

Abstract
Activating mutants of EGFR have been identified in a subset of non-small-cell lung cancers. To investigate mutant-driven signaling, we focused on Y869, a residue in the same activation loop where the L858R and L861Q mutations are located. We observed ligand-independent phosphorylation of Y869 in 32D cells EGFRL858R and EGFRL861Q. The EGFR tyrosine kinase inhibitor (TKI) erlotinib inhibited Y869 P-EGFR in intact cells as well as in a cell-free kinase reaction. Expression of kinase domain of EGFRL858R and EGFRL861Q exhibited auto-phosphorylation of Y869; this was inhibited by EGFR TKIs but not by Src kinase inhibitor. P-Y859 of EGFR-mediated downstream component, STAT5, was also analyzed. Y694 P-STAT5 was eliminated by erlotinib treatment. Analysis of immune-complexes showed constitutive association of mutant EGFRs with STAT5 and Src which was unaffected by erlotinib or PP1. On the other hand, 32D-EGFRWT exhibited constitutive STAT5 phosphorylation and association of EGFR with JAK2. In these cells, a JAK2 inhibitor abrogated P-STAT5 whereas mutant EGFRs did not associate with JAK2. Expression of c-myc was regulated by EGFR/STAT5 signaling in cells expressing EGFRL858R and EGFRL861Q. Our results suggest that ligand-independent and Src activity-independent phosphorylation of Y869 in mutant EGFR regulates STAT5 activation and c-myc expression.
INTRODUCTION
Somatic mutations in kinase domain of epidermal growth factor receptor (EGFR) have been identified in a subset of patients with non-small-cell lung cancer (NSCLC) [1–3]. Although recent studies have started to unveil the cellular phenotype induced by expression of mutant EGFR [3, 4], the signaling pathways utilized by mutant EGFRs remain incompletely characterized. Following ligand-induced receptor dimerization and activation, the wild-type (WT) EGFR initiates signaling by recruiting adaptor proteins and signal transducers to phosphorylated tyrosine residues in the receptor’s C-terminus. Regulation of the receptor’s tyrosine phosphorylation is critical for the modulation of the cellular effects of activated EGFR.
Among the tyrosine residues that are auto-phosphorylated by EGFR activation, Y869 is distinct from others that are localized in the receptor’s C-terminus. The location of Y869 in kinase domain is close to L858 and L861, residues that are substituted in EGFRL858R and EGFRL861Q found in NSCLC with exquisitely sensitivity against the EGFR tyrosine kinase inhibitors (TKIs) gefitinib and erlotinib [1, 2]. Y869 can undergo Src-dependent phosphorylation and this modification has been found to result in EGF-dependent mitogenesis via signal transducer and activator of transcription 5 (STAT5) [5]. STAT5 is a member of STAT family of transcription factors that are involved in a variety of cellular processes including mitogenesis, differentiation, and apoptosis [6]. Activation of STAT5 involves phosphorylation of tyrosines at its C-terminus such as Y694 and translocation to the nucleus where it regulates the transcription of genes involved in cell proliferation and survival such as c-myc and cyclin D1 [7]. Constitutive activation of STAT5 has been observed in lung cancer cells expressing mutant EGFR [4], suggesting it might be important for the maintenance and survival of transformed cells.
We previously reported that expression of mutant EGFR in 32D cells results in ligand-independent receptor phosphorylation and survival [8]. Here we show that phosphorylation of Y869 in mutant EGFR does not require ligand stimulation or Src kinase activity but is eliminated by inhibition of the EGFR tyrosine kinase with gefitinib and erlotinib. Further investigation identified that transcription factor STAT5 as a mediator of c-myc expression in 32D cells expressing mutant EGFR. Mutant EGFRs were constitutively associated with Src and STAT5 while WT EGFR bound to JAK2 in the absence of added ligand. Our results suggest that STAT5 is a critical mediator of signaling by mutant EGFR and thus, a potential therapeutic target in NSCLC.
MATERIALS AND METHODS
Plasmid constructs and stable transfection
The expression constructs of full-length EGFRs were described previously [8]. The EGFRK745R cDNA was provided by James Staros (Vanderbilt University). To construct expression vectors encoding the kinase domain of EGFRs, the coding regions (residues 672 to 998) of these cDNAs were subcloned into the XhoI-SmaI site of pAcHLT-C baculovirus transfer vector (BD Biosciences).
Cell culture and reagents
The establishment and maintenance in culture of 32D cells expressing full-length EGFRs were described previously [8]. The human lung cancer cell lines; NCI H3255 was provided by Bruce Johnson (Dana-Farber Cancer Institute) [1]. PC9 cells were a gift from Kazuto Nishio (Shien-Lab, National Cancer Center Hospital, Tokyo, Japan). A431 cells were purchased from the American Tissue Culture Collection (ATCC; Manassas, VA). Gefitinib was obtained from AstraZeneca Pharmaceuticals. Erlotinib was provided by Mark Sliwkowski (Genentech). Cetuximab was a gift from Dan Hicklin (Imclone). TGFα was from R&D Systems (Minneapolis, MN). PP1 and PP2 were from Calbiochem (San Diego, CA).
Western blot and immunoprecipitation
Western blot analysis and immunoprecipitation were performed as described previously [8]. We used the following antibodies for immunoprecipitation and/or western blot analysis: Y1068 P-EGFR and Y416 P-Src from Cell Signaling Technology (Beverly, MA); Src and c-myc from Santa Cruz Biotechnology (Santa Cruz, CA); actin from Sigma (St.Louis, MO); JAK2 from Chemicon (Temecula, CA); phosphotyrosine (P-Tyr, 4G10) from the Molecular Recognition Core Laboratory at Vanderbilt University; total EGFR from NeoMarkers (Freemont, CA); Y684 P-STAT5 and total STAT5 from Invitrogen (Carlsbad, CA). The Y869 P-EGFR monoclonal antibody was from nanoTool (Wetzlar, Germany).
In vitro kinase assay
Cells were starved in serum-free medium for 2 h in the presence of erlotinib and then lysed in a buffer same as immunoprecipitation described previously [8]. Cell lysates were precipitated overnight at 4°C with the EGFR antibody cetuximab followed by the addition of protein G-Sepharose (Amersham Biosciences) for 3 h with rocking. Precipitates were washed three times with lysis buffer and once with kinase buffer (20 mM HEPES [pH 7.5], 10 mM MgCl2, 10 mM MnCl2, 1 mM dithiothreitol, 0.1 mM Na3VO4). An in vitro kinase assay was initiated by addition of ATP (0-32 mM in 50 mM Tris/HCl, pH 7.5, 20 mM MgCl2 and phosphatase inhibitors) followed by incubation for 5 min at 37 °C. The same inhibitors with which the cells had been preincubated were added at similar concentrations to the kinase reaction before the addition of ATP. The kinase reaction was terminated by adding 5X loading buffer and boiling for 3 min before separation by SDS-PAGE followed by western blot analysis using P-EGFR and total EGFR antibodies.
RESULTS
Ligand-independent phosphorylation of Y869
We used 32D cells stably expressing EGFRWT, EGFRL858R, and EGFRL861Q [8]. In EGFRL858R and EGFRL861Q, Y869 was constitutively phosphorylated whereas in EGFRWT phosphorylation at this residue required the addition of TGFα (Fig. 1A). Addition of ligand further increased Y869 phosphorylation in 32D-EGFRL858R and 32D-EGFRL861Q cells. Since Y869 in EGFR can be a substrate to the Src kinase [9], we examined the effect of Src inhibitor PP1. Low dose (<0.1 μM) of PP1 did not inhibit phosphorylation of Y869 in all EGFR expressing cells while 1 μM of PP1 reduced P-Y869 in 32D-EGFRL858R cells (Fig. 1B). Interestingly, 32D-EGFRWT cells were less sensitive to PP1 than mutant expressing cells. We further investigated the effect of EGFR inhibitor erlotinib and the Src kinase inhibitors PP1 and PP2. Treatment with erlotinib of cells expressing mutant EGFR markedly inhibited basal and ligand-stimulated P-Y869. In 32D-EGFRWT cells, erlotinib blocked ligand-induced P-Y869. To detect active Src we used an antibody against Src phosphorylated in Y416. At concentrations that inhibited (active) Y416 P-Src, PP1 and PP2 did not affect basal P-Y869 in 32D-EGFRL861Q cells and modestly reduced it in 32D-EGFRL858R cells (Fig. 1C).
Fig. 1.
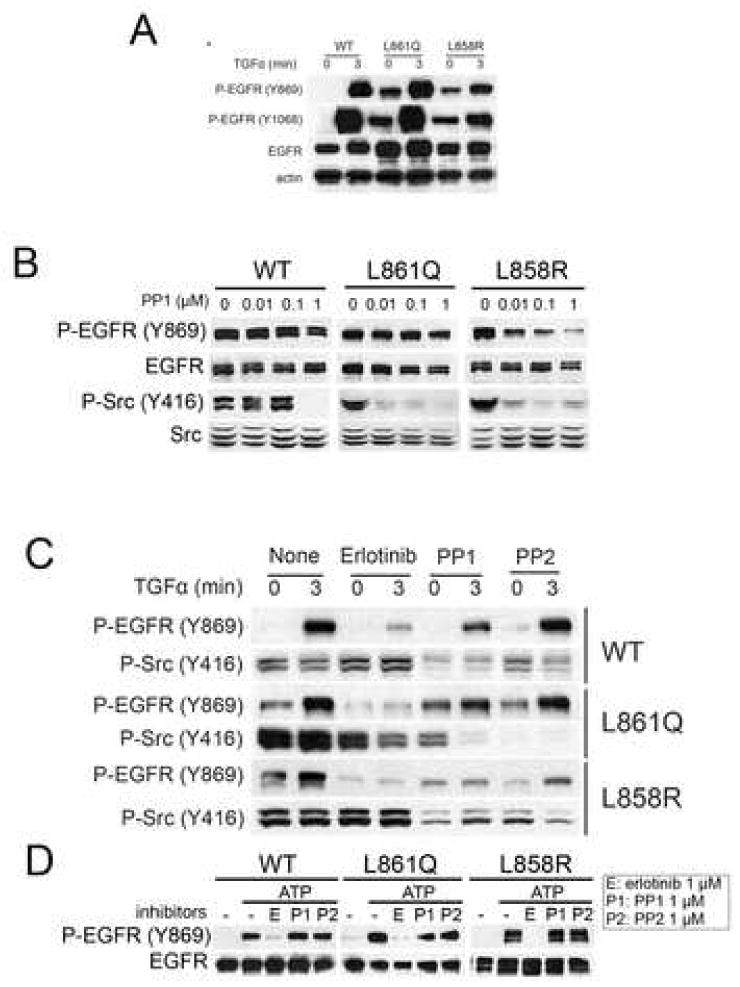
Ligand-independent phosphorylation of Y869 in EGFR. (A, B and C) 32D-EGFRWT, 32D-EGFRL858R, or 32-EGFRL861Q were serum-starved for 2 h in the presence or absence of erlotinib (1 μM for C), PP1 (0.01, 0.1, and 1 μM for B, and 1 μM for C), or PP2 (1 μM for C) where indicated and then stimulated by TGFα (20 ng/ml) for 3 min. Cells were next harvested and subjected to western blot analysis with the indicated antibodies. (D) Cells were grown in serum-free media for 2 hours with 2 μM of erlotinib. EGFRs were precipitated from whole cell lysates with the EGFR antibody cetuximab as indicated in Methods. Immune complexes were tested in an in vitro kinase reaction using cold ATP for 3 min at 37 °C. Erlotinib, PP1, and PP2 were added to the reaction mixture. Kinase reaction products were analyzed by western blot with Y869 P-EGFR and total EGFR antibodies.
We next investigated the effect of erlotinib and the Src inhibitors in a cell-free system. To generate unphosphorylated EGFR cells were pretreated with 2 μM of erlotinib for 2 hours in the culture medium. Cell lysates were prepared and precipitated with an EGFR antibody. The immune complex was then tested in a kinase reaction in vitro to which erlotinib, PP1, or PP2 were added. After incubation with ATP, there was robust phosphorylation in Y869 in WT and mutant EGFRs. This was inhibited by erlotinib but not either of the Src antagonists (Fig. 1D). These results further suggest that phosphorylation of Y869 is dependent on EGFR but not on Src kinase activity. The ability of the WT EGFR to phosphorylate Y869 in vitro (Fig. 1D) but not in intact cells (Fig. 1A and C) without ligand stimulation, suggests that additional cellular factors are required for this modification to occur in vivo. Nonetheless, these data also imply that these additional factors are not required for Y869 phosphorylation in vivo in cells expressing mutant EGFRs (Fig. 1A and C).
Our finding of ligand-independent phosphorylation of Y869 in mutant EGFRs leaded us to hypothesize that the kinase domain of mutant EGFR is sufficient to display Y869 phosphorylation. To test this hypothesis we infected sf9 insect cells with vectors encoding His-tagged WT and mutant receptor kinase domains using a baculovirus infection system. P-Y869 was detected by western blot analysis of lysates from cells expressing EGFRL858R and EGFRL861Q kinase domains but not from 32D-EGFRWT cells or cells expressing a kinase-dead version of the receptor in which the ATP binding site had been mutated (K745R; Fig. 2A). Treatment of insect cells with gefitinib or erlotinib reduced the level of P-Y869 in mutant EGFRs (Fig. 2A and B). Consistent with data shown in Fig. 1, treatment with PP1 did not affect P-Y869 in cells expressing the kinase domain of EGFR mutants (Fig. 2C). These results imply that the mutations in the kinase domain alone are enough to phosphorylate Y869 and that the kinase activity of Src is dispensable for this modification.
Fig. 2.
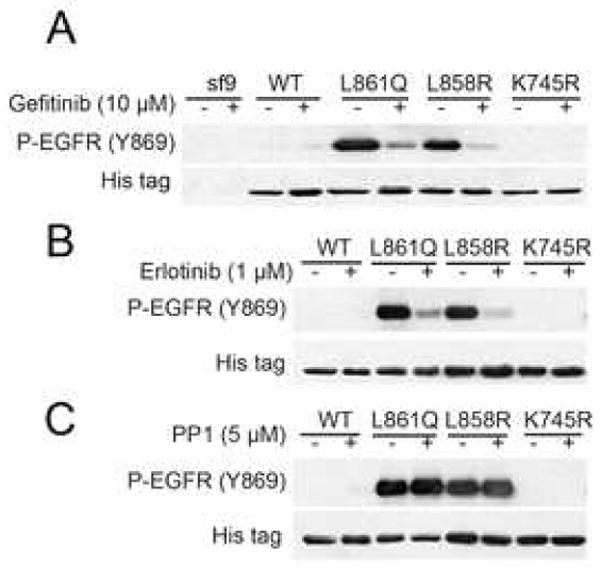
Auto-phosphorylation of Y869 in the kinase domain of EGFR. Sf9 insect cells were transfected with baculovirus vector containing the His-tagged kinase domain of EGFRWT, EGFRL858R, EGFRL861Q, or EGFRK745R using BaculoGold Transfection Kit (BD Biosciences). Virion-containing supernatants were used to infect Sf9 cells in order to amplify viral titer. The infected sf9 cells were cultured with or without gefitinib (A), erlotinib (B), or PP1 (C) for 4 days and then harvested and lysed. Cell lysates were subjected to western blot analysis with the indicated antibodies.
EGFR regulates STAT5 activation
To investigate the biological consequences of autophosphorylation at Y869 in mutant EGFRs, we examined STAT5 phosphorylation. Under conditions of serum deprivation, we detected constitutive phosphorylation of STAT5 at Y694 in 32D cells expressing WT and mutant receptors. In 32D-EGFRWT cells, P-STAT5 was present in the absence of Y869 P-EGFR (Fig. 3A). Treatment with erlotinib reduced P-STAT5 in cells expressing EGFRL858R and EGFRL861Q but not in 32D-EGFRWT cells. The Src inhibitor PP1 modestly reduced P-STAT5 in cells transfected with the L858R mutant but had no effect on cells expressing the L861Q mutant or WT EGFR (Fig. 3B).
Fig. 3.
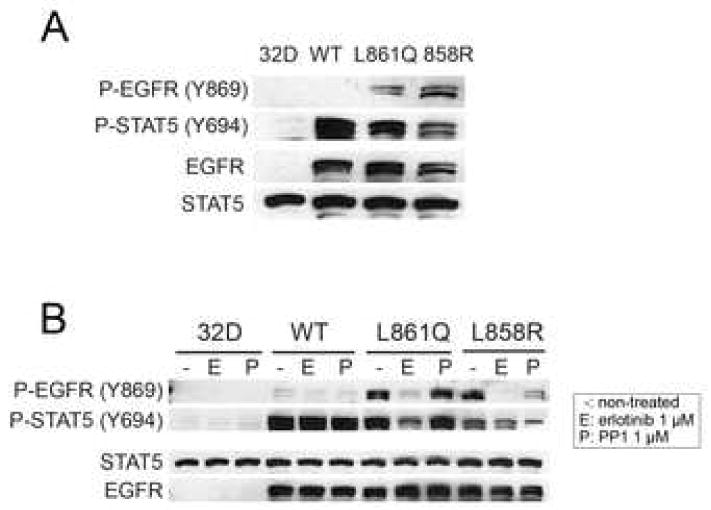
STAT5 phosphorylation by EGFR. (A) Parental 32D cells and 32D-EGFRWT, 32D-EGFRL861Q, 32D-EGFRL858R cells were serum starved for 2 h. Cell lysates were prepared and tested by western blot with indicated antibodies. (B) The same cell lines were treated with erlotinib or PP1 for 2 h at the indicated concentrations and then lysed. Cell lysates were subjected to western blot analysis with antibodies against total and phosphorylated STAT5 and EGFR.
Unexpectedly, the 32D-EGFRWT cells displayed ligand-independent activation of STAT5 in the absence of EGFR phosphorylation (Fig. 3A and 1A). Treatment with EGF of 32D cells expressing a chimera of the tyrosine kinase domain of JAK2 and the extracellular and transmembrane regions of EGFR has been shown to induce tyrosine phosphorylation of the EGFR/JAK2 chimera and STAT5 [10]. Thus, we speculated that JAK2 might mediate the activation of STAT5 in 32D-EGFRWT cells. To test this possibility, we used the JAK kinase inhibitor AG490 [11]. Preincubation of 32D parental cells with AG490 reduced IL-3-induced phosphorylation of JAK2 and STAT5 (Fig. 4A). AG490 also reduced P-STAT5 in 32D-EGFRWT cells (Fig 4B). Since JAK2 is activated by association with receptor tyrosine kinases, we next examined the coupling of JAK2 to EGFR. Under serum-free and in the absence of TGFα, a strong JAK2 band was detectable in EGFR precipitates from 32D-EGFRWT cells but not from cells expressing mutant receptors. EGFR-associated JAK2 was increased by treatment with TGFα (Fig. 4C). In the presence of AG490, treatment with TGFα still induced P-STAT5 but this activation was blocked by erlotinib (Fig. 4D). Neither erlotinib nor PP1 inhibited basal P-STAT5 (Fig 4D), suggesting that the catalytic activity of EGFR and JAK2 but not Src mediate constitutive STAT5 phosphorylation in 32D-EGFRWT cells. Finally, the association of JAK2 with EGFRWT was unaffected by erlotinib or PP1 (Fig 4E). These results also suggest that, in 32D-EGFRWT cells, the activation of STAT5 is dependent on an EGFR-JAK2 complex which does not depend on EGFR or Src catalytic activities.
Fig. 4.
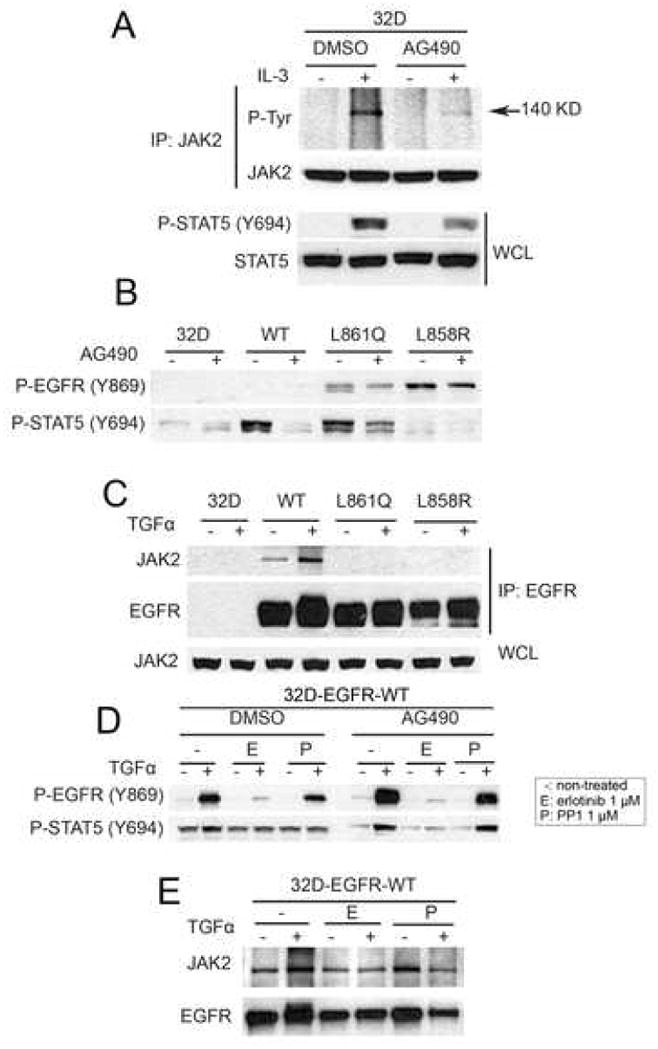
Wild-type EGFR associates with JAK2. (A) Parental 32D cells were switched for 2 h to serum-free medium containing DMSO (control) or AG490 (100 μM) and then treated with medium containing IL-3. Cell lysates were prepared and precipitated with a JAK2 antibody followed by SDS-PAGE and western blot analysis. Phospho-tyrosine band with molecular weight 140 kilodalton was detected using 4G10 antibody. Bottom two rows represent western blot analysis of whole cell lysates (WCL) using Y694 P-STAT5 and total STAT antibodies. (B) Parental 32D cells and 32D-EGFRWT, 32D-EGFRL858R, and 32D-EGFRL861Q cells were switched for 2 h to serum-free medium containing DMSO (control) or AG490. Cell lysates were analyzed by western blot with Y869 P-EGFR and Y694 P-STAT5 antibodies. (C) The indicated cells were kept in serum-free medium for 2 h and then treated with TGFα (20 ng/ml) for 3 min. Cell lysates were prepared and precipitated with cetuximab followed by SDS-PAGE and western blot analysis of immune complexes with antibodies against JAK2 and EGFR. WCL, whole cell lysates probed for JAK2 protein levels by western blot. (D) 32D-EGFRWT cells were switched to serum free medium containing DMSO, AG490, erlotinib, or PP1 where indicated and then treated with TGFα (20 ng/ml) for 3 min. Cell lysates were analyzed by western blot using antibodies against Y1068 P-EGFR and Y694 P-STAT5. (E) 32D-EGFRWT cells were kept in serum-free medium containing erlotinib (1 μM) or PP1 (1 μM) for 2 h and then treated with TGFα (20 ng/ml). Cell lysates were prepared and precipitated with cetuximab followed by SDS-PAGE and western blot analysis with the indicated antibodies.
Mutant EGFR associates with Src and STAT5
STAT5 and Src are involved in EGF-induced mitogenesis and it has been suggested that this requires EGFR phosphorylation of Y869 [12]. Thus, we examined whether WT and mutant receptors in 32D cells associate with Src and STAT5. Mutant EGFRs showed constitutive association with STAT5 and Src whereas this complex was not detected in 32D-EGFRWT cells treated or not with TGFα (Fig. 5A). The association of STAT5 and Src with receptors containing L858R and L861Q was not altered by ligand stimulation nor by treatment with erlotinib (Fig 5A) or PP1 (Fig 5B), suggesting that the kinase activity of EGFR and Src are not required for complex formation. A similar association was observed in H3255 and PC9 lung cancer cells, which contain L858R and an exon 19 deletion in the EGFR gene, respectively [8, 13] (Fig 5C). These results suggest that mutant EGFRs recruit STAT5 and Src for direct activation of STAT5 while EGFRWT uses JAK2 for STAT5 activation.
Fig. 5.
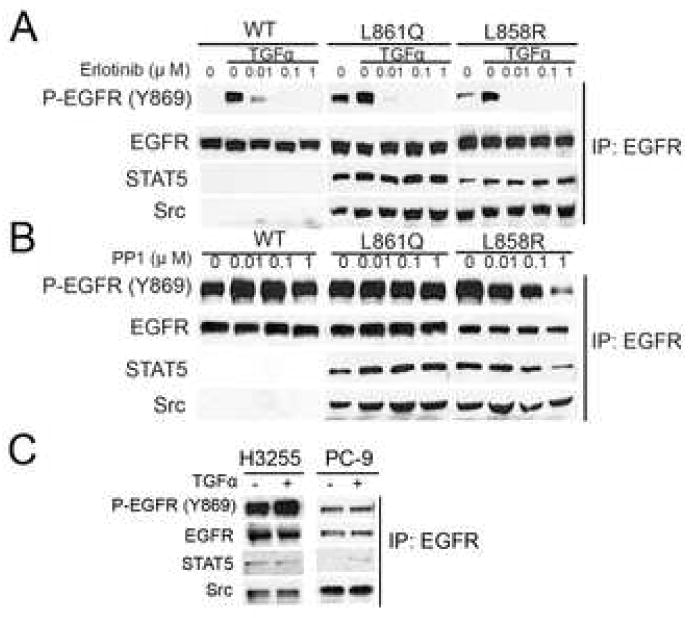
Mutant EGFRs constitutively associate with Src and STAT5. The indicated cells were grown in serum-free medium containing erlotinib (1 μM) or PP1 (1 μM) for 2 h and then treated with TGFα (20 ng/ml) for 3 min where indicated. Cell lysates were prepared and precipitated with cetuximab followed by separation of immune complex by SDS-PAGE and western blot analysis using the indicated antibodies. (A) Erlotinib or (B) PP1-treated 32D-EGFRWT, 32D-EGFRL861Q, or 32D-EGFRL858R cells. (C) H3255 and PC9 human lung cancer cells.
Mutant EGFR regulates expression of c-myc
Transcription of c-myc has been shown to be regulated by STAT5 [14]. Therefore, we examined the levels of c-myc in cells expressing mutant or WT EGFRs and their modulation by TKIs. Expression of c-myc was reduced by erlotinib in 32D-EGFRL858R and 32D-EGFRL861Q cells but not in 32D-EGFRWT cells. Treatment with PP1 had no effect on c-myc levels in 32D-EGFRWT and 32D-EGFRL861Q while P-STAT5 and c-myc expression were reduced by PP1 in 32D-EGFRL858R cells (Fig 6A). The content of c-myc correlated with detectable levels of P-STAT5. In cells expressing EGFRL861Q, erlotinib coordinately downregulated both P-STAT5 and c-myc. Based on these data, we propose the following model in Fig. 6B. In transfected 32D cells, EGFRWT recruits JAK2 in the absence of ligand stimulation resulting in activation of STAT5 and induction of c-myc. This activation can be enhanced by ligand stimulation but, under basal conditions, it does not require EGFR tyrosine kinase activity. On the other hand, mutant EGFRs recruit Src and STAT5 to the receptor, activate STAT5, and upregulate c-myc expression. 32D-EGFRL858R cells also utilize the Src kinase for STAT5 activation whereas 32D-EGFRWT and 32D-EGFRL861Q cells do not.
Fig. 6.
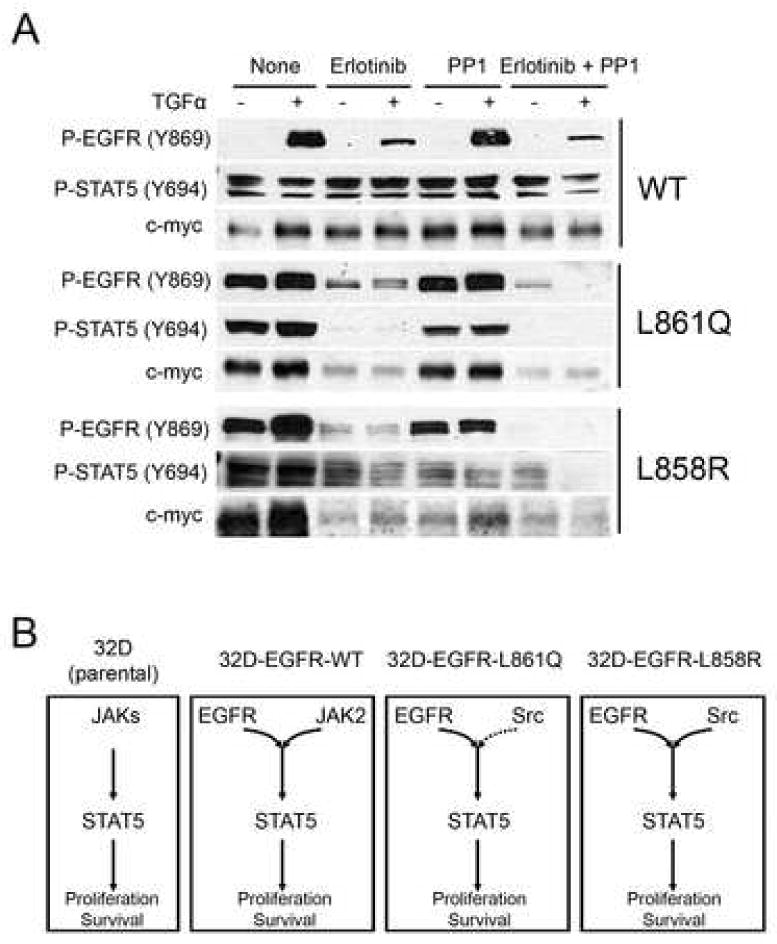
Regulation of c-myc expression by EGFR-STAT5 signaling. (A) 32D-EGFRWT ,32D-EGFRL861Q , or 32D-EGFRL858R cells were switched for 2 h to serum-free medium for 2 h containing erlotinib (1 μM), PP1 (1 μM ), or both compounds followed by the addition of TGFα (20 ng/ml) for 3 min. Cell lysates were prepared and analyzed by western blot with indicated antibodies. (B) Diagram of proposed mechanisms of EGFR-mediated STAT5 signal transduction in parental 32D cells and 32D cells expressing WT and mutant EGFRs. Solid and dashed arrows indicate weak and strong activation for downstream signaling, respectively.
DISCUSSION
Mutations in the tyrosine kinase domain of the EGFR have been identified in a variety of epithelial cancers, mainly in NSCLC. These mutations endow the receptor with ligand-independent catalytic activity and are oncogenic in a number of model systems [8, 15–18]. Most tumors harboring EGFR kinase domain mutations are dependent on them as EGFR tyrosine kinase inhibitors induce a marked antitumor effect [reviewed in [19, 20]]. The molecular mechanisms of ‘gain-of-function’ by these oncogenic mutants remain incompletely characterized. To study these mechanisms, we generated 32D hematopoietic cells stably expressing EGFRWT, EGFRL858R, and EGFRL861Q. One benefit of these cells to study EGFR signaling is the fact that they lack endogenous EGFR as well as its co-receptors in the erbB network, HER2, HER3, and HER4. However, it should be noted that 32D cells are suspension cells and their growth is highly dependent on cytokine signaling which may not reflect cellular/molecular events happened in epithelial tumor cells. Using tranduced 32D cells, we show herein ligand-independent phosphorylation of Y869 in mutant EGFRs and its functional consequences.
Tyrosine 869 (Y869) in the kinase domain of the EGFR exhibits unique features compared to conserved tyrosines in the activation loop of other receptor tyrosine kinases (RTKs) such as the insulin-like growth factor-I receptor (IGF-IR). For example, phosphorylation of Y869 at EGFR does not contribute to its kinase activity [21] while autophosphorylation of the equivalent tyrosines, Y1131, Y1135, and Y1136 in IGF-IR is required for maximal ligand-induced receptor’s catalytic activity [22]. Instead, the EGFR utilizes phosphorylation of this residue for mitogenesis by providing a docking site for the SH2 domain of STAT5 [12]. In 32D cells, EGFRL858R and EGFRL861Q displayed ligand-independent phosphorylation of Y869 in EGFR and of STAT5 (Fig. 1). Whereas the EGFRWT required Src for phosphorylation at Y869 [23, 24], both mutant receptors catalyzed the phosphorylation at this site by their intrinsic kinase activity but not Src activity (Fig. 1D and 2C).
The kinase domain of EGFRL858R and EGFRL861Q but not of the WT receptor was also able to auto-phosphorylate Y869 (Fig. 2). These results imply, first, that ligand binding to the extracellular domain of mutant receptors and their dimerization are dispensable for Y869 phosphorylation. Second, the C-terminus of the receptor does not contribute to phosphorylation at Y869 and, therefore, SH2 domain-containing adaptor proteins that bind to phosphorylated C-terminal tyrosines are not required either. Structural analysis recently reported by Zhang et al. [25] suggest that mutations in the activation loop of EGFR kinase domain destabilize its inactive conformation potentially explaining how the EGFRL858R and EGFRL861Q kinase domains maintain an active conformation. A thermodynamically stable conformation would then enable auto-phosphorylation of Y869 in mutant EGFRs. Phosphorylation of Y869 in mutant EGFR kinase domains was inhibited by erlotinib. Although the Src inhibitors PP1 and PP2 reduced P-Y869 in 32D cells transfected with full-length EGFRL858R and EGFRL861Q (Fig 1B), they did not inhibit P-Y869 in insect cells expressing receptor kinase domains (Fig 2C). These data suggest that mutation L861Q or L858R in kinase domain of EGFR is enough to catalyze the phosphorylation of Y869 and that the kinase activity of Src is not required for this reaction.
Although not abundant, tyrosine phosphorylation is a key post-translational modification induced by kinases on cellular substrates for the activation of signal transduction pathways. The data shown here reflect the dual functions of RTKs on signal transduction. First, their kinase activity can induce their autophosphorylation as well as that of other substrates which, in turn, act as ‘upstream’ kinases in the next round phosphorylation/kinase reactions. For example, mutant EGFRs can auto-phosphorylate Y869 and other tyrosines in the receptor’s C-terminus [4, 8, 26, 27]. A second function of oncogenic RTKs is kinase activity-independent but conformation-dependent and consists of providing docking sites for adaptors and protein complexes which can themselves transmit signals to downstream. EGFRWT is associated with JAK2 (Fig. 4) whereas mutant EGFRs recruit STAT5 and Src in a ligand-independent manner (Fig 5). In this case, the wild-type vs. mutant conformation dictated the preference of its signaling partner. For STAT5 activation, EGFRWT recruits JAK2 while EGFRL861Q and EGFRL858R recruit STAT5 and Src, all in ligand-independent manner. Furthermore, kinase-inactivated mutant receptors (upon treatment with erlotinib) still associated stably with Src and STAT5 (Fig5A). Taken together, these findings support a ‘gain-of-function’ effect of the mutant EGFR kinase that is distinct from its increased catalytic activity.
Acknowledgments
This work was supported by NIH R01 grants CA80195 and CA62212 (CLA), Breast Cancer Specialized Program of Research Excellence (SPORE) P50 CA98131, and Vanderbilt-Ingram Comprehensive Cancer Center Support Grant P30 CA68485.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 2.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 3.Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, Singh B, Heelan R, Rusch V, Fulton L, et al. EGF receptor gene mutations are common in lung cancers from "never smokers" and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004;101:13306–13311. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sordella R, Bell DW, Haber DA, Settleman J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science. 2004;305:1163–1167. doi: 10.1126/science.1101637. [DOI] [PubMed] [Google Scholar]
- 5.Kloth MT, Laughlin KK, Biscardi JS, Boerner JL, Parsons SJ, Silva CM. STAT5b, a Mediator of Synergism between c-Src and the Epidermal Growth Factor Receptor. J Biol Chem. 2003;278:1671–1679. doi: 10.1074/jbc.M207289200. [DOI] [PubMed] [Google Scholar]
- 6.Wittig I, Groner B. Signal transducer and activator of transcription 5 (STAT5), a crucial regulator of immune and cancer cells. Curr Drug Targets Immune Endocr Metabol Disord. 2005;5:449–463. doi: 10.2174/156800805774912999. [DOI] [PubMed] [Google Scholar]
- 7.Calo V, Migliavacca M, Bazan V, Macaluso M, Buscemi M, Gebbia N, Russo A. STAT proteins: from normal control of cellular events to tumorigenesis. J Cell Physiol. 2003;197:157–168. doi: 10.1002/jcp.10364. [DOI] [PubMed] [Google Scholar]
- 8.Yang S, Qu S, Perez-Tores M, Sawai A, Rosen N, Solit DB, Arteaga CL. Association with HSP90 inhibits Cbl-mediated down-regulation of mutant epidermal growth factor receptors. Cancer Res. 2006;66:6990–6997. doi: 10.1158/0008-5472.CAN-06-1042. [DOI] [PubMed] [Google Scholar]
- 9.Sato K, Nagao T, Iwasaki T, Nishihira Y, Fukami Y. Src-dependent phosphorylation of the EGF receptor Tyr-845 mediates Stat-p21waf1 pathway in A431 cells. Genes Cells. 2003;8:995–1003. doi: 10.1046/j.1356-9597.2003.00691.x. [DOI] [PubMed] [Google Scholar]
- 10.Nakamura N, Chin H, Miyasaka N, Miura O. An epidermal growth factor receptor/Jak2 tyrosine kinase domain chimera induces tyrosine phosphorylation of Stat5 and transduces a growth signal in hematopoietic cells. J Biol Chem. 1996;271:19483–19488. doi: 10.1074/jbc.271.32.19483. [DOI] [PubMed] [Google Scholar]
- 11.De Vos J, Jourdan M, Tarte K, Jasmin C, Klein B. JAK2 tyrosine kinase inhibitor tyrphostin AG490 downregulates the mitogen-activated protein kinase (MAPK) and signal transducer and activator of transcription (STAT) pathways and induces apoptosis in myeloma cells. Br J Haematol. 2000;109:823–828. doi: 10.1046/j.1365-2141.2000.02127.x. [DOI] [PubMed] [Google Scholar]
- 12.Boerner JL, Demory ML, Silva C, Parsons SJ. Phosphorylation of Y845 on the epidermal growth factor receptor mediates binding to the mitochondrial protein cytochrome c oxidase subunit II. Mol Cell Biol. 2004;24:7059–7071. doi: 10.1128/MCB.24.16.7059-7071.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perez-Torres M, Guix M, Gonzalez A, Arteaga CL. Epidermal growth factor receptor (EGFR) antibody down-regulates mutant receptors and inhibits tumors expressing EGFR mutations. J Biol Chem. 2006;281:40183–40192. doi: 10.1074/jbc.M607958200. [DOI] [PubMed] [Google Scholar]
- 14.Turkson J. STAT proteins as novel targets for cancer drug discovery. Expert Opin Ther Targets. 2004;8:409–422. doi: 10.1517/14728222.8.5.409. [DOI] [PubMed] [Google Scholar]
- 15.Jiang J, Greulich H, Janne PA, Sellers WR, Meyerson M, Griffin JD. Epidermal growth factor-independent transformation of Ba/F3 cells with cancer-derived epidermal growth factor receptor mutants induces gefitinib-sensitive cell cycle progression. Cancer Res. 2005;65:8968–8974. doi: 10.1158/0008-5472.CAN-05-1829. [DOI] [PubMed] [Google Scholar]
- 16.Politi K, Zakowski MF, Fan PD, Schonfeld EA, Pao W, Varmus HE. Lung adenocarcinomas induced in mice by mutant EGF receptors found in human lung cancers respond to a tyrosine kinase inhibitor or to down-regulation of the receptors. Genes Dev. 2006;20:1496–1510. doi: 10.1101/gad.1417406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ji H, Li D, Chen L, Shimamura T, Kobayashi S, McNamara K, Mahmood U, Mitchell A, Sun Y, Al-Hashem R, et al. The impact of human EGFR kinase domain mutations on lung tumorigenesis and in vivo sensitivity to EGFR-targeted therapies. Cancer Cell. 2006;9:485–495. doi: 10.1016/j.ccr.2006.04.022. [DOI] [PubMed] [Google Scholar]
- 18.Mulloy R, Ferrand A, Kim Y, Sordella R, Bell DW, Haber DA, Anderson KS, Settleman J. Epidermal growth factor receptor mutants from human lung cancers exhibit enhanced catalytic activity and increased sensitivity to gefitinib. Cancer Res. 2007;67:2325–2330. doi: 10.1158/0008-5472.CAN-06-4293. [DOI] [PubMed] [Google Scholar]
- 19.Janne PA, Johnson BE. Effect of epidermal growth factor receptor tyrosine kinase domain mutations on the outcome of patients with non-small cell lung cancer treated with epidermal growth factor receptor tyrosine kinase inhibitors. Clin Cancer Res. 2006;12:4416s–4420s. doi: 10.1158/1078-0432.CCR-06-0555. [DOI] [PubMed] [Google Scholar]
- 20.Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer. 2007;7:169–181. doi: 10.1038/nrc2088. [DOI] [PubMed] [Google Scholar]
- 21.Tice DA, Biscardi JS, Nickles AL, Parsons SJ. Mechanism of biological synergy between cellular Src and epidermal growth factor receptor. Proc Natl Acad Sci U S A. 1999;96:1415–1420. doi: 10.1073/pnas.96.4.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li W, Miller WT. Role of the activation loop tyrosines in regulation of the insulin-like growth factor I receptor-tyrosine kinase. J Biol Chem. 2006;281:23785–23791. doi: 10.1074/jbc.M605269200. [DOI] [PubMed] [Google Scholar]
- 23.Sato K, Sato A, Aoto M, Fukami Y. c-Src phosphorylates epidermal growth factor receptor on tyrosine 845. Biochem Biophys Res Commun. 1995;215:1078–1087. doi: 10.1006/bbrc.1995.2574. [DOI] [PubMed] [Google Scholar]
- 24.Biscardi JS, Maa MC, Tice DA, Cox ME, Leu TH, Parsons SJ. c-Src-mediated phosphorylation of the epidermal growth factor receptor on Tyr845 and Tyr1101 is associated with modulation of receptor function. J Biol Chem. 1999;274:8335–8343. doi: 10.1074/jbc.274.12.8335. [DOI] [PubMed] [Google Scholar]
- 25.Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell. 2006;125:1137–1149. doi: 10.1016/j.cell.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 26.Greulich H, Chen TH, Feng W, Janne PA, Alvarez JV, Zappaterra M, Bulmer SE, Frank DA, Hahn WC, Sellers WR, Meyerson M. Oncogenic Transformation by Inhibitor-Sensitive and -Resistant EGFR Mutants. PLoS Med. 2005;2:e313. doi: 10.1371/journal.pmed.0020313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carey KD, Garton AJ, Romero MS, Kahler J, Thomson S, Ross S, Park F, Haley JD, Gibson N, Sliwkowski MX. Kinetic analysis of epidermal growth factor receptor somatic mutant proteins shows increased sensitivity to the epidermal growth factor receptor tyrosine kinase inhibitor, erlotinib. Cancer Res. 2006;66:8163–8171. doi: 10.1158/0008-5472.CAN-06-0453. [DOI] [PubMed] [Google Scholar]