t test was used to compare the variables between the groups. The Pearson correlation coefficient r was used to evaluate simple linear relationships between variables. The level of statistical significance was set at P < 0.05.
RESULTS
High-Met diet increases plasma N-Hcy-protein levels in mice
We found that plasma N-Hcy-protein levels increased 11.7-fold in C57BL/6J mice fed a high-Met diet relative to the animals fed a normal chow diet. The increase in plasma N-Hcy-protein reflected 36- and 14-fold increases in plasma tHcy and Hcy-thiolactone levels (Table 1). After replacing the high-Met diet with a normal chow diet, plasma Hcy-thiolactone and tHcy returned to normal values, whereas plasma N-Hcy-protein, although decreased 6-fold, remained 2-fold higher than that in mice fed only a normal chow diet (Table 1). In mice fed a high-Met diet for 8 and 18 wk, plasma N-Hcy-protein levels remained elevated 6.1- and 8.2-fold, respectively (not shown).
TABLE 1.
Plasma N-Hcy-protein, Hcy-thiolactone, and tHcy levels in wild-type C57BL/6J mice
| Diet (8 mice/group) | N-Hcy-protein (μM) | Hcy-thiolactone (nM) | tHcy (μMm) |
|---|---|---|---|
| Control, 0 wk | 1.89 ± 0.70 | 5.74 ± 2.42 | 3.45 ± 0.30 |
| High Met, 2 wk | 22.1 ± 13.5 | 79.4 ± 11.9 | 126 ± 76 |
| High Met, 2 wk; control, 4 wk | 3.60 ± 1.11 | 4.47 ± 0.97 | 5.47 ± 2.83 |
Plasma N-Hcy-protein levels are elevated in Cbs-deficient mice
Plasma N-Hcy-protein levels were 8.1-fold higher in Cbs−/− mice than in Cbs+/+ animals (Table 2). In transgenic Tg-S466L mice, plasma N-Hcy-protein levels were 6.4- to 12.8-fold higher in Tg-S466L Cbs−/− than in Tg-S466L Cbs+/− or Tg-S466L Cbs+/+ animals (Table 2). Significantly more plasma N-Hcy-protein was present in Tg-S466L Cbs+/− than in Tg-S466L Cbs+/+ animals.
TABLE 2.
Plasma N-Hcy-protein and tHcy levels in Cbs-deficient mice
| Genotype | N-Hcy-protein (μM) | tHcy (μM) |
|---|---|---|
| Cbs−/− (n=3) | 15.3 ± 2.6 | 204 ± 62 |
| Cbs+/+ (n=8) | 1.89 ± 0.7 | 3.45 ± 0.30 |
| Tg-S466L Cbs−/− (n=4) | 12.8 ± 4.5 | 201 ± 46 |
| Tg-S466L Cbs+/− (n=4) | 2.00 ± 0.63 | 5.65 ± 0.21 |
| Tg-S466L Cbs+/+ (n=4) | 1.01 ± 0.26 | 3.25 ± 0.49 |
Plasma tHcy concentrations in Cbs−/− mice were severely elevated relative to those of the wild-type Cbs+/+ animals (Table 2), as expected. In Tg-S466L mice, plasma tHcy concentrations were also severely elevated in the Tg-S466L Cbs−/− mice compared with those in the Tg-S466L Cbs+/− or Tg-S466L Cbs+/+ animals (Table 2).
Serum and liver N-Hcy-protein levels are elevated in Tg-hCBS and Tg-I278T Cbs-deficient mice
In transgenic Tg-I278T mice, serum N-Hcy-protein levels were 6.3- to 10.6-fold higher in homozygous Tg-I278T Cbs−/− than in heterozygous Tg-I278T Cbs+/− or homozygous Tg-I278T Cbs+/+ animals (Table 3). Liver N-Hcy-protein levels were 11.5-fold higher in Tg-I278T Cbs−/− than in Tg-I278T Cbs+/− and Tg-I278T Cbs+/+ animals (Table 3).
TABLE 3.
Serum and liver N-Hcy-protein and tHcy levels in Tg-I278T and Tg-hCBS mice
| Genotype |
N-Hcy-protein |
tHcy |
||
|---|---|---|---|---|
| Plasma (μM) | Liver (pmol/mg of protein) | Plasma (μM) | Liver (pmol/mg of protein) | |
| Tg-I278T Cbs−/− (n=4) | 16.6 ± 4.1 | 1857 ± 452 | 272 ± 50 | 5680 ± 1777 |
| Tg-I278T Cbs+/− (n=2) | 2.62 ± 1.73 | 161 ± 19 | 5.0 ± 2.6 | 110 |
| Tg-I278T Cbs+/+ (n=2) | 1.56 ± 0.25 | 163 ± 0.0 | 1.9 ± 1.6 | 75 ± 7.1 |
| Tg-hCBS Cbs−/− (n=4) | 10.8 ± 2.1 | 562 ± 132 | 167 ± 40 | 582 ± 326 |
| Tg-hCBS Cbs+/− (n=4) | 2.6 ± 0.8 | 165 ± 24 | 6.1 ± 2.4 | 153 ± 21 |
Serum tHcy concentrations were extremely elevated (54- to 143-fold) in the Tg-I278T Cbs−/− mice compared with Tg-I278T Cbs+/− and Tg-I278T Cbs+/+ animals (Table 3). Liver tHcy concentrations were elevated 51- to 75-fold, to a lower degree than serum tHcy (Table 3), as previously reported (31).
In transgenic Tg-hCBS mice, plasma N-Hcy-protein levels were 4.2-fold higher in homozygous Tg-hCBS Cbs−/− than in heterozygous Tg-hCBS Cbs+/− animals (Table 3). Liver N-Hcy-protein levels were 3.4-fold higher in homozygous Tg-hCBS Cbs−/− than in heterozygous Tg-hCBS Cbs+/− animals (Table 3).
There was a 3.3-fold greater accumulation of N-Hcy-protein in Tg-I278T Cbs−/− mice than in Tg-hCBS Cbs−/− mice (Table 3). There were significant linear relationships between N-Hcy-protein and tHcy in the serum and in the liver (Fig. 1).
Figure 1.
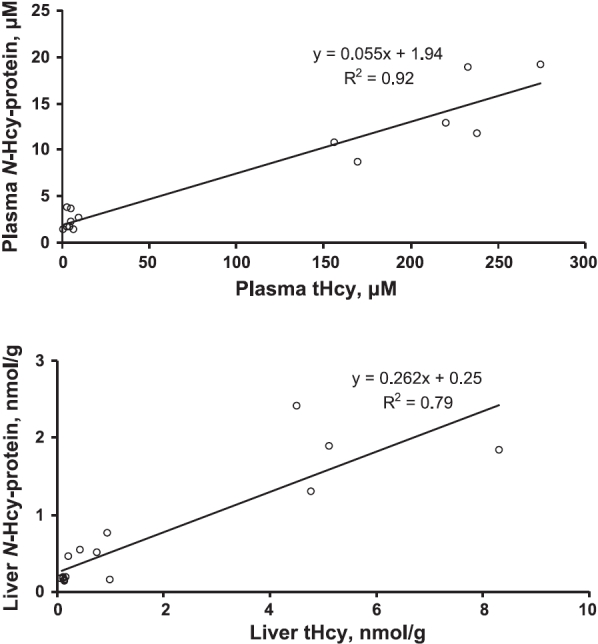
Relations between N-Hcy-protein and tHcy in plasma (top panel) and livers (bottom panel) of Tg-hCBS and Tg-I278T mice.
Plasma but not liver or heart N-Hcy-protein levels are elevated in Mthfr-deficient mice
Plasma N-Hcy-protein levels were significantly higher in homozygous Mthfr−/− than in heterozygous Mthfr+/− or wild-type Mthfr+/+ mice (Table 4). Significantly more plasma N-Hcy-protein was present in heterozygous Mthfr+/− than in wild-type Mthfr+/+ mice. Liver N-Hcy-protein levels were similar in homozygous Mthfr−/− and in wild-type Mthfr+/+ animals (Table 4).
TABLE 4.
Plasma and liver N-Hcy-protein and tHcy levels in Mthfr-deficient mice
| Genotype |
N-Hcy-protein |
tHcy |
||
|---|---|---|---|---|
| Plasma (μM) | Liver (pmol/mg protein) | Plasma (μM) | Liver (pmol/mg protein) | |
| Mthfr−/− (n=7) | 7.48 ± 2.55 | 49.8 ± 38.2 | 37.5 ± 48.6 | 184 ± 71 |
| Mthfr+/− (n=6) | 2.03 ± 0.44 | 9.15 ± 3.33 | ||
| Mthfr+/+ (n=7) | 1.18 ± 0.39 | 34.7 ± 12.7 | 4.42 ± 0.46 | 170 ± 43 |
To determine whether Mthfr gene inactivation affects N-Hcy-protein accumulation in other tissues, protein extracts from mouse hearts were prepared and assayed. We found no differences in heart N-Hcy-protein between homozygous Mthfr−/− (28.3±11.9 pmol/mg of heart protein, n=4) and wild-type Mthfr+/+ mice (26.9±7.3 pmol/mg of heart protein, n=4).
Plasma tHcy in homozygous Mthfr−/− mice was mildly elevated relative to that in heterozygous Mthfr+/− and wild-type Mthfr+/+ animals (Table 4), as previously reported (28). However, liver tHcy levels were similar in Mthfr−/− and Mthfr+/+ mice (Table 4). There was a linear relationship between N-Hcy-protein and tHcy in the plasma; however, liver N-Hcy-protein essentially did not correlate with liver tHcy in these mice (Fig. 2).
Figure 2.
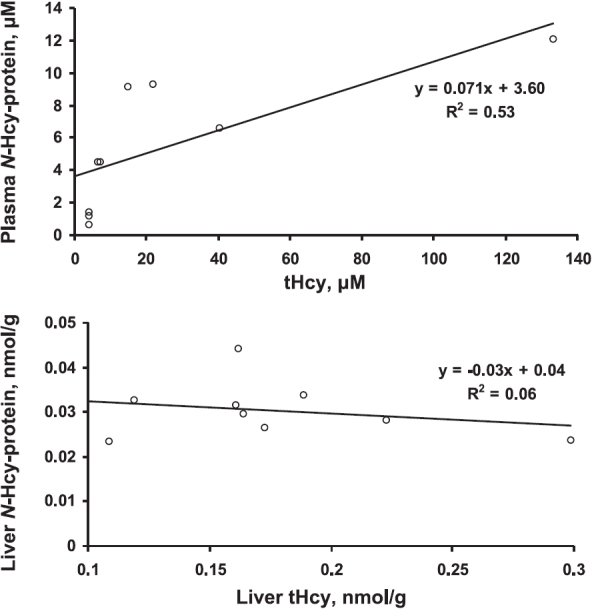
Relations between N-Hcy-protein and tHcy in plasma (top panel) and livers (bottom panel) of Mthfr−/− and Mthfr+/+ mice.
Plasma and liver N-Hcy-protein levels are elevated in Pcft-deficient mice
Plasma N-Hcy-protein levels were ∼30-fold higher in homozygous Pcft−/− mice compared with those in heterozygous Pcft+/− and wild-type Pcft+/+ animals (Table 5). Plasma tHcy was ∼65-fold higher in Pcft−/− mice compared with that in Pcft+/− and Pcft+/+ animals (Table 5). Liver N-Hcy-protein and tHcy levels were 3.6- and 5.1-fold, respectively, higher in homozygous Pcft−/− mice than in wild type Pcft+/+ animals (Table 5).
TABLE 5.
Plasma and liver N-Hcy-protein and tHcy levels in Pcft-deficient mice
| Genotype |
N-Hcy-protein |
tHcy |
||
|---|---|---|---|---|
| Plasma (μM) | Liver (pmol/mg protein) | Plasma (μM) | Liver (pmol/mg protein) | |
| Pcft−/− (n=4) | 56.6 ± 13.2 | 318 ± 98 | 134.8 ± 28.3 | 1529 ± 390 |
| Pcft+/− (n=4) | 1.79 ± 0.30 | 2.44 ± 0.71 | ||
| Pcft+/+ (n=4) | 2.28 ± 0.58 | 87 ± 32 | 1.79 ± 0.56 | 299 ± 92 |
DISCUSSION
Genetic and nutritional models have been generated in mice to approximate human disorders of Hcy or folate metabolism. Among these models, transgene treatments allowed generation of Cbs-deficient mice in which the presence of liver pathological changes depends on the extent of hyperhomocysteinemia (31). In the present work, we have used these models to address the question of the pathophysiological significance of N-Hcy-protein. We found that 1) N-Hcy-protein is present in the mouse, 2) wild-type mice fed a high-Met diet have significantly elevated plasma N-Hcy-protein levels, 3) mice with genetic deficiencies in folate metabolism (Mthfr or Pcft deficiency) or Hcy metabolism (Cbs deficiency) have significantly elevated plasma or serum N-Hcy-protein levels, and 4) although N-Hcy-protein is elevated in the plasma of mildly hyperhomocysteinemic mice (having plasma tHcy of 37.5 μM), these animals have a capacity to prevent the elevation of N-Hcy-protein in their organs: N-Hcy-protein is not affected in the liver or heart of mildly hyperhomocysteinemic mice (compared with animals with normal plasma tHcy of 4.4 μM) but is significantly elevated in severely or extremely hyperhomocysteinemic animals (serum tHcy of 167 or 272 μM, respectively). These results provide evidence that N-Hcy-protein is an important Hcy metabolite associated with mouse pathological conditions.
We showed previously that N-Hcy-protein is a significant metabolite in humans (23, 33). The findings of the present work extend these observations to mice. Similar to humans (32), in wild-type mice plasma N-Hcy-protein occurs at micromolar concentrations and increases 10-fold in Cbs- or Mthfr-deficient animals. Mouse plasma N-Hcy-protein levels are increased 25-fold by a deficiency in folate transport (in Pcft−/− animals) and 10-fold by a high-Met diet in wild-type animals. Hcy-thiolactone, a metabolic precursor of N-Hcy-protein, is also present in mice (Table 1) (7) and humans (7, 21, 22).
Diets supplemented with high Met are used to induce experimental hyperhomocysteinemia and vascular pathological changes in animal models (12, 34, 35). Our present findings that a high-Met, proatherogenic diet elevates plasma levels of N-Hcy-protein, in addition to those of Hcy-thiolactone and tHcy (Table 1), suggest that the pathway involving methionyl-tRNA synthetase-catalyzed conversion of Hcy to Hcy-thiolactone followed by protein N-homocysteinylation by Hcy-thiolactone, deduced from the in vitro and ex vivo human and rodent tissue culture studies (2, 6, 9, 19), is active in the mouse. This conclusion is strengthened by our findings that hyperhomocysteinemia caused by the inactivation of genes involved in Hcy (Cbs) or folate metabolism (Mthfr and Pcft) results in greatly increased accumulation of N-Hcy-protein in mice (Tables 2345). The lowering of N-Hcy-protein levels to close to normal values and normalization of Hcy-thiolactone and tHcy levels after return to a standard chow diet (Table 1) indicate that N-Hcy-protein is metabolized in the mouse, albeit somewhat slower than Hcy-thiolactone and tHcy. These findings also demonstrate that the plasma level of N-Hcy-protein, in addition to those of Hcy-thiolactone and tHcy, can be reversibly modified by the diet.
Although the biological significance of N-Hcy-proteins is not fully understood, accumulating evidence suggests that protein N-homocysteinylation plays a role in pathophysiology. For example, N-Hcy-proteins are autoimmunogenic (18), and titers of anti N-Hcy-protein autoantibodies are elevated in human stroke (36) and coronary artery disease patients (37, 38). N-Hcy-protein levels are elevated in coronary heart disease (24) and uremia (39, 40) in humans. Furthermore, prothrombotic N-Hcy-fibrinogen accumulates in human CBS deficiency (23), which could contribute to the increased atherothrombosis observed in these patients (1, 13). Because nutritionally (34, 35) or genetically (27, 28, 31, 41, 42) modified mice exhibit pathological changes in their vasculature, brain, or livers, the findings of the present work are consistent with a concept that N-Hcy-protein plays a role in these pathological changes.
Hereditary folate malabsorption is a rare autosomal recessive disorder caused by loss-of-function mutations in the human PCFT gene, which impair intestinal folate absorption and transport into the central nervous system (29). Affected infants develop anemia, immunoglobulin deficiency, seizures, and neurodevelopmental defects. Our present findings that N-Hcy-protein is elevated in plasma and livers of Pcft−/− mice suggest that N-Hcy-protein contributes to the pathological changes of hereditary folate malabsorption syndrome.
Our findings that N-Hcy-protein accumulates in livers of transgenic Cbs-deficient mice provide a possible mechanism for the induction of endoplasmic reticulum (ER) stress and unfolded protein response (UPR) in hyperhomocysteinemic animals. N-Hcy-proteins have a propensity to form aggregates (10, 19), which can induce the ER stress and UPR. The finding that N-Hcy-low-density lipoprotein, similar to other N-Hcy-proteins, has the propensity to aggregate (10, 19) and induce cell death in cultured human endothelial cells (43) is consistent with this concept. The ER stress response is observed in the livers of extremely hyperhomocysteinemic Tg-I278T Cbs−/− mice (serum tHcy of 296 μM) (31), which accumulate 1857 ± 452 pmol of N-Hcy-protein/mg of liver protein (Table 3), but not in the livers of severely hyperhomocysteinemic Tg-hCBS Cbs−/− mice (serum tHcy of 169 μM) (31), which accumulate 3.3 times less N-Hcy-protein in the liver (Table 3). The involvement of N-Hcy-protein in the ER stress is also supported by our previous findings showing that immunohistochemical staining for N-Hcy-protein is increased in aortic lesions from ApoE−/− mice fed a high-Met diet (26). The staining for N-Hcy-protein is similar to the staining for markers of the ER stress in these mice (26, 34). It is possible that Hcy, which accumulates in livers of Tg-I278T Cbs−/− mice (Table 3), might contribute to ER stress and UPR by forming S-homocysteinylated proteins (12, 31). However, this possibility is unlikely because, in contrast to N-Hcy-protein, S-Hcy-protein is not known to aggregate or be toxic to cells.
Our finding that plasma N-Hcy-protein levels are increased in Mthfr−/− mice compared with those in wild-type animals (Table 4) is consistent with a concept that N-Hcy-protein plays a role in brain and vascular pathological changes observed in these animals (28). The findings that N-Hcy-protein does not increase in the livers and hearts of Mthfr−/− mice, relative to Mthfr+/+ animals, suggest that the mildly hyperhomocysteinemic Mthfr−/− animals have a capacity to prevent N-Hcy-protein accumulation in these organs. This suggestion is supported by our findings showing that N-Hcy-protein levels increase to a significantly lower extent in the liver than in the plasma of Pcft-deficient mice (3.6-fold vs. 30-fold, respectively) (Table 5). Our data also suggest that this capacity can be overwhelmed and that the extremely elevated N-Hcy-protein levels (Table 3) could lead to liver pathological changes that are observed in Tg-I278T CBS−/− mice (31). Furthermore, the data shown in Table 3 are consistent with a threshold effect of liver N-Hcy-protein accumulation on liver damage and the ER stress phenotype, which are observed in Tg-I278T Cbs−/− mice but not in Tg-hCBS Cbs−/− animals (31), which have 3.3-fold less liver N-Hcy-protein. Similar liver damage that is observed in 8- to 32-week-old Cbs−/− mice fed a choline-supplemented diet (42) could be linked to the accumulation of N-Hcy-protein as well; however, N-Hcy-protein has not yet been studied in that model.
In conclusion, our findings provide evidence that hyperhomocysteinemia due to genetic deficiencies in Hcy or folate metabolism or induced by a diet leads to significant increases in the propathogenic N-Hcy-protein in the mouse.
Acknowledgments
This work was supported in part by grants from the American Heart Association and the Ministry of Science and Higher Education, Poland. We thank Rima Rozen (McGill University, Montreal, QC, Canada) for providing plasma and tissue samples from Mthfr-deficient mice. We also thank anonymous reviewers for their helpful comments.
References
- Mudd S. H., Levy H. L., Kraus J. P. Disorders of transsulfuration. Scriver C. R., Beaudet A. L., Sly W. S., Valle D., Childs B., Kinzler K. W., Vogelstein B., editors. McGraw-Hill; New York: The Metabolic and Molecular Bases of Inherited Disease. 2001;2:2007–2056. [Google Scholar]
- Jakubowski H., Goldman E. Synthesis of homocysteine thiolactone by methionyl-tRNA synthetase in cultured mammalian cells. FEBS Lett. 1993;317:237–240. doi: 10.1016/0014-5793(93)81283-6. [DOI] [PubMed] [Google Scholar]
- Jakubowski H. Translational accuracy of aminoacyl-tRNA synthetases: implications for atherosclerosis. J Nutr. 2001;131:2983S–2987S. doi: 10.1093/jn/131.11.2983S. [DOI] [PubMed] [Google Scholar]
- Jakubowski H. Molecular basis of homocysteine toxicity in humans. Cell Mol Life Sci. 2004;61:470–487. doi: 10.1007/s00018-003-3204-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakubowski H. Accuracy of aminoacyl-tRNA synthetases: proofreading of amino acids. Ibba M., Francklyn C., Cusack S., editors. Landes Bioscience/Eurekah.com; Georgetown, TX, USA.: The Aminoacyl-tRNA Synthetases. 2005:384–396. [Google Scholar]
- Jakubowski H. Metabolism of homocysteine thiolactone in human cell cultures: possible mechanism for pathological consequences of elevated homocysteine levels. J Biol Chem. 1997;272:1935–1942. [PubMed] [Google Scholar]
- Chwatko G., Boers G. H., Strauss K. A., Shih D. M., Jakubowski H. Mutations in methylenetetrahydrofolate reductase or cystathionine β-synthase gene, or a high-methionine diet, increase homocysteine thiolactone levels in humans and mice. FASEB J. 2007;21:1707–1713. doi: 10.1096/fj.06-7435com. [DOI] [PubMed] [Google Scholar]
- Jakubowski H. Pathophysiological consequences of homocysteine excess. J Nutr. 2006;136:1741S–1749S. doi: 10.1093/jn/136.6.1741S. [DOI] [PubMed] [Google Scholar]
- Jakubowski H., Zhang L., Bardeguez A., Aviv A. Homocysteine thiolactone and protein homocysteinylation in human endothelial cells: implications for atherosclerosis. Circ Res. 2000;87:45–51. doi: 10.1161/01.res.87.1.45. [DOI] [PubMed] [Google Scholar]
- Jakubowski H. Homocysteine thiolactone: metabolic origin and protein homocysteinylation in humans. J Nutr. 2000;130:377S–381S. doi: 10.1093/jn/130.2.377S. [DOI] [PubMed] [Google Scholar]
- Maron B. A., Loscalzo J. The treatment of hyperhomocysteinemia. Annu Rev Med. 2008;60:39–54. doi: 10.1146/annurev.med.60.041807.123308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lentz S. R. Mechanisms of homocysteine-induced atherothrombosis. J Thromb Haemost. 2005;3:1646–1654. doi: 10.1111/j.1538-7836.2005.01364.x. [DOI] [PubMed] [Google Scholar]
- Mudd S. H., Skovby F., Levy H. L., Pettigrew K. D., Wilcken B., Pyeritz R. E., Andria G., Boers G. H., Bromberg I. L., Cerone R., Fowler B., Gröbe H., Schmidt H., Schweitzer L. The natural history of homocystinuria due to cystathionine β-synthase deficiency. Am J Hum Genet. 1985;37:1–31. [PMC free article] [PubMed] [Google Scholar]
- Yap S., Boers G. H., Wilcken B., Wilcken D. E., Brenton D. P., Lee P. J., Walter J. H., Howard P. M., Naughten E. R. Vascular outcome in patients with homocystinuria due to cystathionine β-synthase deficiency treated chronically: a multicenter observational study. Arterioscler Thromb Vasc Biol. 2001;21:2080–2085. doi: 10.1161/hq1201.100225. [DOI] [PubMed] [Google Scholar]
- Rosenblatt D., Fenton W. Disorders of transsulfuration. Scriver C. R., Beaudet A. L., Sly W. S., Valle D., Childs B., Kinzler K. W., Vogelstein B., editors. McGraw-Hill; New York: The Metabolic and Molecular Bases of Inherited Disease. 2001:3897–3933. [Google Scholar]
- Strauss K. A., Morton D. H., Puffenberger E. G., Hendrickson C., Robinson D. L., Wagner C., Stabler S. P., Allen R. H., Chwatko G., Jakubowski H., Niculescu M. D., Mudd S. H. Prevention of brain disease from severe 5,10-methylenetetrahydrofolate reductase deficiency. Mol Genet Metab. 2007;91:165–175. doi: 10.1016/j.ymgme.2007.02.012. [DOI] [PubMed] [Google Scholar]
- Jakubowski H. The molecular basis of homocysteine thiolactone-mediated vascular disease. Clin Chem Lab Med. 2007;45:1704–1716. doi: 10.1515/CCLM.2007.338. [DOI] [PubMed] [Google Scholar]
- Jakubowski H. Anti-N-homocysteinylated protein autoantibodies and cardiovascular disease. Clin Chem Lab Med. 2005;43:1011–1014. doi: 10.1515/CCLM.2005.177. [DOI] [PubMed] [Google Scholar]
- Jakubowski H. Protein homocysteinylation: possible mechanism underlying pathological consequences of elevated homocysteine levels. FASEB J. 1999;13:2277–2283. [PubMed] [Google Scholar]
- Jakubowski H. The determination of homocysteine-thiolactone in biological samples. Anal Biochem. 2002;308:112–119. doi: 10.1016/s0003-2697(02)00224-5. [DOI] [PubMed] [Google Scholar]
- Chwatko G., Jakubowski H. The determination of homocysteine-thiolactone in human plasma. Anal Biochem. 2005;337:271–277. doi: 10.1016/j.ab.2004.11.035. [DOI] [PubMed] [Google Scholar]
- Chwatko G., Jakubowski H. Urinary excretion of homocysteine-thiolactone in humans. Clin Chem. 2005;51:408–415. doi: 10.1373/clinchem.2004.042531. [DOI] [PubMed] [Google Scholar]
- Jakubowski H., Boers G. H., Strauss K. A. Mutations in cystathionine β-synthase or methylenetetrahydrofolate reductase gene increase N-homocysteinylated protein levels in humans. FASEB J. 2008;22:4071–4076. doi: 10.1096/fj.08-112086. [DOI] [PubMed] [Google Scholar]
- Yang X., Gao Y., Zhou J., Zhen Y., Yang Y., Wang J., Song L., Liu Y., Xu H., Chen Z., Hui R. Plasma homocysteine thiolactone adducts associated with risk of coronary heart disease. Clin Chim Acta. 2006;364:230–234. doi: 10.1016/j.cccn.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Gu W., Lu J., Yang G., Dou J., Mu Y., Meng J., Pan C. Plasma homocysteine thiolactone associated with risk of macrovasculopathy in Chinese patients with type 2 diabetes mellitus. Adv Ther. 2008;25:914–924. doi: 10.1007/s12325-008-0097-8. [DOI] [PubMed] [Google Scholar]
- Perla-Kajan J., Stanger O., Luczak M., Ziolkowska A., Malendowicz L. K., Twardowski T., Lhotak S., Austin R. C., Jakubowski H. Immunohistochemical detection of N-homocysteinylated proteins in humans and mice. Biomed Pharmacother. 2008;62:473–479. doi: 10.1016/j.biopha.2008.04.001. [DOI] [PubMed] [Google Scholar]
- Watanabe M., Osada J., Aratani Y., Kluckman K., Reddick R., Malinow M. R., Maeda N. Mice deficient in cystathionine β-synthase: animal models for mild and severe homocyst(e)inemia. Proc Natl Acad Sci U S A. 1995;92:1585–1589. doi: 10.1073/pnas.92.5.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z., Karaplis A. C., Ackerman S. L., Pogribny I. P., Melnyk S., Lussier-Cacan S., Chen M. F., Pai A., John S. W., Smith R. S., Bottiglieri T., Bagley P., Selhub J., Rudnicki M. A., James S. J., Rozen R. Mice deficient in methylenetetrahydrofolate reductase exhibit hyperhomocysteinemia and decreased methylation capacity, with neuropathology and aortic lipid deposition. Hum Mol Genet. 2001;10:433–443. doi: 10.1093/hmg/10.5.433. [DOI] [PubMed] [Google Scholar]
- Qiu A., Min S. H., Jansen M., Malhotra U., Tsai E., Cabelof D. C., Matherly L. H., Zhao R., Akabas M. H., Goldman I. D. Rodent intestinal folate transporters (SLC46A1): secondary structure, functional properties, and response to dietary folate restriction. Am J Physiol Cell Physiol. 2007;293:C1669–C1678. doi: 10.1152/ajpcell.00202.2007. [DOI] [PubMed] [Google Scholar]
- Gupta S., Wang L., Hua X., Krijt J., Kozich V., Kruger W. D. Cystathionine β-synthase p S466L mutation causes hyperhomocysteinemia in mice. Hum Mutat. 2008;29:1048–1054. doi: 10.1002/humu.20773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S., Kuhnisch J., Mustafa A., Lhotak S., Schlachterman A., Slifker M. J., Klein-Szanto A., High K. A., Austin R. C., Kruger W. D. Mouse models of cystathionine β-synthase deficiency reveal significant threshold effects of hyperhomocysteinemia. FASEB J. 2009;23:883–893. doi: 10.1096/fj.08-120584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakubowski H. New method for the determination of protein N-linked homocysteine. Anal Biochem. 2008;380:257–261. doi: 10.1016/j.ab.2008.05.049. [DOI] [PubMed] [Google Scholar]
- Jakubowski H. Homocysteine is a protein amino acid in humans: implications for homocysteine-linked disease. J Biol Chem. 2002;277:30425–30428. doi: 10.1074/jbc.C200267200. [DOI] [PubMed] [Google Scholar]
- Zhou J., Werstuck G. H., Lhotak S., de Koning A. B., Sood S. K., Hossain G. S., Moller J., Ritskes-Hoitinga M., Falk E., Dayal S., Lentz S. R., Austin R. C. Association of multiple cellular stress pathways with accelerated atherosclerosis in hyperhomocysteinemic apolipoprotein E-deficient mice. Circulation. 2004;110:207–213. doi: 10.1161/01.CIR.0000134487.51510.97. [DOI] [PubMed] [Google Scholar]
- Hofmann M. A., Lalla E., Lu Y., Gleason M. R., Wolf B. M., Tanji N., Ferran L. J., Jr, Kohl B., Rao V., Kisiel W., Stern D. M., Schmidt A. M. Hyperhomocysteinemia enhances vascular inflammation and accelerates atherosclerosis in a murine model. J Clin Invest. 2001;107:675–683. doi: 10.1172/JCI10588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Undas A., Perla J., Lacinski M., Trzeciak W., Kazmierski R., Jakubowski H. Autoantibodies against N-homocysteinylated proteins in humans: implications for atherosclerosis. Stroke. 2004;35:1299–1304. doi: 10.1161/01.STR.0000128412.59768.6e. [DOI] [PubMed] [Google Scholar]
- Undas A., Jankowski M., Twardowska M., Padjas A., Jakubowski H., Szczeklik A. Antibodies to N-homocysteinylated albumin as a marker for early-onset coronary artery disease in men. Thromb Haemost. 2005;93:346–350. doi: 10.1160/TH04-08-0493. [DOI] [PubMed] [Google Scholar]
- Undas A., Stepien E., Glowacki R., Tisonczyk J., Tracz W., Jakubowski H. Folic acid administration and antibodies against homocysteinylated proteins in subjects with hyperhomocysteinemia. Thromb Haemost. 2006;96:342–347. doi: 10.1160/TH06-04-0228. [DOI] [PubMed] [Google Scholar]
- Uji Y., Motomiya Y., Hanyu N., Ukaji F., Okabe H. Protein-bound homocystamide measured in human plasma by HPLC. Clin Chem. 2002;48:941–944. [PubMed] [Google Scholar]
- Perna A. F., Satta E., Acanfora F., Lombardi C., Ingrosso D., De Santo N. G. Increased plasma protein homocysteinylation in hemodialysis patients. Kidney Int. 2006;69:869–876. doi: 10.1038/sj.ki.5000070. [DOI] [PubMed] [Google Scholar]
- Wang G., Woo C. W., Sung F. L., Siow Y. L., O K. Increased monocyte adhesion to aortic endothelium in rats with hyperhomocysteinemia: role of chemokine and adhesion molecules. Arterioscler Thromb Vasc Biol. 2002;22:1777–1783. doi: 10.1161/01.atv.0000035404.18281.37. [DOI] [PubMed] [Google Scholar]
- Robert K., Nehme J., Bourdon E., Pivert G., Friguet B., Delcayre C., Delabar J. M., Janel N. Cystathionine beta synthase deficiency promotes oxidative stress, fibrosis, and steatosis in mice liver. Gastroenterology. 2005;128:1405–1415. doi: 10.1053/j.gastro.2005.02.034. [DOI] [PubMed] [Google Scholar]
- Ferretti G., Bacchetti T., Moroni C., Vignini A., Nanetti L., Curatola G. Effect of homocysteinylation of low density lipoproteins on lipid peroxidation of human endothelial cells. J Cell Biochem. 2004;92:351–360. doi: 10.1002/jcb.20069. [DOI] [PubMed] [Google Scholar]