

Abstract
Tulp1 is a protein of unknown function exclusive to rod and cone photoreceptor cells. Mutations in the gene cause autosomal recessive retinitis pigmentosa in humans and photoreceptor degeneration in mice. In tulp1−/− mice, rod and cone opsins are mislocalized, and rhodopsin-bearing extracellular vesicles accumulate around the inner segment, indicating that Tulp1 is involved in protein transport from the inner segment to the outer segment. To investigate this further, we sought to define which outer segment transport pathways are Tulp1-dependent. We used immunohistochemistry to examine the localization of outer segment proteins in tulp1−/− photoreceptors, prior to retinal degeneration. We also surveyed the condition of inner segment organelles and rhodopsin transport machinery proteins. Herein, we show that guanylate cyclase 1 and guanylate cyclase activating proteins 1 and 2 are mislocalized in the absence of Tulp1. Furthermore, arrestin does not translocate to the outer segment in response to light stimulation. Additionally, data from the tulp1−/− retina adds to the understanding of peripheral membrane protein transport, indicating that rhodopsin kinase and transducin do not co-transport in rhodopsin carrier vesicles and phosphodiesterase does not co-transport in guanylate cyclase carrier vesicles. These data implicate Tulp1 in the transport of selective integral membrane outer segment proteins and their associated proteins, specifically, the opsin and guanylate cyclase carrier pathways. The exact role of Tulp1 in outer segment protein transport remains elusive. However, without Tulp1, two rhodopsin transport machinery proteins exhibit abnormal distribution, Rab8 and Rab11, suggesting a role for Tulp1 in vesicular docking and fusion at the plasma membrane near the connecting cilium.
Keywords: photoreceptor, retinal degeneration, Tulp1, rhodopsin, protein transport, retinitis pigmentosa, mouse mutant
1. Introduction
Disc membranes in the photoreceptor cells of the vertebrate retina are continually renewed by the addition of membranes at the base of the outer segment (OS) and the removal of older discs from the distal end. OS-bound proteins are sorted and trafficked from the endoplasmic reticulum (ER) and trans-Golgi network (TGN) as cargo in vesicles from the inner segment (IS) toward the OS on a continual basis (Papermaster et al., 1985; Besharse and Wetzel, 1995). Rhodopsin, the photopigment molecule in rod photoreceptors, and other OS-resident proteins are reliant upon this polarized protein trafficking, which involves interactions between proteins of the transport machinery and proteins present on the cargo vesicles (Papermaster et al., 1985; Besharse and Wetzel, 1995; Aroeti et al., 1998). The complexity of this interaction is underscored by the fact that a defect in only one of these components can result in a loss of polarity in protein trafficking and ultimately photoreceptor cell death (Hagstrom et al., 1999; Deretic, 2006).
Retinitis pigmentosa incorporates a group of heterogeneous inherited retinal degenerations (Berson, 1993). One early onset form of retinitis pigmentosa is caused by mutations in TULP1 (Banerjee et al., 1998; Gu et al., 1998; Hagstrom et al., 1998; Paloma et al., 2000). Tulp1 is a cytoplasmic protein expressed exclusively in photoreceptors, localizing to the IS, connecting cilium (CC), perikarya, and terminals (Hagstrom et al., 1999). Tulp1 associates with cellular membranes and the cytoskeletal elements (Hagstrom et al., 1999; Ikeda et al., 2000; Hagstrom et al., 2001; Xi et al., 2005) and appears to play a role in intracellular protein trafficking (Hagstrom et al., 1999; Hagstrom et al., 2001; Xi et al., 2005; Xi et al., 2007; Grossman et al., 2009). Tulp1−/− mice develop a rapid photoreceptor degeneration, similar to that in patients with retinitis pigmentosa due to TULP1 mutations (Hagstrom et al., 1999). In the young tulp1−/− retina prior to photoreceptor degeneration, there is an accumulation of vesicles in the interphotoreceptor matrix surrounding the IS. These vesicles show rhodopsin immunoreactivity, likely representing misrouted transport carriers for opsin (Hagstrom et al., 1999). Indeed, rod and cone opsins are mislocalized throughout the photoreceptor. Importantly, this mistargeting occurs early in development, indicating a primary defect, and not a secondary effect due to a generalized degenerative process (Hagstrom et al., 1999; Grossman et al., 2009). However, other OS proteins are not mislocalized in the tulp1−/− retina, indicating a specialized role for Tulp1 in OS protein transport (Hagstrom et al., 1999).
Proposed OS protein transport pathways have been deduced from a handful of mutant mouse models (Yang et al., 1999; Baehr et al., 2007; Zhang et al., 2007; Karan et al., 2008b). Therefore, it is critical to study additional mouse mutants which present phenotypes showing OS protein transport defects. To define the transport pathways affected in the Tulp1 mutant mouse, we analyzed the localization of several categories of OS-resident proteins in tulp1−/− retinas as compared to wild-type (wt) retinas. We also surveyed several IS proteins known to be critical in the transport of OS proteins.
2. Materials and methods
2.1. Mice
The generation of tulp1−/− mice has been described previously (Hagstrom et al., 1999) and is maintained on a C57BL/6 background. Mice were euthanized by CO2 inhalation followed by cervical dislocation. All experiments on animals were approved by the Institutional Animal Care and Use Committee of the Cleveland Clinic and were performed in compliance with the National Institutes of Health guidelines.
2.2. Preparation of retinal sections
Mouse eyes were prepared as previously described (Xi et al., 2007). Briefly, after removal of the cornea and lens, the posterior poles were fixed in 4% paraformaldehyde in PBS for 3 hrs. The eyes cups were then immersed through a graded series of sucrose solutions as follows: 10% for 1 hr, 20% for 1 hr and 30% overnight. The posterior pole was embedded in OCT freezing medium, flash frozen on powderized dry ice and immediately transferred to −80°C. The tissue was sectioned at 10-μm thickness using a cryostat (Leica, Wetzlar, Germany) at −30°C.
2.3. Immunofluorescent staining
For each genotype (wt and tulp1−/−), a minimum number of eight sections from six different mice were examined for each antibody. Retinal sections were blocked in 5% bovine serum albumin and 1% normal goat serum with 0.1% Triton X-100 for 1 hr before incubation with primary antibodies overnight at 4°C. Primary antibodies, their source and the dilutions used in this study are listed in table 1. After washing 3 times in PBS, sections were incubated in fluorescent secondary antibodies at room temperature for 1 hr. A panel of well-characterized antibodies against retinal proteins was used for immunostaining. Table 1 contains a complete list of antibodies, immunogens, sources, host species and dilutions used. Secondary antibodies were: Alexa Fluor® 488 goat anti-rabbit IgG and goat anti-mouse IgG; Alexa Fluor® 594 goat anti-rabbit IgG and goat anti-mouse IgG (Invitrogen, Carlsbad, California). The sections were then rinsed 3 times with PBS followed by a light rinse in dH2O and coverslipped with Vectashield® mounting media with DAPI (Vector Laboratories, Burlingame, California). Sections were imaged using an Olympus BX-61 fluorescent microscope (Olympus, Tokyo, Japan) equipped with a CCD monochrome camera (Hamamatsu Photonics, Bridgewater, New Jersey).
Table 1.
Primary Antibodies
| Antigen | Immunogen | Source | Host | Dilution | Reference |
|---|---|---|---|---|---|
| Arrestin | Amino acid residues 301–320 of bovine S- arrestin1 (HEDTNLASSTIIKEGIDKTV) | Dr. Clay W. Smith, Univ. Florida, Gainesville, FL (SCT-128) | mouse monoclonal | 1:200 | Orisme et al., 2010 |
| Rhodopsin | Membrane preparation from adult rat retina | Abcam (Cambridge, MA), (#ab3267–500, lot #786120) (RET-P1) | mouse monoclonal | 1:200 | Meira et al., 2009 |
| Peripherin/rds | Amino acid residues of the C-terminus of mouse Peripherin/rds (CVEAEGADAGPAPEAG) | Dr. Andrew F.X. Goldberg, Oakland Univ., Rochester, MI (pAbMPCT) | rabbit polyclonal | 1:500 | Goldberg et al., 2007 |
| ROM1 | Full-length human Rom1 | Dr. Roderick McInnes, The Hospital for Sick Children, University of Toronto, Toronto, Canada | rabbit polyclonal | 1:1000 | Clarke et al., 2000 |
| GRK1 | Full-length human GRK1a | Dr. Krzysztof Palczewski, Case Western Reserve Univ., Cleveland, OH (G8) | mouse monoclonal | 1:500 | Zhao et al., 1998; Baehr et al., 2007; Zhang et al., 2007 |
| PDE6-β | Amino acid residues 20–36 of mouse PDE6- β (HQYFG(K/R)KLSPENVAGAC) | Abcam (Cambridge, MA), rabbit polyclonal (#ab5663, lot #595592) | rabbit polyclonal | 1:1000 | Sanges et al., 2006; Giordano et al., 2007 |
| CNGB1 | Amino acid residues of the C-terminus of mouse CNGB1 (EAAGPPEPSVRIRVSPGP) | Dr. Martin Biel, Ludwig-maximilians-universität, München, Germany (C-AbmCNGB1) | rabbit polyclonal | 1:500 | Huttl et al., 2005 |
| GCAP1 | Amino acid residues 26–205 of bovine GCAP1 | Dr. Krzysztof Palczewski, Case Western Reserve Univ., Cleveland, OH (UW14) | rabbit polyclonal | 1:3000 | Howes et al., 1998 |
| GCAP2 | Full-length bovine GCAP2 | Dr. Krzysztof Palczewski, Case Western Reserve Univ., Cleveland, OH (UW50) | rabbit polyclonal | 1:2000 | Howes et al., 1998 |
| GC1 | Amino acid residues of the C-terminus of bovine GC1 (RQKLEKARPGQFSGK) | Dr. Krzysztof Palczewski, Case Western Reserve Univ., Cleveland, OH (IS4) | mouse monoclonal | 1:10,000 | Baehr et al., 2007 |
| Blue cone opsin | Full-length human blue opsin | Millipore (Billerica, MA), (#ab5407, lot #24121205) | rabbit polyclonal | 1:400 | Corbo and Cepko, 2005; Haverkamp et al., 2005; Raven et al., 2008; Ng et al., 2010 |
| Transducin-α | Amino acid residues 60–110 of human transducin-α | Santa Cruz Biotechnology (Santa Cruz, CA), (#sc-389, lot #B2508) | rabbit polyclonal | 1:1000 | Elias et al., 2004; Coleman and Semple-Rowland, 2005 |
| Rab6 | Amino acid residues 158–208 of human Rab6 | Santa Cruz Biotechnology (Santa Cruz, CA), (#sc-310, lot #J0608) | rabbit polyclonal | 1:500 | Zhang and Townes-Anderson, 2002; Mazelova et al., 2009 |
| Rab8 | Amino acid residues 84–205 of human Rab8 | BD Transduction Laboratories (San Diego, CA), (#610844, lot #39862) (4/Rab4) | mouse monoclonal | 1:50 | Moritz et al., 2001; Deretic et al., 2004 |
| Rab11 | C-terminus of recombinant human Rab11 | Invitrogen (Carlsbad, California), (#71–5300, lot #646681A) | rabbit polyclonal | 1:50 | Mazelova et al., 2009 |
| PrBP/δ | Full-length bovine PrBP/δ | Dr. Rick H. Cote, University of New Hampshire, Durham, NH (FL δ) | rabbit polyclonal | 1:500 | Norton et al., 2005; Zhang et al., 2007 |
| GM130 | Amino acid residues 869–982 of rat GM130 | BD Transduction Laboratories (San Diego, CA), (#610822, lot #14868) (35/GM130) | mouse monoclonal | 1:100 | Kerov et al., 2005; Krajacic et al., 2006 |
| Calreticulin | Amino acid residues 412–417 of human Calreticulin (QAKDEL) | Millipore (Billerica, MA), (#06–661, lot #29709) | rabbit polyclonal | 1:100 | Michalak et al., 1996 |
| COXIV | Full-length human COXIV isoform 1 | Invitrogen (Carlsbad, California), (#A21348) (20E8C12) | mouse monoclonal | 1:500 | Johnson et al., 2007 |
2.4. Light adaptation
To study the translocation of OS proteins in response to light exposure, eight mice were separated into two groups. At the onset of lights off in the vivarium, both groups were transferred into a chamber under constant darkness. All animals were anesthetized with an intraperitoneal injection of ketamine (80 mg/kg) and xylazine (16 mg/kg) and the pupils were dilated with a mixture of 1% cyclopentolate-HCl, 2.5% phenylephrine, and 0.25% tropicamide. At approximately 1 hr prior to the time of lights on, one group was transferred to a lighting chamber and exposed to 1,000 lux for 60 min, while the control group remained in the lightproof chamber. After 60 min, both groups were sacrificed, and the eyes extracted and processed as described above.
3. Results
3.1. Tulp1−/− mice form outer segments
The retinas of postnatal day (P)16 tulp1−/− mice contain the normal complement of retinal cell types including photoreceptors. However, at this age, the OS of the tulp1−/− photoreceptor is slightly disorganized (Fig. 1E) as compared to wt (Fig. 1A) (Hagstrom et al., 1999). The presence of OSs in tulp1−/− photoreceptors is confirmed by the expression and confinement of peripherin/rds to the OS (Fig. 1C and G). In contrast, rhodopsin is severely mislocalized to all cellular compartments in tulp1−/− photoreceptors (Fig. 1F) as compared to wt (Fig. 1B). Peripherin/rds and rhodopsin are both integral membrane proteins that are incorporated into nascent OS discs (Lee et al., 2006); however, unlike rhodopsin, peripherin/rds is clearly able to be targeted to the OS in the absence of Tulp1 (Fig. 1G). These results reveal that OS proteins are affected differently in tulp1−/− mice, providing evidence that Tulp1 functions in selective OS transport pathways.
Figure 1.

Tulp1−/− mice form OSs but exhibit defects in OS protein transport. A–D: wt retina at P16. E–H: tulp1−/− retina at P16. Differential interference contrast (DIC) images reveal that the wt retina (A) and the tulp1−/− retina (E) form OSs. Immunofluorescent staining indicates that rhodopsin, normally localized exclusively to the OS (B), is distributed throughout the OS, IS, ONL and photoreceptor synapses of the OPL in the tulp1−/− retina (F). In the wt retina, peripherin/rds is a structural protein that is also confined to the OS (C), but unlike rhodopsin, is localized normally in the tulp1−/− retina (G). The far right column shows a merge of the channels in wt (D) and tulp1−/− (H) retinal sections. Nuclei are labeled with DAPI (blue). Scale bars = 20μm.
3.2. Outer segment proteins that are normally localized in tulp1−/− retinas
Since rhodopsin is mislocalized in tulp1−/− photoreceptors, we sought to determine whether additional OS proteins rely on the presence of Tulp1 for their transport to the OS compartment. To this end, we surveyed a panel of OS-resident proteins in the tulp1−/− retina as compared to the wt retina. Figure 2 presents the results of the localization of several well-studied phototransduction and structural OS proteins. All studies were conducted at P16, an age at which photoreceptor development is complete in wt mice, but precedes photoreceptor cell death in tulp1−/− mice (Hagstrom et al., 1999; Grossman et al., 2009).
Figure 2.
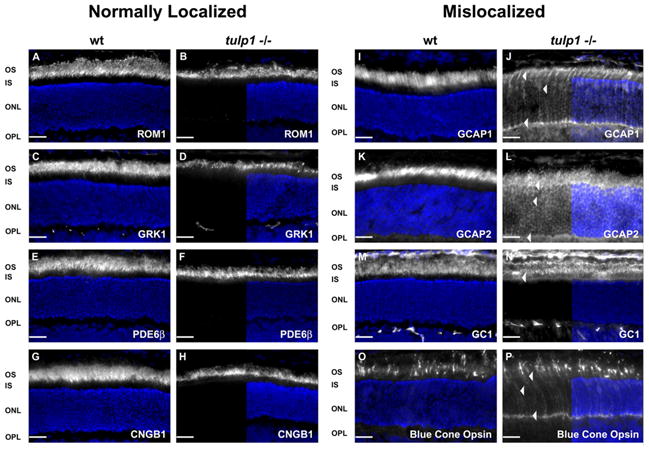
Differential trafficking of OS resident proteins in the tulp1−/− retina. Immunofluorescent images of proteins (white) and nuclei (DAPI; blue) compare the compartmental localization of OS proteins in wt and tulp1−/− retinal sections at P16. Panels in the second and fourth columns show a split screen view. On the right half, the protein of interest and nuclei are co-labeled, while the left half displays only the immunofluorescent signal of the targeted protein. A–H: OS proteins localized normally in wt and tulp1−/− retinas. We identified several OS resident proteins that retain their normal localization in the tulp1−/− retina. These proteins include: ROM1 (B), GRK1 (D), PDE6 (F) and CNGB1 (H). I–P: OS proteins mislocalized in tulp1−/− retinas. In addition to rhodopsin (Fig. 1F), four additional OS-resident proteins are mislocalized to other photoreceptor compartments in the tulp1−/− retina. Although staining of GCAP1 (J) and 2 (L), GC1 (N) and blue cone opsin (P) is still apparent in the OS, arrowheads highlight the mislocalization to other cellular compartments. Scale bars = 20μm.
Figure 2A shows a wt mouse retinal section stained with antibodies against rod OS membrane protein 1 (ROM1). ROM1 is an integral membrane protein localized to the rim of the OS discs similar to peripherin/rds, where the two proteins form heterotetramers (Lee et al., 2006). In the tulp1−/− retina, ROM1 staining is restricted to the OS, indicating that Tulp1 is not involved in the transport of this disc structural protein (Fig. 2B). Rhodopsin kinase (GRK1) is a G-protein coupled receptor kinase that participates in the inactivation of rhodopsin through phosphorylation (Smith et al., 1983). It is a peripheral membrane protein that acquires its membrane association from posttranslational processing in the ER (Inglese et al., 1992). In the wt and tulp1−/− retina, GRK1 is clearly confined to the OS (Fig. 2C and D). Also properly localized in the tulp1−/− retina is another peripheral membrane protein, cGMP phosphodiesterase (PDE6). PDE6 is the central effector enzyme in the activation phase of the phototransduction cascade (Chabre et al., 1993). Its primary action is to hydrolyze cGMP to GMP, which causes the closure of cGMP gated ion channels in the OS (Qin et al., 1992). PDE6 is also solely posttranslationally processed in the ER (Karan et al., 2008b). Antibodies against the β subunit of PDE6 reveal that this protein is confined to the OS in the tulp1−/− retina (Fig. 2F), as it is in the wt retina (Fig. 2E). The cGMP-gated cation channel (CNG) is an integral membrane protein, and like all other OS integral membrane proteins, is synthesized in the ER and processed through the Golgi apparatus, eventually exiting the TGN in packaged vesicles (Lippincott-Schwartz et al., 2000). In the dark, the channel maintains a standing cation influx and is subsequently closed by light during the activation phase of vision (Kaupp and Seifert, 2002). CNGB1 is the beta subunit of the channel and is localized properly to the OS in the tulp1−/− retina (Fig. 2G and H).
3.3. Outer segment proteins that are mislocalized in tulp1−/− retinas
We identified several other OS proteins that are mislocalized in the tulp1−/− retina. The guanylate cyclases (GCs) convert GTP to cGMP, which is the second messenger in phototransduction (Gorczyca et al., 1995). Guanylate cyclase activating protein (GCAP)1 is a peripheral membrane-associated protein that acts to activate the GCs. In the wt retina, GCAP1 is restricted to the OS (Fig. 2I); however, in the tulp1−/− retina, it is severely mislocalized. Prominent staining is seen throughout the photoreceptor cell, including the OS, IS, outer nuclear layer (ONL) and outer plexiform layer (OPL) (Fig. 2J). GCAP2, a cytosolic protein that also activates the GCs, has a similar staining pattern to GCAP1 (Gorczyca et al., 1995). In the wt retina, it is restricted to the OS (Fig. 2K); while in the tulp1−/− retina, staining is evident throughout all photoreceptor compartments (Fig. 2L). GC1 is a transmembrane protein that acts to produce cGMP (Gorczyca et al., 1995). In the wt retina, GC1 is located exclusively in the OS (Fig. 2M). In the tulp1−/− retina however, it is localized to both the OS and IS (Fig. 2N). Finally, we investigated blue cone opsin, the photopigment molecule in blue cones. As previously reported, this protein is mislocalized throughout the photoreceptor cell in the tulp1−/− retina (Fig. 2P) as compared to the restricted OS staining seen in the wt retina (Fig. 2O) (Hagstrom et al., 1999).
3.4. Signaling proteins translocate differentially in response to light in tulp1−/− retinas
Next we analyzed whether photoreceptor proteins that exhibit light-dependent movement into and out of the OS are affected in the tulp1−/− retina. Transducin and arrestin are two signal transduction proteins that translocate in response to lighting conditions (McGinnis et al., 1992; Calvert et al., 2006; Slepak and Hurley, 2008). Transducin is the heterotrimeric G protein in rod photoreceptors. It is a peripheral membrane-associated protein that activates PDE6 to hydrolyze cGMP, thereby closing cation channels (Chabre and Deterre, 1989). In the wt retina, transducin is localized to the OS in the dark (Fig. 3A) and translocates to the IS, ONL and OPL upon light stimulation (Fig. 3C). In the tulp1−/− retina, transducin movement appears to occur normally. It is localized to the OS in the dark (Fig. 3B) and the inner compartments of the cell in the light (Fig. 3D). Arrestin is a cytosolic protein that binds to phosphorylated rhodopsin, playing an important role in the recovery phase of phototransduction (Chabre and Deterre, 1989). This protein also translocates with light stimulation, but in the opposite direction to that of transducin. In the wt retina, arrestin is localized to the IS, ONL and OPL in darkness (Fig. 3E & Fig. 4A) and redistributes to the OS in the light (Fig. 3G & Fig. 4C). However, in the tulp1−/− retina, the distribution of arrestin remains confined to the IS, ONL and OPL in both the dark (Fig. 3F & Fig. 4B) and light (Fig. 3H & Fig. 4D). Our data indicates that light dependent translocation of arrestin in the tulp1−/− retina is severely impaired.
Figure 3.

Light-induced translocation of arrestin but not transducin is severely impaired in P16 tulp1−/− retinas. A–D: Immunostaining for transducin in wt and tulp1−/− retinas. Immunolocalization reveals that in the wt retina, transducin is localized to the OS in the dark (A) and translocates to the IS, ONL and synapses of the OPL upon light stimulation (C). (B&D) A similar translocation pattern is seen in the tulp1−/− retina. E–H: Immunostaining for arrestin in P16 wt and tulp1−/− retinas. In contrast to transducin, arrestin is localized in the wt retina to the IS, ONL and OPL in the dark (E), and translocates to the OS after exposure to bright light (G). In the dark, arrestin is normally localized to the IS, ONL and OPL in the tulp1−/− retina (F); however, arrestin does not translocate in response to light, but remains in the IS, ONL and OPL (H). A graphical depiction of transducin and arrestin localizations (red) in the light and dark adapted photoreceptor cell designated by open and closed boxes is shown in between each corresponding image. Scale bars = 20μm.
Figure 4.
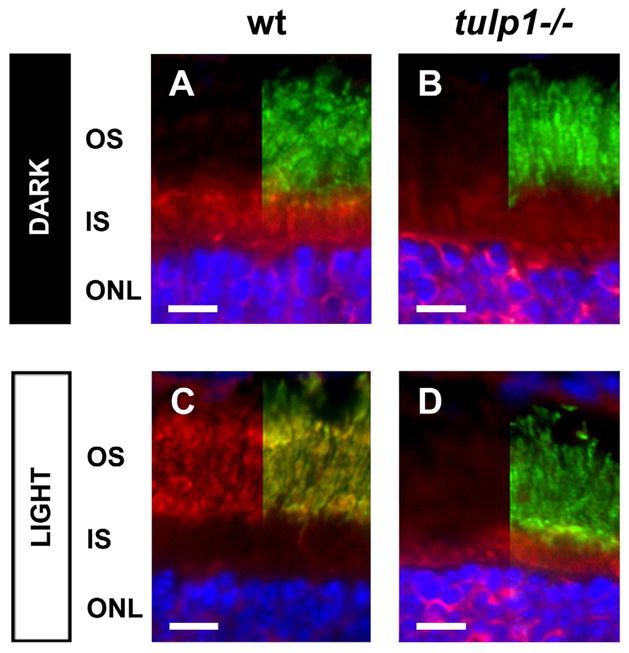
High resolution images highlight the inability of arrestin to translocate to the OS in response to light exposure in the P16 tulp1−/− retina. A–D: Double-labeled immunostaining for arrestin (red) and peripherin/rds (green) in wt and tulp1−/− retinas (DAPI; blue). The right half of each image shows the fluorescent signal of sections co-labeled with antibodies against arrestin and peripherin/rds, while the left half displays only the immunofluorescent signal for arrestin. (A) In the wt retina in the dark, arrestin is localized to the ONL and IS, but is absent from the OS. Upon light stimulation, arrestin staining is no longer present in the IS and ONL, but is present in the OS, where it is tightly co-localized with peripherin/rds (C). (B) In the P16 tulp1−/− retina in the dark, arrestin is present in the ONL and IS, but is absent from the OS. However, arrestin staining remains localized to the ONL and the IS after light exposure, and there is no detectable fluorescent signal in the OS compartment (D). Scale bars = 10μm.
3.5. Outer segment protein transport machinery is disrupted in tulp1−/− retinas
Recent studies have clarified the protein complexes that regulate the trafficking of OS proteins, most notably rhodopsin. These include Rab6, Rab8, Rab11, ADP-ribosylation factor 4 (ARF4) and prenyl binding protein delta (PrBP/δ) (Deretic and Papermaster, 1993; Deretic et al., 1995; Deretic, 1997; Moritz et al., 2001; Deretic, 2006; Zhang et al., 2007; Karan et al., 2008b; Mazelova et al., 2009). Figure 5 compares the distribution of these proteins in wt and tulp1−/− retinas. The left column shows the immunoreactivity of the targeted protein in the wt retina, while the right column presents the immunoreactivity in the tulp1−/− retina. Rab6 has been implicated in the generation and budding of rhodopsin-laden vesicles from the TGN (Deretic and Papermaster, 1993; Deretic, 2006). In the wt retina, Rab6 staining is confined to the IS, immediately adjacent to the outer limiting membrane near the location of the TGN (Fig. 5A). In the tulp1−/− retina, staining is also present in this portion of the IS, but punctate signals extend beyond the boundary of the outer limiting membrane and into the ONL (Fig. 5B; arrowheads indicate mislocalized Rab6 molecules in the ONL). Rab8 is thought to be important in the docking and fusion of rhodopsin-bearing vesicles to the IS plasma membrane, in close proximity to the CC (Deretic et al., 1995; Moritz et al., 2001; Deretic, 2006). In the wt retina, the staining of Rab8 is robust in the ellipsoid region of the IS and extends toward the CC (Fig. 5C). In striking contrast, Rab8 immunoreactivity is absent from the IS adjacent to the CC in the tulp1−/− retina, but is present adjacent to the ONL, in the myoid region of the IS (Fig. 5D; arrowheads indicate mislocalized Rab8 molecules to the ONL). In conjunction with Rab6, Rab11 functions in the sorting and budding of vesicles from the TGN (Deretic et al., 1996; Deretic, 2006). In the wt retina, Rab11 is primarily located in the ellipsoid region of the IS in close proximity to the CC (Fig. 5E). In the tulp1−/− retina, however, it is mislocalized and clustered near the myoid region and extends into the ONL (Fig. 5F; arrowheads indicate mislocalized Rab11 molecules to the ONL).
Figure 5.
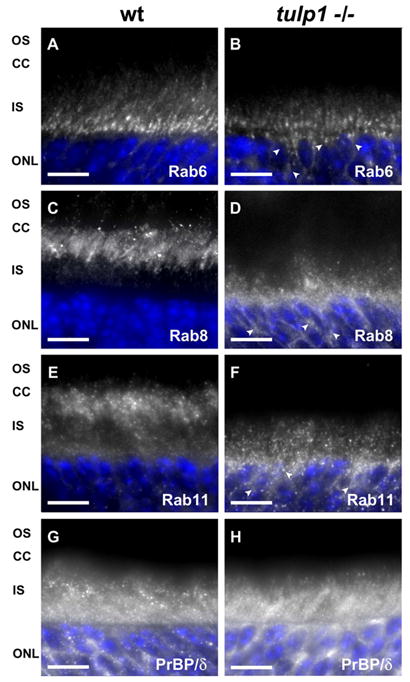
OS protein transport machinery is mislocalized in tulp1−/− retinas. A–F: Immunofluorescent images of three Rab GTPases involved in the transport of rhodopsin. G–H: Immunofluorescent images of PrBP/δ, a protein implicated in the transport of several peripheral membrane-associated proteins. (A) In the wt retina, Rab6 is localized to the IS. (B) In the tulp1−/− retina, although it remains in the IS, arrowheads highlight areas where Rab6 is mislocalized to the ONL. (C) Rab8 is localized immediately adjacent to the CC in the wt retina; while in the tulp1−/− retina, it is positioned far from the CC and extends into the ONL (D). (E) Rab11 follows a similar pattern to that of Rab8. In the wt retina, Rab11 is primarily located in close proximity to the CC; however, in the tulp1−/− retina, it is clustered near and throughout the ONL (F). In contrast to the Rab proteins, PrBP/δ staining appears normal in the tulp1−/− retina. (G) PrBP/δ is localized to the IS and ONL in the wt retina as well as in the tulp1−/− retina (H). Scale bars = 20μm.
We also investigated the localization of a transport protein of peripheral membrane-associated proteins, PrBP/δ. Unlike integral membrane proteins such as rhodopsin, the peripheral membrane-associated proteins are posttranslationally processed solely in the ER and include PDE6, GRK1, transducin and GCAP1 (Karan et al., 2008b). PrBP/δ is thought to participate in the removal of prenylated proteins from the ER, a necessary step prior to transportation (Norton et al., 2005). In wt and tulp1−/− retinas, PrBP/δ has a similar distribution, with the most robust staining in the IS and ONL (Figs. 5G and H).
3.6. Inner segment organelles are distributed normally in tulp1−/− retinas
To examine whether there are abnormalities in the distribution and morphology of the IS organelles responsible for post-translational modification of proteins prior to OS transport, we evaluated the ER, Golgi and mitochondria by immunofluorescence (Fig. 6). The ER is the initial site of protein modification, and with certain OS-resident proteins, is the sole organelle through which they are modified before being transported. This group of OS proteins includes the peripheral membrane-associated proteins such as PDE6, GRK1, transducin and GCAP1 (Karan et al., 2008b). To label the ER, we stained retinal sections with antibodies against calreticulin, an ER chaperone that ensures proper folding of newly synthesized proteins (Smith and Koch, 1989). The ER is normally located in the IS and cell bodies of the ONL. In both the wt (Fig. 6A) and tulp1−/− retinas (Fig. 6B), calreticulin localization was strongest in the IS but extended throughout the ONL. To visualize the Golgi apparatus, we utilized an antibody against golgi matrix protein 130 kd (GM130), a member of the Golgi protein family that is localized to the cis-Golgi complex of the IS. All of the integral membrane proteins (GC1, ROM1, cone opsins, rhodopsin) are processed through the Golgi and transported in vesicles derived from the TGN (Karan et al., 2008b). In the wt retina (Fig. 6C), GM130 labeling was strongly observed in the myoid region of the IS, in close proximity to the ER. The tulp1−/− retina exhibits a similar appearance, indicating that the Golgi network is not disrupted in Tulp1’s absence (Fig. 6D). Finally, we investigated the mitochondria located in the IS. These organelles are critical in meeting the heavy energy demands of the active transport of vesicles in the IS. In the wt retina, immunoreactivity of an antibody against the cytochrome c oxidase subunit IV (COXIV), an enzyme localized to the inner mitochondrial membrane, is observed in the apical IS (Fig. 6E). In the tulp1−/− retina, the staining pattern is similar to wt (Fig. 6F).
Figure 6.
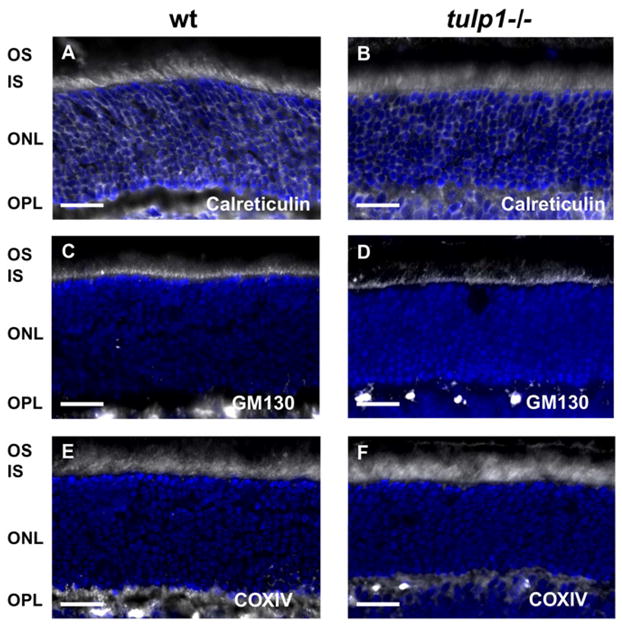
IS organelles are normally distributed in tulp1−/− retinas. A,C,E: P16 wt retinas. B,D,F: P16 tulp1−/− retinas. (A) In the wt retina, calreticulin immunofluorescence reveals that the localization of the ER is confined to the IS and ONL. (B) This pattern is not altered in the tulp1−/− retina. (C) In the wt retina, as well as in the tulp1−/− retina (D), GM130 staining shows that the Golgi apparatus is confined to a narrow band in the IS. (E) COXIV staining indicates IS localization of mitochondria in wt and tulp1−/− retinas (F). Scale bars = 20μm.
4. Discussion
Mouse models in which OSs form, but OS-resident proteins fail to reach their destination have provided critical information regarding the existence of distinct OS trafficking pathways in the photoreceptor cell (Marszalek et al., 2000; Lee et al., 2006; Baehr et al., 2007; Baehr and Palczewski, 2007; Zhang et al., 2007; Avasthi et al., 2009). While recent work has provided information on the trafficking of rhodopsin, little is known about the targeting and transport of other OS-resident proteins. Questions remain regarding how these proteins are sorted into carrier vesicles, which proteins aid in vesicular docking and fusing with the IS plasma membrane and which motor proteins transport the proteins through the CC. Further obscuring the understanding of OS protein transport are conflicting data regarding which OS proteins co-transport in vesicles (Marszalek et al., 2000; Karan et al., 2008a; Karan et al., 2008b; Avasthi et al., 2009). Thus, the analysis of additional mouse models that exhibit differential OS protein targeting is critical to help resolve questions and shed light upon these pathways.
Our previous analysis of tulp1−/− mice has shown that rhodopsin but not peripherin/rds is severely mislocalized (Hagstrom et al., 1999; Hagstrom et al., 2001; Grossman et al., 2009). This phenotypic feature makes the tulp1−/− mouse a model in which to study the processes that underlie the differential trafficking of OS proteins, as compared to other mouse models in which OSs do not form and thus trafficking is completely disrupted (Sanyal et al., 1980; Sanyal and Jansen, 1981; Lem et al., 1999; Westfall et al., 2010). In the present study, we surveyed the localization of several OS-specific proteins in the tulp1−/− retina as compared to the wt retina to define Tulp1-dependent OS transport pathways. Representative OS proteins of different functional classes (phototransduction and structural) and different membrane affiliations (integral membrane, peripheral membrane-associated and cytosolic) were analyzed. In addition, we also evaluated several known IS proteins that function as part of the machinery for OS protein transport in an attempt to pinpoint the defective step.
4.1. Peripheral membrane-associated proteins in tulp1−/− retinas
Our results indicate that the trafficking of several peripheral membrane-associated proteins, including PDE, GRK1 and transducin, are not affected in tulp1−/− retinas (Figs. 2 and 3). These proteins are posttranslationally processed solely in the ER and involve protein transfer to vesicular carriers (Inglese et al., 1992; Qin et al., 1992; Karan et al., 2008b). The transfer of these proteins between membranes is mediated by cytosolic prenyl binding proteins. In the retina, PrBP/δ is a prenyl binding protein known to interact in vitro with the prenyl chains of GRK1 and PDE (Li and Baehr, 1998; Zhang et al., 2004). In a mouse model lacking PrBP/δ, GRK1 as well as cone PDE fail to be transported to the OS (Zhang et al., 2007). Given our results that several peripheral membrane-associated proteins are not mislocalized, it was not surprising that PrBP/δ was correctly distributed to the IS and ONL in tulp1−/− retinas (Fig. 5H). These results indicate that Tulp1 does not have a direct role in peripheral membrane-associated protein transport. However, GCAP1 is a peripheral membrane-associated protein that is mislocalized in the tulp1−/− retina (Fig. 2J), but this mislocalization may be due to a secondary effect. It has been postulated that non-transmembrane proteins are transported in vesicles with their integral membrane protein partners (Karan et al., 2008b). Thus, the mislocalization of GCAP1 may be the result of traveling in vesicles with its integral membrane binding partner, GC1. This hypothesis is supported by our current results showing that GC1 is mislocalized to the IS in Tulp1’s absence (Fig. 2N). In addition, the cytosolic protein GCAP2 was also mistrafficked in Tulp1’s absence (Fig. 2L). Based on the analysis of several mouse mutant phenotypes, it has been proposed that GCAP1, GCAP2, GC1 and GC2 are co-transported, and that their transport is independent of the rhodopsin trafficking system (Baehr et al., 2007; Karan et al., 2008b). Unfortunately, we were not able to obtain a reliable GC2 antibody for localization in the tulp1−/− retina.
4.2. Integral membrane proteins in tulp1−/− retinas
Integral membrane proteins are posttranslationally processed in the Golgi and bud from vesicles derived from the TGN surface. Although two OS-specific integral membrane proteins, rhodopsin and GC1, are mistargeted in Tulp1’s absence (Figs. 1F and 2N), there are other OS integral membrane proteins that are able to localize properly without Tulp1, specifically peripherin/rds, ROM1 and CNGB1 (Figs. 1G, 2B and 2H). Therefore, Tulp1 appears to be involved in the transport of integral membrane proteins, and the proteins that co-transport with them, specifically, the rhodopsin and GC transport pathways (Fig. 7). Curiously, the localization of GC1 in the tulp1−/− retina did not follow the pattern of the other mistargeted proteins which appeared to be uniformly mislocalized throughout the photoreceptor cell. Instead, the mislocalization of GC1 was confined to the IS (Fig. 2N). It is possible that in the tulp1−/− retina, GC1 remains associated with transport vesicles that become bottle-necked in the IS.
Figure 7.

A graphical model of vesicular-mediated OS protein transport pathways which are affected in the tulp1−/− retina. This simplified schematic focuses on the normal flow of the pathways, shown in green dashed lines, and the segment in the pathways that are affected in the tulp1−/− retina, shown with red Xs. The pathway on the right shows a simplified depiction of the guanylate cyclase transport system. Peripheral membrane-associated proteins are posttranslationally processed solely in the ER and extracted for transport with the help of proteins such as PrBP/δ. In the tulp1−/− photoreceptor, PrBP/δ is correctly localized along with several peripheral membrane-associated proteins, indicating that Tulp1 is not directly involved in the transport of peripheral membrane-associated proteins. Although GCAP1 is a mislocalized peripheral membrane-associated protein, its disruption in the tulp1−/− photoreceptor is proposed to be caused by its co-transport in guanylate cyclase carriers. The left pathway shows a simplified depiction of rhodopsin transport through the IS. In the wt IS, rhodopsin molecules are posttranslationally modified in the Golgi apparatus, and are inserted into carrier vesicles generated from the TGN surface. Rab6 and Rab11 are involved in the budding of these rhodopsin-laden vesicles. In the tulp1−/− photoreceptor, Rab11 is mislocalized. The cytosolic protein arrestin may be co-transported in the rhodopsin transport pathway in the wt photoreceptor; however, it translocates to the OS in response to light. In the tulp1−/− photoreceptor, arrestin is not localized to the OS after light exposure. Rab8 is responsible for the docking and fusion of vesicles with the IS plasma membrane, which is necessary for the subsequent transport of rhodopsin through the CC to the OS discs. It is mislocalized in the tulp1−/− photoreceptor. Therefore, these data point to a role for Tulp1 at a step between vesicular budding at the TGN and transport of specific proteins through the CC.
4.3. Signaling proteins in tulp1−/− retinas
The lack of arrestin localization in the tulp1−/− photoreceptor OS in response to light may be attributed to the disruption of the transport carrier vesicles in which rhodopsin and arrestin co-transport. This idea was proposed in a conditional KO of the Kif3a subunit of the motor protein Kinesin-II, in which both rhodopsin and arrestin were mislocalized (Marszalek et al., 2000; Mendez et al., 2003). Underlying this, there is evidence that the translocation of arrestin to the OS is energy-dependent and requires an intact cytoskeleton (McGinnis et al., 2002; Reidel et al., 2008; Orisme et al., 2010). In light of this, it is not surprising that arrestin’s translocation is disrupted, given that rhodopsin is no longer confined to the OS in tulp1−/− mice. However, there is also evidence to suggest that arrestin does not co-transport with rhodopsin in post-Golgi vesicles but instead translocates by diffusion (Nair et al., 2005; Slepak and Hurley, 2008). In this scenario, the light-driven compartmentalization of arrestin is attributed to the temporary trapping of molecules by binding “sinks” in the IS and OS, and the affinity of either sink is modulated by the presence or absence of light (Slepak and Hurley, 2008). The OS sink would be activated rhodopsin, while the IS sink would likely be microtubules (Slepak and Hurley, 2008). In support of this, it has been shown that arrestin and microtubules interact more substantially in the dark. In addition, mutations in arrestin that enhance binding to microtubules decrease its ability to translocate to the OS (Nair et al., 2005). Interestingly, we have identified two proteins involved in the polymerization and stabilization of microtubules, microtubule associated protein 1B and tubulin, as potential Tulp1 binding partners (Xi et al., 2003). Without Tulp1, arrestin may bind more efficiently with the microtubules thereby inhibiting arrestin’s ability to diffuse and bind with rhodopsin in the OS. A separate possibility for the lack of arrestin in the OS is that the diminished amount of rhodopsin present in the tulp1−/− OS is not in the active form, which has been suggested to be necessary for the translocation of arrestin to the OS (Mendez et al., 2003).
4.4. Rhodopsin transport machinery in tulp1−/− retinas
In tulp1−/− photoreceptors, the small GTPase Rab8 staining is no longer present in the ellipsoid region of the IS in close proximity to the CC. Instead, it is clustered in the myoid region of the IS close to the ONL (Fig. 5D). This shift in sub-compartmental localization may indicate that Rab8 is unable to interact with cargo thereby preventing the vesicles from docking and fusing with the plasma membrane. Moreover, mutations in Rab8 induce an accumulation of rhodopsin-laden vesicles by inhibiting vesicular fusion, resulting in retinal degeneration (Moritz et al., 2001; Deretic, 2006). Interestingly, we have identified rab geranyl geranyl transferase as a potential Tulp1 interacting protein (Xi et al., 2003). Rab geranyl geranyl transferase is a prenyl transferase that transfers two geranylgeranyl groups to the C-terminal of Rab proteins, reducing their solubility and allowing them to become anchored to the plasma membrane near the CC. It is possible that if rab geranyl geranyl transferase is negatively affected in Tulp1’s absence, Rab8 is unable to associate with the plasma membrane. Furthermore, phosphatidylinositol-4,5-bisphosphate, actin and moesin, in concert with Rab8, appear to be required for actin cytoskeleton remodeling associated with vesicle fusion (Deretic et al., 1995; Deretic et al., 2004; Deretic, 2006). The association between Tulp1, phosphatidylinositols, actin and microtubules (Xi et al., 2003; Xi et al., 2005), together with the displacement of Rab8 in Tulp1’s absence, makes it tempting to assign a role for Tulp1 in vesicle fusion with the plasma membrane at the base of the CC.
The other two Rab proteins investigated in this study, Rab6 and Rab11, have been implicated in the generation and budding of vesicles from the TGN (Deretic and Papermaster, 1993; Deretic et al., 1996; Deretic, 1997; Deretic, 2006). Although there is some displacement of Rab6 in the tulp1−/− retina (Fig. 5B), Rab11 staining is dramatically affected (Fig. 5F). In the wt photoreceptor, Rab11 staining extends throughout the IS but appears most concentrated adjacent to the CC (Fig. 5E). In the tulp1−/− photoreceptor, Rab11 is severely mislocalized, similar to Rab8. This pattern makes sense given that Rab11 remains attached to the vesicles after budding from the TGN (Deretic, 2006). Therefore, the alteration in Rab11 staining may be indicative of a transport deficiency, rather than a defect in vesicle generation at the TGN.
4.5. The Tulp1 mouse model adds to our understanding of photoreceptor OS transport pathways
A transport model of membrane associated phototransduction proteins was put forth based on the analysis of data generated largely in PrBP/δ knockout (KO) mice (Zhang et al., 2007), GC1 KO mice (Coleman et al., 2004) and GC1/GC2 double KO mice (Baehr et al., 2007; Karan et al., 2008a). While it was proposed that the subunits of PDE co-transport in carriers with GC1 and GC2, our results indicate that PDE6-β is able to be targeted exclusively to the OS while GC1 is not. Additionally, it was proposed that GRK1 and transducin are co-transported in rhodopsin transport carriers. In the tulp1−/− retina, although rhodopsin carriers are severely mistrafficked, GRK1 and transducin localize normally. These differences may indicate that PDE, GRK1, and transducin, are endogenous cargo of another yet-unidentified carrier type, or in Tulp1’s absence, they are sorted into alternative carrier vesicles. These discrepancies may also arise from the comparison between mutant mouse lines in which OS proteins are affected differently, i.e., mislocalized vs. downregulated. In GC1 KO mice, GCAP1 protein levels are reduced (Coleman et al., 2004). In GC1/GC2 double KO mice, GCAP1, GCAP2 and rod PDE6 subunits are also posttranslationally downregulated (Baehr et al., 2007). It was proposed that in the absence of GC-bearing vesicles, PDE6 and GCAPs are not transported but degraded, suggesting a role for the GCs in protein trafficking (Baehr et al., 2007; Karan et al., 2008b; Karan et al., 2010). In tulp1−/− mice, GCAP1 and GCAP2 do not appear to be downregulated but are mislocalized. In addition, PDE6-β is unaffected. It is possible that in the absence of an integral membrane protein, its binding partners are not extracted from the membranes of the synthetic machinery and are eventually degraded. This is the case in the GC1 KO retina. In contrast, the misrouting of an integral membrane protein may result in the mislocalization of its binding partners in the tulp1−/− retina.
5. Conclusion
These new findings indicate that Tulp1 is essential for normal photoreceptor function by participating in the polarized transport of a subset of OS phototransduction proteins in post-Golgi vesicles. Our data suggest that both the rhodopsin and GC carrier pathways are Tulp1-dependent, while alternative pathways are not. A graphical model of disrupted protein transport pathways are shown in Figure 7. Our present work adds to the limited understanding regarding the transport of peripheral membrane proteins, suggesting that GRK1 and transducin are not endogenous cargo of the rhodopsin carrier pathway and that PDE is not co-transported in the GC carrier vesicles. A phenotypic analysis of individual carrier types, where the cargo constituents can be directly identified, will be critical in defining these OS transport pathways. Finally, the displacement of Rab8 and Rab11 may denote a role for Tulp1 in vesicular docking and fusion at the plasma membrane of the rhodopsin transport pathway, although future studies are required to investigate this relationship.
We surveyed localization of outer segment proteins in tulp1−/− mice.
Tulp1 is required in the rhodopsin and guanylate cyclase transport pathways.
Rhodopsin kinase and transducin are not cargo of the rhodopsin carrier pathway.
Phosphodiesterase is not co-transported in the guanylate cyclase carrier pathway.
Disruption of Rab8 and 11 may denote a vesicle docking and fusion role for Tulp1.
Acknowledgments
We thank Drs. W. Clay Smith, Andrew F.X. Goldberg, Roderick R. McInnes, Krzysztof Palczewski, Martin Biel and Rick H. Cote for their generous donations of antibodies and Dr. Neal S. Peachey for his insightful comments on this manuscript. Supported by NIH EY15638 (SAH) and EY16072, Foundation Fighting Blindness (SAH), Hope for Vision (SAH), Prevent Blindness Ohio (RFW), Fight For Sight (GHG), Research to Prevent Blindness Center Grant, and a Research to Prevent Blindness Sybil B. Harrington Special Scholar Award (SAH).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aroeti B, Okhrimenko H, Reich V, Orzech E. Polarized trafficking of plasma membrane proteins: emerging roles for coats, SNAREs, GTPases and their link to the cytoskeleton. Biochim Biophys Acta. 1998;1376(1):57–90. doi: 10.1016/s0304-4157(98)00005-7. [DOI] [PubMed] [Google Scholar]
- Avasthi P, Watt CB, Williams DS, Le YZ, Li S, Chen CK, Marc RE, Frederick JM, Baehr W. Trafficking of membrane proteins to cone but not rod outer segments is dependent on heterotrimeric kinesin-II. J Neurosci. 2009;29(45):14287–14298. doi: 10.1523/JNEUROSCI.3976-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baehr W, Karan S, Maeda T, Luo DG, Li S, Bronson JD, Watt CB, Yau KW, Frederick JM, Palczewski K. The function of guanylate cyclase 1 and guanylate cyclase 2 in rod and cone photoreceptors. J Biol Chem. 2007;282(12):8837–8847. doi: 10.1074/jbc.M610369200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baehr W, Palczewski K. Guanylate cyclase-activating proteins and retina disease. Subcell Biochem. 2007;45:71–91. doi: 10.1007/978-1-4020-6191-2_4. [DOI] [PubMed] [Google Scholar]
- Banerjee P, Xu XJ, Poulter LW, Rustin MH. Changes in CD23 expression of blood and skin in atopic eczema after Chinese herbal therapy. Clin Exp Allergy. 1998;28(3):306–314. doi: 10.1046/j.1365-2222.1998.00233.x. [DOI] [PubMed] [Google Scholar]
- Berson EL. Retinitis pigmentosa. The Friedenwald Lecture. Invest Ophthalmol Vis Sci. 1993;34(5):1659–1676. [PubMed] [Google Scholar]
- Besharse JC, Wetzel MG. Immunocytochemical localization of opsin in rod photoreceptors during periods of rapid disc assembly. J Neurocytol. 1995;24(5):371–388. doi: 10.1007/BF01189064. [DOI] [PubMed] [Google Scholar]
- Calvert PD, Strissel KJ, Schiesser WE, Pugh EN, Jr, Arshavsky VY. Light-driven translocation of signaling proteins in vertebrate photoreceptors. Trends Cell Biol. 2006;16(11):560–568. doi: 10.1016/j.tcb.2006.09.001. [DOI] [PubMed] [Google Scholar]
- Chabre M, Antonny B, Bruckert F, Vuong TM. The G protein cascade of visual transduction: kinetics and regulation. Ciba Found Symp. 1993;176:112–120. doi: 10.1002/9780470514450.ch8. discussion 121–114. [DOI] [PubMed] [Google Scholar]
- Chabre M, Deterre P. Molecular mechanism of visual transduction. Eur J Biochem. 1989;179(2):255–266. doi: 10.1111/j.1432-1033.1989.tb14549.x. [DOI] [PubMed] [Google Scholar]
- Coleman JE, Zhang Y, Brown GA, Semple-Rowland SL. Cone cell survival and downregulation of GCAP1 protein in the retinas of GC1 knockout mice. Invest Ophthalmol Vis Sci. 2004;45(10):3397–3403. doi: 10.1167/iovs.04-0392. [DOI] [PubMed] [Google Scholar]
- Deretic D. Rab proteins and post-Golgi trafficking of rhodopsin in photoreceptor cells. Electrophoresis. 1997;18(14):2537–2541. doi: 10.1002/elps.1150181408. [DOI] [PubMed] [Google Scholar]
- Deretic D. A role for rhodopsin in a signal transduction cascade that regulates membrane trafficking and photoreceptor polarity. Vision Res. 2006;46(27):4427–4433. doi: 10.1016/j.visres.2006.07.028. [DOI] [PubMed] [Google Scholar]
- Deretic D, Huber LA, Ransom N, Mancini M, Simons K, Papermaster DS. rab8 in retinal photoreceptors may participate in rhodopsin transport and in rod outer segment disk morphogenesis. J Cell Sci. 1995;108 (Pt 1):215–224. doi: 10.1242/jcs.108.1.215. [DOI] [PubMed] [Google Scholar]
- Deretic D, Papermaster DS. Rab6 is associated with a compartment that transports rhodopsin from the trans-Golgi to the site of rod outer segment disk formation in frog retinal photoreceptors. J Cell Sci. 1993;106 (Pt 3):803–813. doi: 10.1242/jcs.106.3.803. [DOI] [PubMed] [Google Scholar]
- Deretic D, Puleo-Scheppke B, Trippe C. Cytoplasmic domain of rhodopsin is essential for post-Golgi vesicle formation in a retinal cell-free system. J Biol Chem. 1996;271(4):2279–2286. doi: 10.1074/jbc.271.4.2279. [DOI] [PubMed] [Google Scholar]
- Deretic D, Traverso V, Parkins N, Jackson F, Rodriguez de Turco EB, Ransom N. Phosphoinositides, ezrin/moesin, and rac1 regulate fusion of rhodopsin transport carriers in retinal photoreceptors. Mol Biol Cell. 2004;15(1):359–370. doi: 10.1091/mbc.E03-04-0203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorczyca WA, Polans AS, Surgucheva IG, Subbaraya I, Baehr W, Palczewski K. Guanylyl cyclase activating protein. A calcium-sensitive regulator of phototransduction. J Biol Chem. 1995;270(37):22029–22036. doi: 10.1074/jbc.270.37.22029. [DOI] [PubMed] [Google Scholar]
- Grossman GH, Pauer GJ, Narendra U, Peachey NS, Hagstrom SA. Early synaptic defects in tulp1−/− mice. Invest Ophthalmol Vis Sci. 2009;50(7):3074–3083. doi: 10.1167/iovs.08-3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu S, Lennon A, Li Y, Lorenz B, Fossarello M, North M, Gal A, Wright A. Tubby-like protein-1 mutations in autosomal recessive retinitis pigmentosa. Lancet. 1998;351(9109):1103–1104. doi: 10.1016/S0140-6736(05)79384-3. [DOI] [PubMed] [Google Scholar]
- Hagstrom SA, Adamian M, Scimeca M, Pawlyk BS, Yue G, Li T. A role for the Tubby-like protein 1 in rhodopsin transport. Invest Ophthalmol Vis Sci. 2001;42(9):1955–1962. [PubMed] [Google Scholar]
- Hagstrom SA, Duyao M, North MA, Li T. Retinal degeneration in tulp1−/− mice: vesicular accumulation in the interphotoreceptor matrix. Invest Ophthalmol Vis Sci. 1999;40(12):2795–2802. [PubMed] [Google Scholar]
- Hagstrom SA, North MA, Nishina PL, Berson EL, Dryja TP. Recessive mutations in the gene encoding the tubby-like protein TULP1 in patients with retinitis pigmentosa. Nat Genet. 1998;18(2):174–176. doi: 10.1038/ng0298-174. [DOI] [PubMed] [Google Scholar]
- Ikeda S, Shiva N, Ikeda A, Smith RS, Nusinowitz S, Yan G, Lin TR, Chu S, Heckenlively JR, North MA, Naggert JK, Nishina PM, Duyao MP. Retinal degeneration but not obesity is observed in null mutants of the tubby-like protein 1 gene. Hum Mol Genet. 2000;9(2):155–163. doi: 10.1093/hmg/9.2.155. [DOI] [PubMed] [Google Scholar]
- Inglese J, Koch WJ, Caron MG, Lefkowitz RJ. Isoprenylation in regulation of signal transduction by G-protein-coupled receptor kinases. Nature. 1992;359(6391):147–150. doi: 10.1038/359147a0. [DOI] [PubMed] [Google Scholar]
- Karan S, Frederick JM, Baehr W. Involvement of guanylate cyclases in transport of photoreceptor peripheral membrane proteins. Adv Exp Med Biol. 2008a;613:351–359. doi: 10.1007/978-0-387-74904-4_41. [DOI] [PubMed] [Google Scholar]
- Karan S, Frederick JM, Baehr W. Novel functions of photoreceptor guanylate cyclases revealed by targeted deletion. Mol Cell Biochem. 2010;334(1–2):141–155. doi: 10.1007/s11010-009-0322-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karan S, Zhang H, Li S, Frederick JM, Baehr W. A model for transport of membrane-associated phototransduction polypeptides in rod and cone photoreceptor inner segments. Vision Res. 2008b;48(3):442–452. doi: 10.1016/j.visres.2007.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaupp UB, Seifert R. Cyclic nucleotide-gated ion channels. Physiol Rev. 2002;82(3):769–824. doi: 10.1152/physrev.00008.2002. [DOI] [PubMed] [Google Scholar]
- Lee ES, Burnside B, Flannery JG. Characterization of peripherin/rds and rom-1 transport in rod photoreceptors of transgenic and knockout animals. Invest Ophthalmol Vis Sci. 2006;47(5):2150–2160. doi: 10.1167/iovs.05-0919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lem J, Krasnoperova NV, Calvert PD, Kosaras B, Cameron DA, Nicolo M, Makino CL, Sidman RL. Morphological, physiological, and biochemical changes in rhodopsin knockout mice. Proc Natl Acad Sci U S A. 1999;96(2):736–741. doi: 10.1073/pnas.96.2.736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Baehr W. Expression and characterization of human PDEdelta and its Caenorhabditis elegans ortholog CEdelta. FEBS Lett. 1998;440(3):454–457. doi: 10.1016/s0014-5793(98)01501-4. [DOI] [PubMed] [Google Scholar]
- Lippincott-Schwartz J, Roberts TH, Hirschberg K. Secretory protein trafficking and organelle dynamics in living cells. Annu Rev Cell Dev Biol. 2000;16:557–589. doi: 10.1146/annurev.cellbio.16.1.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marszalek JR, Liu X, Roberts EA, Chui D, Marth JD, Williams DS, Goldstein LS. Genetic evidence for selective transport of opsin and arrestin by kinesin-II in mammalian photoreceptors. Cell. 2000;102(2):175–187. doi: 10.1016/s0092-8674(00)00023-4. [DOI] [PubMed] [Google Scholar]
- Mazelova J, Astuto-Gribble L, Inoue H, Tam BM, Schonteich E, Prekeris R, Moritz OL, Randazzo PA, Deretic D. Ciliary targeting motif VxPx directs assembly of a trafficking module through Arf4. EMBO J. 2009;28(3):183–192. doi: 10.1038/emboj.2008.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinnis JF, Matsumoto B, Whelan JP, Cao W. Cytoskeleton participation in subcellular trafficking of signal transduction proteins in rod photoreceptor cells. J Neurosci Res. 2002;67(3):290–297. doi: 10.1002/jnr.10120. [DOI] [PubMed] [Google Scholar]
- McGinnis JF, Whelan JP, Donoso LA. Transient, cyclic changes in mouse visual cell gene products during the light-dark cycle. J Neurosci Res. 1992;31(3):584–590. doi: 10.1002/jnr.490310325. [DOI] [PubMed] [Google Scholar]
- Mendez A, Lem J, Simon M, Chen J. Light-dependent translocation of arrestin in the absence of rhodopsin phosphorylation and transducin signaling. J Neurosci. 2003;23(8):3124–3129. doi: 10.1523/JNEUROSCI.23-08-03124.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moritz OL, Tam BM, Hurd LL, Peranen J, Deretic D, Papermaster DS. Mutant rab8 Impairs docking and fusion of rhodopsin-bearing post-Golgi membranes and causes cell death of transgenic Xenopus rods. Mol Biol Cell. 2001;12(8):2341–2351. doi: 10.1091/mbc.12.8.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair KS, Hanson SM, Mendez A, Gurevich EV, Kennedy MJ, Shestopalov VI, Vishnivetskiy SA, Chen J, Hurley JB, Gurevich VV, Slepak VZ. Light-dependent redistribution of arrestin in vertebrate rods is an energy-independent process governed by protein-protein interactions. Neuron. 2005;46(4):555–567. doi: 10.1016/j.neuron.2005.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norton AW, Hosier S, Terew JM, Li N, Dhingra A, Vardi N, Baehr W, Cote RH. Evaluation of the 17-kDa prenyl-binding protein as a regulatory protein for phototransduction in retinal photoreceptors. J Biol Chem. 2005;280(2):1248–1256. doi: 10.1074/jbc.M410475200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orisme W, Li J, Goldmann T, Bolch S, Wolfrum U, Smith WC. Light-dependent translocation of arrestin in rod photoreceptors is signaled through a phospholipase C cascade and requires ATP. Cell Signal. 2010;22(3):447–456. doi: 10.1016/j.cellsig.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paloma E, Hjelmqvist L, Bayes M, Garcia-Sandoval B, Ayuso C, Balcells S, Gonzalez-Duarte R. Novel mutations in the TULP1 gene causing autosomal recessive retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2000;41(3):656–659. [PubMed] [Google Scholar]
- Papermaster DS, Schneider BG, Besharse JC. Vesicular transport of newly synthesized opsin from the Golgi apparatus toward the rod outer segment. Ultrastructural immunocytochemical and autoradiographic evidence in Xenopus retinas. Invest Ophthalmol Vis Sci. 1985;26(10):1386–1404. [PubMed] [Google Scholar]
- Qin N, Pittler SJ, Baehr W. In vitro isoprenylation and membrane association of mouse rod photoreceptor cGMP phosphodiesterase alpha and beta subunits expressed in bacteria. J Biol Chem. 1992;267(12):8458–8463. [PubMed] [Google Scholar]
- Reidel B, Goldmann T, Giessl A, Wolfrum U. The translocation of signaling molecules in dark adapting mammalian rod photoreceptor cells is dependent on the cytoskeleton. Cell Motil Cytoskeleton. 2008;65(10):785–800. doi: 10.1002/cm.20300. [DOI] [PubMed] [Google Scholar]
- Sanyal S, De Ruiter A, Hawkins RK. Development and degeneration of retina in rds mutant mice: light microscopy. J Comp Neurol. 1980;194(1):193–207. doi: 10.1002/cne.901940110. [DOI] [PubMed] [Google Scholar]
- Sanyal S, Jansen HG. Absence of receptor outer segments in the retina of rds mutant mice. Neurosci Lett. 1981;21(1):23–26. doi: 10.1016/0304-3940(81)90051-3. [DOI] [PubMed] [Google Scholar]
- Slepak VZ, Hurley JB. Mechanism of light-induced translocation of arrestin and transducin in photoreceptors: interaction-restricted diffusion. IUBMB Life. 2008;60(1):2–9. doi: 10.1002/iub.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MJ, Koch GL. Multiple zones in the sequence of calreticulin (CRP55, calregulin, HACBP), a major calcium binding ER/SR protein. EMBO J. 1989;8(12):3581–3586. doi: 10.1002/j.1460-2075.1989.tb08530.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith VC, Pokorny J, van Norren D. Densitometric measurement of human cone photopigment kinetics. Vision Res. 1983;23(5):517–524. doi: 10.1016/0042-6989(83)90126-8. [DOI] [PubMed] [Google Scholar]
- Westfall JE, Hoyt C, Liu Q, Hsiao YC, Pierce EA, Page-McCaw PS, Ferland RJ. Retinal degeneration and failure of photoreceptor outer segment formation in mice with targeted deletion of the Joubert syndrome gene, Ahi1. J Neurosci. 2010;30(26):8759–8768. doi: 10.1523/JNEUROSCI.5229-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi Q, Pauer GJ, Ball SL, Rayborn M, Hollyfield JG, Peachey NS, Crabb JW, Hagstrom SA. Interaction between the photoreceptor-specific tubby-like protein 1 and the neuronal-specific GTPase dynamin-1. Invest Ophthalmol Vis Sci. 2007;48(6):2837–2844. doi: 10.1167/iovs.06-0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi Q, Pauer GJ, Marmorstein AD, Crabb JW, Hagstrom SA. Tubby-like protein 1 (TULP1) interacts with F-actin in photoreceptor cells. Invest Ophthalmol Vis Sci. 2005;46(12):4754–4761. doi: 10.1167/iovs.05-0693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi Q, Pauer GJ, West KA, Crabb JW, Hagstrom SA. Retinal degeneration caused by mutations in TULP1. Adv Exp Med Biol. 2003;533:303–308. doi: 10.1007/978-1-4615-0067-4_37. [DOI] [PubMed] [Google Scholar]
- Yang RB, Robinson SW, Xiong WH, Yau KW, Birch DG, Garbers DL. Disruption of a retinal guanylyl cyclase gene leads to cone-specific dystrophy and paradoxical rod behavior. J Neurosci. 1999;19(14):5889–5897. doi: 10.1523/JNEUROSCI.19-14-05889.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Li S, Doan T, Rieke F, Detwiler PB, Frederick JM, Baehr W. Deletion of PrBP/delta impedes transport of GRK1 and PDE6 catalytic subunits to photoreceptor outer segments. Proc Natl Acad Sci U S A. 2007;104(21):8857–8862. doi: 10.1073/pnas.0701681104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Liu XH, Zhang K, Chen CK, Frederick JM, Prestwich GD, Baehr W. Photoreceptor cGMP phosphodiesterase delta subunit (PDEdelta) functions as a prenyl-binding protein. J Biol Chem. 2004;279(1):407–413. doi: 10.1074/jbc.M306559200. [DOI] [PubMed] [Google Scholar]