

Abstract
Molecular analysis of brain tissue is greatly complicated by having many different classes of neurons and glia interspersed throughout the brain. Fluorescence-activated cell sorting (FACS) has been used to purify selected cell types from brain tissue. However, its use has been limited to brain tissue from embryos or transgenic mice with promoter-driven reporter genes. To overcome these limitations, we developed a FACS procedure for dissociating intact cell bodies from adult wild-type rat brains and sorting them using commercially available antibodies against intracellular and extracellular proteins. As an example, we isolated neurons using a NeuN antibody and confirmed their identity using microarray and real time PCR of mRNA from the sorted cells. Our FACS procedure allows rapid, high-throughput, quantitative assays of molecular alterations in identified cell types with widespread applications in neuroscience.
Keywords: glia, genes, microarray, qPCR
1. INTRODUCTION
The brain is the most heterogeneous organ in the body, with many cell types that respond differently to drugs, disease states, and stimuli in behaving animals. These cell types are intermingled, making it difficult to assess molecular characteristics of individual cell types, particularly in homogenates of brain regions. Thus, a cell type-specific technique that is rapid, quantitative, and has high molecular throughput (easily assesses many different molecules from each sample), would be essential for identifying molecular changes in individual brain cell types.
Among current methods, immunohistochemistry is not quantitative and has relatively low throughput, while in situ hybridization is semi-quantitative with even lower throughput. Although laser capture microdissection can have high throughput once cells are selected, it is laborious and provides very low levels of molecular product for analysis. Recently developed ribosomal tagging techniques are useful for obtaining mRNA expressed in specific cell types; however they require the use of transgenic mice, permit collection of mRNA from only one cell population at a time, and do not provide the potential for protein analysis (Doyle et al., 2008; Heiman et al., 2008; Sanz et al., 2009).
Fluorescence-activated cell sorting (FACS) from brain can overcome many of these problems. FACS separates cells based on their size and molecular phenotype. While FACS is commonly used in the immunology and cancer fields, its use in neuroscience has largely been limited to embryonic brain tissue, cultured cells, stem cells, or synaptosomes (Arlotta et al., 2005; John et al., 1986; Maric and Barker, 2005; Wolf and Kapatos, 1989a, b, c) because these cells or organelles lack or have fewer processes and connections than adult neurons. Sorting of adult neurons has been done using transgenic mice expressing green fluorescent protein (GFP) controlled by cell-specific promoters (Lobo et al., 2006); however this procedure also requires production of transgenic mice, which prohibits use of many existing drug and disease models developed in rats.
We have developed a novel FACS procedure that overcomes these limitations. It uses commercially available antibodies that label intracellular and extracellular markers to identify and efficiently purify cell types in adult wild-type rat brains. Our protocol provides analyzable RNA from specific cell types, is rapid and quantitative, and has high molecular throughput. In the present study, we use our FACS procedure to purify neurons from adult rat striatum and analyze their transcriptome.
2. METHODS
2.1 Animals
Sprague-Dawley rats (Charles River, Raleigh, NC) weighing 300–850g were housed in plastic cages in a temperature and humidity controlled room maintained on a 12:12 hr reverse light/dark cycle (lights on at 8:00 PM) with free access to food and water. Experimental procedures were approved by the NIDA Animal Care and Use Committee.
2.2 Dissociation of striatal tissue to obtain single cell suspension
For each run where RNA was extracted for analysis, 4–6 rats were rapidly decapitated and striata extracted within two minutes. For runs where tissue was analyzed for protein expression only, 2 rats were rapidly decapitated and either striata or midbrains were similarly extracted. Each striatum or midbrain was minced with razor blades on an ice-cold glass plate and placed in a microfuge tube with 1 ml of Hibernate A (Brewer, 1997; Brewer et al., 1993) (HA-LF; Brain Bits, Springfield, IL) on ice. Hibernate A was replaced with 1 ml of Accutase (SCR005; Millipore, Temecula, CA), a mixture of proteolytic and collagenolytic enzymes, and tubes were rotated for 30 min at 4°C. Tubes were centrifuged at 425×g for two minutes and the pellet was resuspended in 250 μl of ice-cold Hibernate A. All centrifugation until FACS was at 4°C.
To dissociate cells, two to four striata or two midbrains were combined in 1 ml of Hibernate A and triturated 10 times with a large diameter fire-polished Pasteur pipet (~1.3 mm). Tubes were placed on ice, large tissue pieces were allowed to settle, and ~600 μl of cloudy supernatant containing dissociated cells were transferred to a 15 ml Falcon tube on ice. 600 μl of Hibernate A were added to the original tube, and the process was repeated with medium- and small-diameter pipets (~0.8 mm and 0.4 mm). Both supernatants were collected and pooled with the first supernatant in the 15 ml Falcon tube. The last cells were removed by adding 750 μl of Hibernate A to the original tube, inverting several times and settling on ice. Approximately 800 μl of cloudy supernatant was finally added to the 15 ml Falcon tube.
2.3 Removal of cell clusters and debris from the cell suspension
Large debris and cell clusters were removed from the cell suspension by serial filtration through pre-wetted 100 μm (Falcon 352360; BD Biosciences, San Jose, CA) and then 40 μm (Falcon 352340; BD Biosciences) cell strainers into 50 ml Falcon tubes on ice. Small cellular debris was reduced by density centrifugation through a three-density step gradient of Percoll (P1644; Sigma, St. Louis, MO). One milliliter of each solution (high density solution: 3.426 ml Hibernate A + 824.5 μl Percoll + 97.8 μl of 1M NaCl; medium density solution: 3.600 ml Hibernate A + 650.5 μl Percoll + 76.5 μl of 1M NaCl; low density solution: 3.770 ml Hibernate A + 480.3 μl Percoll + 59.5 μl of 1M NaCl) was carefully layered in a 15 ml Falcon tube, with the highest density solution on the bottom. The filtered cell suspension was applied to the top of this gradient and centrifuged at 430×g for 3 minutes; longer centrifugation times did not produce the desired separation. The cloudy top layer (~2 ml) containing debris was removed and discarded. Cells in the remaining layers were pelleted by centrifugation at 550×g for 5 min.
2.4 Immunolabeling of dissociated cells
Cell pellets were resuspended in 1 ml of Hibernate A and divided into microfuge tubes as required for labeling. An equal volume of cold absolute ethanol was added to each tube, gently vortexed, and kept on ice for 15 min with occasional inversion. Cells were pelleted by centrifugation at 425×g for 2 min and resuspended in phosphate-buffered saline (PBS).
Fixed cells were incubated with biotinylated primary antibody against NeuN (MAB377B; Millipore) diluted 1:1000 in PBS and rotated end-over-end for 30 min at 4°C. Cells studied for flow cytometry alone were similarly labeled for NeuN along with a second marker: midbrain cells were co-labeled for NeuN and tyrosine hydroxylase (TH; AB1542; Millipore) diluted 1:250, and striatal cells for NeuN and the D1 dopamine receptor (D1R; NB100–79930; Novus Biologicals, Littleton, CO) diluted 1:250 in PBS. After incubation with primary antibody, cells were washed once by adding 800 μl of PBS to each tube and shaking vigorously, then centrifuged at 950×g for 3 min before resuspending in PBS. Cells were then incubated with fluorescent labels and rotated end-over-end for 15 min at 4°C. NeuN-labeled cells were incubated with phycoerythrin (PE)-labeled streptavidin (Streptavidin, R-PE, SA1004-1; Invitrogen, Carlsbad, CA) diluted 1:1000 in PBS. TH- and D1R-labeled cells were incubated with Alexa Fluor 488-labeled anti-sheep (A11015; Invitrogen) and anti-rabbit (A21206, Invitrogen) secondary antibodies diluted 1:1000 in PBS. Cells co-labeled with NeuN and either TH or D1R were also labeled with DAPI (4′,6-diamidino-2-phenylindole, 1:4000 dilution; D3571, Invitrogen). After incubation with fluorescent labels, cells were washed again with 800 μl of PBS, centrifuged at 950×g for 3 min, and resuspended in 1 mL PBS on ice. For FACS purification of endothelial cells, mouse anti-CD71 antibody (CBL1518; Millipore) was diluted 1:1000 in PBS and the secondary antibody chicken anti-mouse Alexa Fluor 488-labeled secondary antibody (A21200; Invitrogen) was diluted 1:1000.
2.5 Flow cytometry
A FACS Aria (BD Biosciences) instrument was used for cell sorting, and both the FACS Aria and FACS Calibur (BD Biosciences) instruments were used for analysis without sorting (flow cytometry core facility of Dr. Mark Soloski, Johns Hopkins Bayview campus). A small portion of the fixed cells was incubated without antibodies to gate cells according to their light scattering characteristics (see Results section 3.4 for detailed description). Another small portion of the fixed cells was mixed with streptavidin-PE or Alexa Fluor 488 conjugates without primary antibody to set the maximum threshold for non-specific binding. Finally, test samples labeled with primary antibodies and fluorescent labels were scanned or sorted at 4°C. Sorted cells were always kept at 4°C and collected into low-binding microfuge tubes (022431081; Eppendorf, Westbury, NY) with 100 μl of PBS in each tube.
NeuN-positive sorted cells were centrifuged at 2650×g for 8 min at 18°C, and NeuN-negative sorted cells were centrifuged at 6000×g for 8 min at 18°C. Total RNA was extracted from pellets using the RNeasy Micro kit (74004; Qiagen, Valencia, CA) according to manufacturer’s instructions, including DNase treatment. RNA concentration and purity was determined using the Nanodrop spectrophotometer (Thermo Scientific, Wilmington, DE).
2.6 Microarray analysis of RNA from NeuN-sorted cells
RNA from three biological replicates of NeuN-positive and NeuN-negative sorted cells were hybridized to the RatRef-12 Expression BeadChip array (BD-27–303; Illumina Inc., San Diego, CA), as previously described (Jayanthi et al., 2009). In brief, a 500ng aliquot of total RNA from each striatal sample was amplified using Ambion’s Illumina RNA Amplification kit (IL1791; Ambion, Austin, TX). Single stranded RNA (cRNA) was generated and labeled by incorporating biotin-16-UTP (11388908910; Roche Diagnostics GmbH, Mannheim, Germany). 750ng of each cRNA sample were hybridized to the Illumina array at 55°C overnight according to the Illumina Whole-Genome Gene Expression Protocol for BeadStation (11201828; Illumina Inc.). Hybridized biotinylated cRNA was detected with Cyanine3-streptavidine (146065; Amersham Biosciences, Piscataway, NJ) and quantified using Illumina’s BeadStation 500GX Genetic Analysis Systems scanner. Microarray data were analyzed using DIANE 6.0, a spreadsheet-based microarray analysis program based on SAS JMP 7.0 system as previously described (Garg et al., 2009). Raw microarray data were subjected to filtering by the detection p-value and Z normalization. Sample quality was first analyzed using scatter plots, principal components analysis, and gene sample Z-scores based on hierarchy clustering to exclude possible outliers. ANOVA tests were then used to eliminate genes with larger variances within each comparing group. Genes were identified as differentially expressed after calculating the Z ratio, which indicates the fold-difference between experimental groups, as well as the false discovery rate (fdr), which controls for the expected proportion of false rejected hypotheses. Individual genes with p value ≤ 0.05, absolute value of Z ratio ≥ 1.5 and fdr ≤ 0.3 were considered significantly changed.
2.7 Real Time quantitative PCR
RNA was reverse transcribed into cDNA using RETROscript kit (AM1710; Ambion, Austin, TX) with an oligo(dT) primer, according to manufacturer instructions. cDNA was analyzed using real time qPCR, with each sample repeated in technical triplicates. Each 25 μl reaction included 12.5 μl of Taqman Gene Expression Master Mix (4369514; Applied Biosystems, Foster City, CA), 1.125 μl each of 20 μM forward and reverse primers, 0.25 μl of 100 μM FAM-labeled probe, 5 μl of water, and 5 μl of 1 ng/μl cDNA. Primer and probe combinations were designed using the Roche Universal Probe Library Assay Design Center to be intron-spanning (https://www.roche-applied-science.com/sis/rtpcr/upl/index.jsp?id=UP030000). DNA sequences are shown in Supplemental Table 1. PCR reactions were monitored using the Opticon Light Cycler (BioRad, Hercules, CA). The program began with 20 plate reads at 50°C for photobleaching, followed by 5 minutes at 95°C, and then 40 cycles of 20 seconds at 94°C, 1 minute at 60°C, and a plate read.
Standard curve reactions were first run for each gene to determine amplification efficiency (Supplemental Table 1). For evaluation of reference genes we calculated expression as efficiency^ΔCq, where ΔCq = Cq(NeuN−) − Cq(NeuN+) (Bustin et al., 2009; Pfaffl, 2001). Technical assay triplicate Cq values were averaged before calculating ΔCq for each gene in a sample. Relative expression levels for neural and glial genes were calculated using efficiency^ΔCq where ΔCq = Cq(experimental gene) − Cq(reference gene) (Pfaffl, 2001). For each mRNA measured in qPCR, gene expression values were averaged across 4–8 biological replicates. To reflect fold-change values in Figure 4, we normalized the neural marker genes to the NeuN-negative level of expression, and normalized the glial marker genes to the NeuN-positive level of expression. These values are indicated as means and standard errors in Figure 4.
Figure 4.
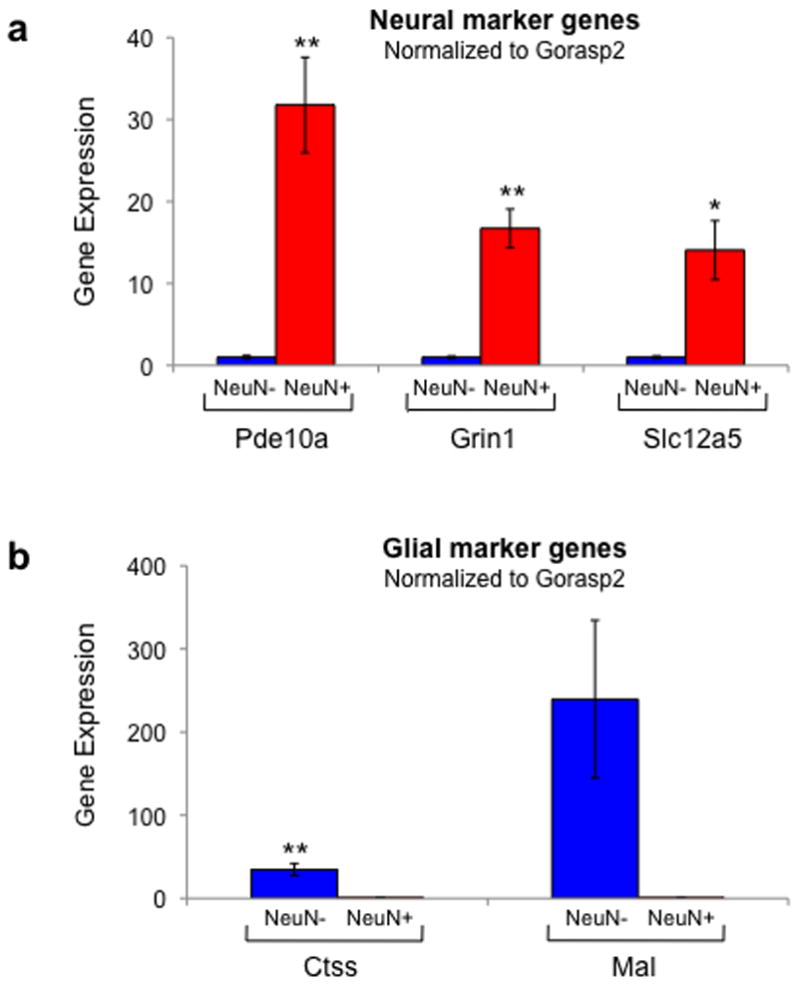
Real time PCR confirms the microarray results, showing that NeuN-positive sorted cells have the expression pattern of neurons and NeuN-negative sorted cells have the expression pattern of glia. (a) The neuronal markers Pde10a, Grin1, and Slc12a5 were more strongly expressed in NeuN-positive cells than NeuN-negative cells (n=7,7,4). Data were normalized to the reference gene Gorasp2. (b) The glial markers Mal and Ctss were more strongly expressed in NeuN-negative cells than NeuN-positive cells (n=5,5). Data were normalized to the reference gene Gorasp2. Data represent mean ± s.e.m. * = p<0.05; ** = p<0.01.
2.8 Statistics
For each mRNA measured in qPCR, biological replicate values were averaged to obtain the mean and standard error of the mean. A paired two-tailed t-test was performed to determine whether the expression was different between the NeuN-negative and NeuN-positive groups. Statistical significance was set at p <0.05.
3. RESULTS
We describe five steps in the development of our FACS procedure [Fig. 1a] using striata obtained from adult rat brains: 1-dissociation of brain tissue to a cell suspension; 2-filtration of the cell suspension; 3-fixation and immunolabeling; 4-FACS analysis and sorting; 5-RNA extraction and validation of FACS with microarray and real-time quantitative PCR. Adult rat striata and midbrains used for protein analysis alone were not subject to step 5.
Figure 1.
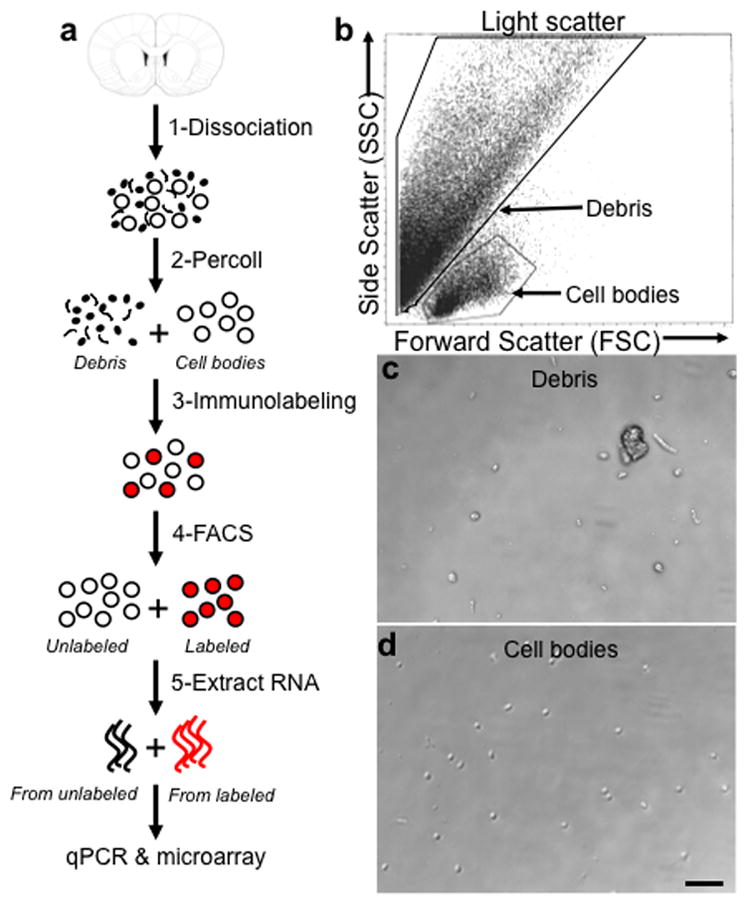
FACS involves dissociating brain tissue into a single cell suspension. (a) Schematic of FACS protocol including 1-dissociation of tissue to a cell suspension, 2-removal of debris by filtration and Percoll gradient centrifugation, 3-fixation and immunolabeling of cells, 4-purification of cells using FACS, and 5-extraction of RNA from sorted cell populations for downstream use in microarray or quantitative PCR. (Coronal brain section adapted with permission from Paxinos and Watson, 2005 (Paxinos and Watson, 2005).) (b) A typical light scatter plot showing distinct clusters for debris and cells. Each dot represents one event detected by the laser. Forward scatter (FSC) represents size of the event, side scatter (SSC) represents granularity of the event. The boxes around cell bodies and debris indicate events that were sorted for microscopic analyses shown in panels (c,d) as well as cell bodies “gated” for subsequent fluorescent analysis. (c) Debris collected from sorting based on light scatter. (d) Cell bodies collected from sorting based on light scatter. Scale bar, 50 μm.
3.1 Dissociation of brain tissue
A variety of enzymes were tested at 4°C and 37°C for 5 to 60 min for their ability to digest connective tissue with minimal cell damage. Papain and trypsin produced excessive lysing of cells and nuclei, indicated by DAPI staining of DNA. Accutase, a mixture of proteolytic and collagenolytic enzymes, was the only enzyme solution that produced high numbers of single cells with minimal nuclear lysis and was effective at 4°C. Once tissue was digested, cells were triturated through Pasteur pipets similar to preparation of primary cell cultures (Nunez, 2008).
3.2 Purification of single cell suspension
The cell dissociation process tears dendrites and matrix from cells and produced a mixture of larger, round cell bodies (~7–10μm in diameter) and large amounts of cellular debris. Excess debris leads to difficulty finding relevant cells in light scatter plots, increased FACS time, and can clog the FACS instrument. Larger debris was removed with 100 μm and 40 μm cell strainers. To remove smaller cellular debris, we assessed density gradients of Optiprep, Ficoll, Percoll, or sucrose at various concentrations. Fractions from these gradients were sorted and visually inspected under a microscope (described below; Fig. 1c and d). A step gradient of Percoll was most effective at removing debris without significant loss of cell bodies (data not shown).
3.3 Fixation and immunolabeling
Immunolabeling of intracellular markers requires fixation and permeabilization so that antibodies have access to their antigens. Fixation of cells with 0.25–1% paraformaldehyde and permeabilization with Triton X-100 allowed immunolabeling and FACS; however insufficient amounts of RNA could be extracted from these sorted cells. We subsequently found that 50% ethanol fixed and permeabilized cells without detergent. This procedure allowed immunolabeling and FACS, and produced sufficient levels of RNA for microarray and PCR (see below). Some extracellular markers, such as the neuronal marker Thy-1/CD90.1, were also used to label fixed and unfixed cells for FACS; however large amounts of debris were labeled using this extracellular marker (data not shown).
For neuronal immunolabeling, samples were incubated with a biotinylated primary antibody against NeuN which is a neuron-specific intracellular protein in the nuclear membrane, or with primary antibodies against tyrosine hydroxylase (TH) and the dopamine D1 receptor (D1R) which are proteins that identify sub-populations of neurons in midbrain and striatum, respectively. After washing, samples were incubated with their respective fluorescent labels. One wash with vigorous shaking removed unbound antibody from the cell suspension. Repeated washes caused loss of cells without affecting baseline fluorescence in FACS. Antibodies bound to their antigens at 4°C sufficiently well for FACS; this temperature likely improved RNA preservation.
3.4 FACS analysis and sorting of NeuN-labeled striatal neurons
Prior to processing the bulk of the sample with FACS, small portions (<10%) of the sample were used to determine sorting criteria, including selection of cell bodies and exclusion of non-specific labeling by the fluorescent marker. During FACS, cells pass single-file through the path of a laser; light passing through the sample (forward scatter: FSC) represents size of the event, and light reflected at a 90° angle (side scatter: SSC) represents granularity of the event. In our typical light scatter plots [Fig. 1b], each dot represents one event (one cell or piece of debris) that crossed the path of the laser. Events of a similar type tend to cluster due to their unique combination of FSC and SSC. To distinguish between cell bodies and debris, we sorted, collected and examined with light microscopy two fractions from the Percoll gradient: one that contained higher levels of cells, and one that was mostly debris. In both fractions, debris formed the majority of events, and corresponded with the upper left part of the plot [Fig. 1b,c]. Events corresponding with cell bodies were found below debris-related events and comprised approximately 7% of total events [Fig. 1b,d]. Based on these analyses, we used a small portion of the sample to establish the region of the light scatter plot corresponding to cell bodies. When the bulk of the sample was processed, only events in the cell body portion were selected or “gated” by the software for further fluorescence analysis.
We then established thresholds for non-specific labeling by the fluorescent marker by incubating a small portion of the sample with streptavidin-phycoerythrin (PE) without primary antibody [Fig. 2a]. Each axis on a fluorescence plot represents fluorescence intensity at a specific wavelength. The maximum threshold for non-specific fluorescence was selected depending on the acceptable rate of false positives (typically 0.1% of cell body-related events). In the example shown in Figure 2a, the threshold was set very conservatively to produce few false-positive NeuN-immunoreactive cells.
Figure 2.
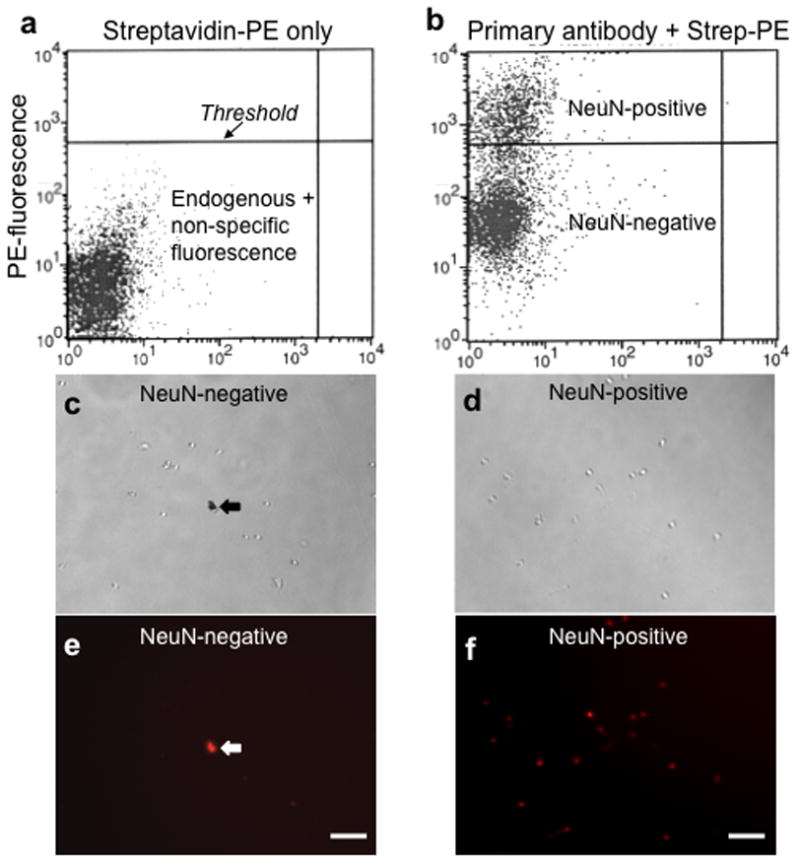
Sorting neurons from glia using the neuron-specific antibody, NeuN. (a) A fluorescence plot showing cells treated with streptavidin-phycoerythrin (PE) only. Each dot represents one event or one cell. The y-axis measures fluorescence intensity in the PE channel. This control is used to determine the upper threshold for endogenous and non-specific fluorescence (from PE). (b) A fluorescence plot showing cells treated with both the primary antibody against NeuN (which is conjugated to biotin), and streptavidin-PE. Note the cluster of events that lies above the threshold set in panel (a); these are considered NeuN-positive. Light (c) and fluorescence (e) photographs of NeuN-negative cells after sorting; light (d) and fluorescence (f) photographs of NeuN-positive cells after sorting. (c,e) include debris that is autofluorescent, which is indicated with the arrows. Scale bars, 50 μm.
The bulk of the test sample was incubated with NeuN primary antibody and streptavidin-PE and analyzed using FACS. Cell body-related events above the maximum threshold for non-specific fluorescence were considered positive for NeuN. Using these criteria, ~30% of all cell bodies (gated by FSC vs. SSC) were positively labeled for NeuN [Fig. 2b], and thus presumed to be neurons. We have used this procedure for NeuN-labeled FACS approximately 150 times with very consistent and reproducible results. Under a microscope, NeuN-positive events were larger round cell bodies (~7–10μm in diameter) labeled with PE (red) with little unlabeled debris [Fig. 2d,f]. None of the NeuN-negative events were labeled with PE [Fig. 2c,e], although occasional clusters of smaller objects were observed with some debris labeled in all fluorescent channels. Typically, cellular yield was approximately 200,000 NeuN-positive cells and 350,000 NeuN-negative cells per adult rat striatum.
3.5 Validation of FACS using microarrays and qPCR
We assessed the validity of this FACS procedure in adult rat brain by analyzing RNA expression in the NeuN-positive and NeuN-negative populations. Sorted cells were collected into 1.5 ml Eppendorf low protein-binding tubes for RNA extraction. We typically obtained 0.5–1.5 μg total RNA from NeuN-positive cells and 0.25–0.75 μg RNA from NeuN-negative cells when starting with 8 striata from 4 rats. A260/280 ratios were typically between 1.8–2.0.
cDNA was reverse transcribed from the RNA of three biological replicates of NeuN-positive and NeuN-negative cells and then hybridized to Illumina arrays for rat gene expression. Both principle component analysis and scatter plot analysis showed that biological replicates were very similar to each other (r2 values between 0.968 and 0.994), but more variable between NeuN-positive and NeuN-negative samples (r2 values between 0.683 and 0.721). The NeuN-positive sorted cells had higher mRNA levels of neuron-specific genes, including those encoding ion channels and synaptic proteins [Fig. 3c; Supplemental table 2]. The NeuN-negative sorted cells had higher mRNA levels of glial-specific genes, including those encoding myelin and immune system proteins [Fig. 3a; Supplemental table 2].
Figure 3.
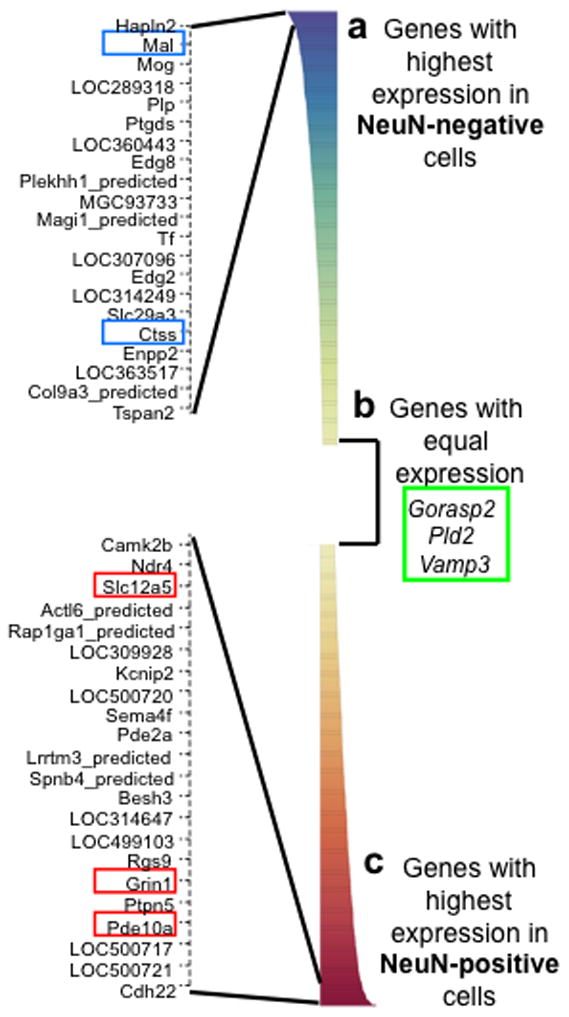
The Illumina rat microarray validates FACS by showing that NeuN-positive sorted cells have the expression pattern of neurons and NeuN-negative sorted cells have the expression pattern of glia. 736 genes are expressed significantly differently between NeuN-positive and NeuN-negative sorted cells. (a) Genes that have significantly higher expression in NeuN-negative cells include many glial markers such as Mal and Ctss. The top differentially expressed genes are shown in the expanded view on the left. (b) Many genes are not differently expressed in NeuN-positive and NeuN-negative cells. These are potential reference genes for qPCR, and they include Gorasp2, Pld2, and Vamp3. (c) Genes that have significantly higher expression in NeuN-positive cells include many neuronal markers such as Pde10a, Grin1, and Slc12a5. The top differentially expressed genes are shown in the expanded view on the left.
We used real-time quantitative PCR to confirm the microarray results. Gene expression for potential reference genes was calculated as efficiency ^ΔCq, where ΔCq = Cq(NeuN−) −Cq(NeuN+) and Cq is the quantification cycle of the PCR (Bustin et al., 2009; Pfaffl, 2001). An appropriate reference gene has the same expression in neurons and glia; thus, ΔCq is 0, and expression is 1. The commonly used reference genes RNA polymerase II and GAPDH were differentially expressed between NeuN-positive and -negative cells (data not shown). These housekeeping genes are often used in qPCR experiments to compare gene expression in samples of the same cell types but following different treatments; the housekeeping genes RNA polymerase and GAPDH are not expected to change following most treatments. However we were comparing gene expression in different cell types (i.e., neurons vs. glia) following the same treatment. Different cell types are likely to have inherently different levels of these same housekeeping genes. We thus used the microarray data to select three different candidate reference genes, Gorasp2, Pld2, and Vamp3 with apparently equal expression in NeuN-positive and NeuN-negative cells [Fig. 3b]. Although qPCR indicated levels of Pld2 and Vamp3 expression were relatively similar between samples, and much closer to parity than expression of neuronal or glial markers [Supplemental Fig. 1], the ratios of NeuN-positive to NeuN-negative expression were still significantly different from parity (Pld2 0.24 ± 0.06-fold, n=8, p<0.001; Vamp3 0.43 ± 0.17-fold, n=6, p<0.05) [Supplemental Fig. 2]. Only Gorasp2 levels were not statistically different between samples (0.96 ± 0.20-fold, n=8, p=0.85), and thus Gorasp2 was used as the reference gene for all subsequent comparisons of cell-specific genes. Gorasp2 encodes Golgi reassembly stacking protein 2, which is found in many different cell types in various species (Kinseth et al., 2007; Shorter et al., 1999).
We then used qPCR to assess expression of several neuronal and glial markers that were most differentially expressed in the microarray data [Fig. 3]. The neuronal markers included Pde10a (phosphodiesterase 10a) (Seeger et al., 2003; Xie et al., 2006), Slc12a5 (or Kcc2, a potassium-chloride transporter) (Williams et al., 1999), and Grin1 (glutamate receptor, ionotropic NMDA 1 (Conti et al., 1996; Monyer et al., 1994), which is also expressed in some astrocytic processes, but is predominantly a neuronal gene (Conti et al., 1996)). The glial markers included Mal (myelin and lymphocyte protein) (Schaeren-Wiemers et al., 2004) and Ctss (cathepsin S protease) (Petanceska et al., 1994). Using Gorasp2 as the reference gene to normalize cDNA input between samples, expression levels of neuron-specific genes were greater in NeuN-positive cells than in NeuN-negative cells [Fig. 4a]. Pde10a was increased 31.8 ± 5.8-fold (n=7, p<0.005), Grin1 was increased 16.7 ± 2.4-fold (n=7, p<0.0005), and Slc12a5 was increased 14.1 ± 3.6-fold (n=4, p<0.05). Expression levels of the glial-specific genes Mal and Ctss were 239.5 ± 95.0-fold (n=5, p=0.06) and 34.6 ± 7.4-fold (n=5, p<0.01) greater in NeuN-negative cells than in NeuN-positive cells, respectively [Fig. 4b]. The large fold differences in neuronal- and glial-specific gene expression between NeuN-positive and negative samples confirm the microarray data and selectivity of FACS purification.
3.6 Flow cytometry of neurons using plasma membrane and cytoplasmic markers
To determine if FACS could be used to sort sub-populations of neurons based on membrane or non-nuclear intracellular proteins, we co-immunolabeled striatal cells for NeuN and D1R and midbrain cells for NeuN and TH. To verify labeling in intact cells only, we also labeled all cells with DAPI. Parameters for non-specific fluorescent labeling were set as described in Section 3.4. Striatal cells were gated as previously described. Midbrain DAPI-labeled events were identified on light scatter plots (using back-gating) to determine the location of these cells, and the majority of intact cells were found within the lower left quadrant of the plot. Thus, cells only within a gate encompassing this region were used for midbrain analyses (data not shown).
For FACS analysis, only DAPI-labeled events were included to ensure that analysis was only of intact cells. A typical DAPI plot yielded a tight cell population that was easily identifiable [Fig. 5a]. DAPI-positive events within the cell body gate were considered positive for NeuN labeling if they were above the maximum threshold for non-specific fluorescence. Using these criteria, 18.7% and 17.4% of cell bodies were labeled for NeuN in striatum and midbrain, respectively, and thus presumed to be neurons. To determine if there was an identifiable population of NeuN-labeled events that were double-labeled with D1R or TH, we plotted NeuN-labeling vs. D1R- or TH-labeling [Fig. 5b and 5c]. Thresholds for determining specific labeling were established using the fluorescently labeled secondary antibody only, as described for the NeuN labeling in section 3.4. The top right quadrant of each scatter-plot represents the neuronal population positive for D1R or TH, whereas the top left quadrant represents neurons negative for D1R or TH. To determine the percentage of all neurons that labeled with one of the subtype markers, all NeuN-positive events were plotted onto graphs showing D1R- or TH-labeling [Fig 5d and 5e]. Of striatal neurons, 14.1% were found to be D1R-positive; of midbrain neurons, 30.2% were TH-positive.
Figure 5.
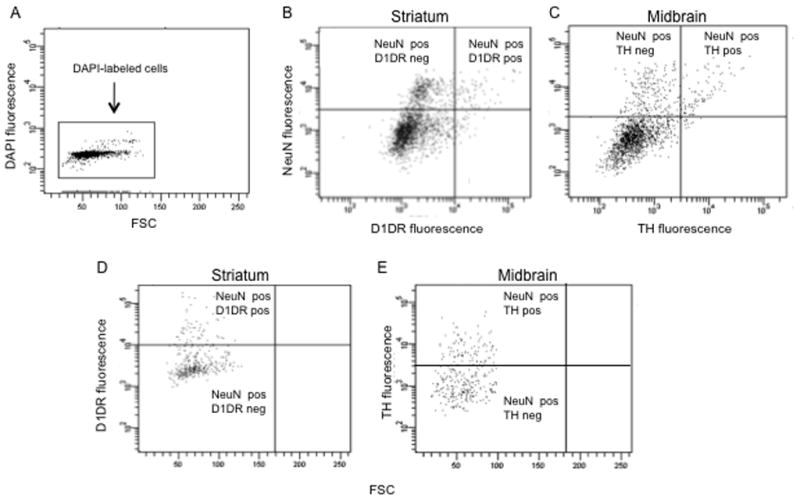
Flow cytometry of striatal cells labeled with D1R and midbrain neurons labeled with TH. (a) A forward light scatter (FSC) vs. fluorescence plot of DAPI fluorescence. Each dot represents one event or one cell. The y-axis measures fluorescence intensity in the DAPI channel. Only events positive for DAPI were used for further FACS analysis. (b) Fluorescence plot of striatal cells colabeled with NeuN and D1R antibodies. (c) Fluorescence plot showing midbrain cells labeled with NeuN and TH antibodies. Quadrant boundaries were determined by thresholds for non-specific labeling using fluorescent labels alone (without primary antibody). Note the top left and top right quadrants, which identify (b) D1R-negative and D1R-positive neurons, respectively and (c) TH-negative and TH-positive neurons, respectively. (d) and (e) Forward scatter (FSC) vs. fluorescence plots showing (d) striatal D1R and (e) midbrain TH labeling of only NeuN-labeled neurons (PE-positive events). These graphs are forward gated so that all events on these plots are NeuN-positive. Note distinct clusters of D1R or TH-labeled events above and below the respective labeling thresholds.
3.7 FACS sorting of endothelial cells from brain using a membrane marker
We also used this FACS procedure to sort a non-neuronal cell type from adult rat brain. Endothelial cells were labeled with a primary antibody against the brain endothelial cell-specific marker CD71 and a FITC-conjugated secondary antibody. CD71 is also known as the transferrin receptor, which is a membrane protein that helps cells take up iron (Jefferies et al., 1984). FACS sorting indicated that ~2% of cell bodies (gated by FSC vs. SSC) were CD71-immunoreactive. When cells were incubated with antibodies for both CD71 and NeuN, FACS indicated no overlap among CD71-positive events and NeuN-positive events [Fig. 6], which confirmed the selectivity of our FACS procedure.
Figure 6.
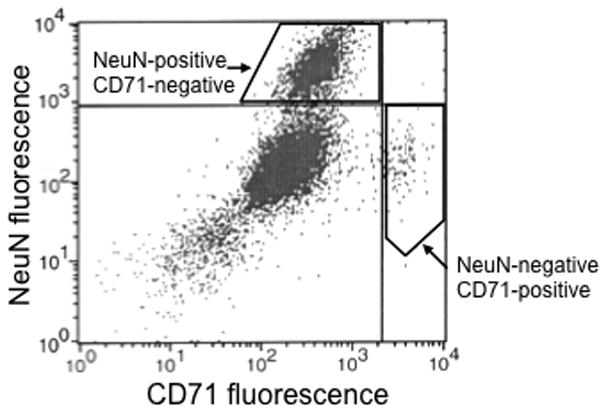
FACS can separate neurons from endothelial cells in adult rat striatum. In this fluorescence plot, each dot represents one event or one cell. The x-axis measures fluorescence in the FITC channel (marking endothelial cells) and the y-axis measures fluorescence in the PE channel (marking neurons). The quadrants are determined using only the fluorescent secondary antibodies to determine the threshold that divides non-specific from specific labeling. In the upper left quadrant are neurons (NeuN-positive, CD71-negative); in the lower right quadrant are endothelial cells (NeuN-negative, CD71-positive). Note the lack of events in the upper right quadrant, as there should not be cells labeled with both NeuN and CD71.
4. DISCUSSION
We developed and validated a rapid, quantitative, high molecular throughput FACS procedure for purifying selected cell types from adult wild-type rat brain. The use of antibodies to label intracellular markers in addition to extracellular markers permits widespread application of this procedure without the requirement for transgenic mice. The ability to separate cells based on intracellular markers is an important advantage, since many established cell markers are intracellular proteins. The ability to separate cell types from adult rat brain also permits immediate application of this technique to many well-established disease and behavioral models in rats, with the additional benefit of providing more brain tissue than from mice.
Tissue is processed within 90 minutes at 4°C, from brain extraction to fixing cell suspensions with ethanol or PFA. Short processing times at low temperatures are important for assaying molecular characteristics that best represent their status in the intact behaving animal. RNA can degrade rapidly and compromise the resulting data, but fixation stops most metabolism and degradation of molecules. However when simply analyzing cell populations (not sorting for RNA collection afterwards), the cells should be stable after preparation for several hours if kept on ice in the interval between cell preparation and flow cytometry analysis.
Our microarray results from NeuN-sorted cells correlate well with results from an analysis of gene coexpression relationships from human brain identifying modules of coexpressed genes corresponding to neurons, oligodendrocytes, astrocytes, and microglia (Oldham et al., 2008). Three of our top 10 neuronal markers from rat striatum were in the top 10% of genes expressed in human caudate neurons. This provides an independent confirmation of the validity of our sorting method. Eight of our top 10 glial markers were in the top 10% of genes in the oligodendrocyte module. Since our striatal samples contain a large amount of white matter, it is expected that oligodendrocytes are prominently represented in the NeuN-negative cell population.
Flow cytometry with D1R labeling of striatal cells indicates that FACS should be possible with plasma membrane markers, but with limitations. Only 14% of all NeuN-labeled striatal neurons were double-labeled with D1R, which is below the expected 53% of striatal neurons shown to express D1R-immunoreactivity on their cell bodies (Hersch et al., 1995). Our interpretation is that the majority of D1R protein is expressed on dendrites and not on cell bodies so that the level of D1R expressed on each cell body is probably very low. While detectable with light microscopy, most of these cell bodies probably have less than the threshold amount of D1R to be identified as D1R-positive in our FACS procedure. Only 14% out of a possible 53% of neurons have enough D1R on their cell bodies to be detected by FACS. This translates to FACS purification of 27% of all possible D1R-positive neurons in our sample. While we believe that the D1R-positive cells we detected are truly D1R-expressing neurons, 73% of the D1R neurons are false negatives in our procedure. Nevertheless 27% may be enough to assess gene expression selectively in D1R striatal neurons.
Using antibodies against the cytoplasmic enzyme TH, we successfully obtained labeling of cell bodies from the ventral third of the midbrain region, which contains many TH-expressing dopaminergic neurons in the substantia nigra and ventral tegmental area. Using our conditions, we found 30% of all NeuN-labeled neurons from this brain region were double-labeled with TH. Not surprisingly, we cannot find a published paper that describes the percent of TH-containing neurons in the particular dissection that we used, but we do know that multiple non-TH-containing cell types are found in this brain region amongst the relatively abundant TH-containing neurons, so 30% of all neurons appears to be a reasonable approximation for the percent of TH neurons.
The FACS procedure has many potential applications in neuroscience that would be overly difficult or labor-intensive using existing techniques. For example, drugs or disease can induce different molecular responses in different brain cell types. FACS purification of specific cell types allows us to distinguish the molecular responses unique to each cell type. We have used this technique to study the striatal neurons specifically activated by cocaine in rats (Guez-Barber et al., 2011). Because it can be used to study any specific set of cells for which a selective antibody is available, FACS is a powerful tool with vast potential applications in neuroscience.
Supplementary Material
Highlights.
Current methods do not allow for efficient gene analysis of specific brain cell populations
FACS purified antibody-labeled specific cell-types from adult wild-type rat brain
FACS purified neurons, glia, and endothelial cells from rat striatum
mRNA from FACS-purified cells were analyzed with microarrays and qPCR
FACS purification of antibody-labeled neurons has wide application in neuroscience
Acknowledgments
This research was supported by the Intramural Research Programs of the NIH, NIDA and NIA. DGB was supported by NIH MSTP TG T32GM07205, Award Number F30DA024931 from NIDA, and the Charles B.G. Murphy Chair in Psychiatry. MRP was supported by DA00436, DA14241 and the State of Connecticut, Department of Mental Health and Addiction Services. We thank Joe Chrest and Lee Blosser for technical assistance with FACS, as well as Sam Golden for assistance with rats. We thank Mary Kay Lobo and Eric Nestler for sharing open communication that helped improve this technique.
Abbreviations
- FACS
fluorescence activated cell sorting
- qPCR
quantitative polymerase chain reaction
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Danielle Guez-Barber, Email: danielle.guez@yale.edu.
Sanya Fanous, Email: fanoussa@mail.nih.gov.
Brandon K Harvey, Email: brandon.harvey@nih.gov.
Yongqing Zhang, Email: yongqing.zhang@nih.gov.
Kevin G Becker, Email: kevin.becker@nih.gov.
Marina R Picciotto, Email: marina.picciotto@yale.edu.
Bruce T Hope, Email: bhope@intra.nida.nih.gov.
References
- Arlotta P, Molyneaux BJ, Chen J, Inoue J, Kominami R, Macklis JD. Neuronal subtype-specific genes that control corticospinal motor neuron development in vivo. Neuron. 2005;45:207–21. doi: 10.1016/j.neuron.2004.12.036. [DOI] [PubMed] [Google Scholar]
- Brewer GJ. Isolation and culture of adult rat hippocampal neurons. Journal of Neuroscience Methods. 1997;71:143–55. doi: 10.1016/s0165-0270(96)00136-7. [DOI] [PubMed] [Google Scholar]
- Brewer GJ, Torricelli JR, Evege EK, Price PJ. Optimized Survival of Hippocampal Neurons in B27-Supplemented Neurobasal (TM), a New Serum-free Medium Combination. Journal of Neuroscience Research. 1993;35:567–76. doi: 10.1002/jnr.490350513. [DOI] [PubMed] [Google Scholar]
- Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT. The MIQE Guidelines: Minimum Information for Publication of Quantitative Real-Time PCR Experiments. Clinical Chemistry. 2009;55:611–22. doi: 10.1373/clinchem.2008.112797. [DOI] [PubMed] [Google Scholar]
- Conti F, DeBiasi S, Minelli A, Melone M. Expression of NR1 and NR2A/B Subunits of the NMDA Receptor in Cortical Astrocytes. Glia. 1996;17:254–8. doi: 10.1002/(SICI)1098-1136(199607)17:3<254::AID-GLIA7>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Doyle JP, Dougherty JD, Heiman M, Schmidt EF, Stevens TR, Ma G, Bupp S, Shrestha P, Shah RD, Doughty ML, Gong S, Greengard P, Heintz N. Application of a translational profiling approach for the comparative analysis of CNS cell types. Cell. 2008;135:749–62. doi: 10.1016/j.cell.2008.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg S, Nichols JR, Esen N, Liu S, Phulwani NK, Syed MM, Wood WH, Zhang Y, Becker KG, Aldrich A, Kielian T. MyD88 expression by CNS-resident cells is pivotal for eliciting protective immunity in brain abscesses. ASN Neuro. 2009;1:e00007. doi: 10.1042/AN20090004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guez-Barber D, Fanous S, Golden SA, Schrama R, Koya E, Stern AL, Bossert JM, Harvey BK, Picciotto MR, Hope BT. FACS identifies unique cocaine-induced gene regulation in selectively activated adult striatal neurons. The Journal of Neuroscience. 2011;31:4251–9. doi: 10.1523/JNEUROSCI.6195-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiman M, Schaefer A, Gong S, Peterson JD, Day M, Ramsey KE, Suarez-Farinas M, Schwarz C, Stephan DA, Surmeier DJ, Greengard P, Heintz N. A translational profiling approach for the molecular characterization of CNS cell types. Cell. 2008;135:738–48. doi: 10.1016/j.cell.2008.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hersch SM, Ciliax BJ, Gutekunst C-A, Rees HD, Heilman CJ, Yung KK, Bolam JP, Ince E, Yi H, Levey AI. Electron microscopic analysis of D1 and D2 dopamine receptor proteins in the dorsal striatum and their synaptic relationships with motor corticostriatal afferents. The Journal of Neuroscience. 1995;15:5222–37. doi: 10.1523/JNEUROSCI.15-07-05222.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayanthi S, McCoy MT, Beauvais G, Ladenheim B, Gilmore K, William Wood I, Becker K, Cadet JL. Methamphetamine induces dopamine D1 receptor-dependent endoplasmic reticulum stress-related molecular events in the rat striatum. PLoS ONE. 2009;4:e6092. doi: 10.1371/journal.pone.0006092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jefferies WA, Brandon MR, Hunt SV, Williams AF, Gatter KC, Mason DY. Transferrin receptor on endothelium of brain capillaries. Nature. 1984;312:162–3. doi: 10.1038/312162a0. [DOI] [PubMed] [Google Scholar]
- Kinseth MA, Anjard C, Fuller D, Guizzunti G, Loomis WF, Malhotra V. The golgi-associated protein GRASP is required for unconventional protein secretion during development. Cell. 2007;130:524–34. doi: 10.1016/j.cell.2007.06.029. [DOI] [PubMed] [Google Scholar]
- Lobo MK, Karsten SL, Gray M, Geschwind DH, Yang XW. FACS-array profiling of striatal projection neuron subtypes in juvenile and adult mouse brains. Nature Neuroscience. 2006;9:443–52. doi: 10.1038/nn1654. [DOI] [PubMed] [Google Scholar]
- Maric D, Barker JL. Fluorescence-based sorting of neural stem cells and progenitors. Current Protocols in Neuroscience. 2005 doi: 10.1002/0471142301.ns0318s33. [DOI] [PubMed] [Google Scholar]
- Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 1994;12:529–40. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- Nunez J. Primary culture of hippocampal neurons from P0 newborn rats. Journal of Visualized Experiments. 2008:19. doi: 10.3791/895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldham MC, Konopka G, Iwamoto K, Langfelder P, Kato T, Horvath S, Geschwind DH. Functional organization of the transcriptome in human brain. Nat Neurosci. 2008;11:1271–82. doi: 10.1038/nn.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. 5. Elsevier Academic Press; Burlington, MA: 2005. [Google Scholar]
- Petanceska S, Burke S, Watson SJ, Devi L. Differential distribution of messenger RNAs for cathepsins B, L and S in adult rat brain: an in situ hybridization study. Neuroscience. 1994;59:729–38. doi: 10.1016/0306-4522(94)90190-2. [DOI] [PubMed] [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Research. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz E, Yang L, Su T, Morris DR, McKnight GS, Amieux PS. Cell-type-specific isolation of ribosome-associated mRNA from complex tissues. Proc Natl Acad Sci USA. 2009;106:13939–44. doi: 10.1073/pnas.0907143106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeren-Wiemers N, Bonnet A, Erb M, Erne B, Bartsch U, Kern F, Mantei N, Sherman D, Suter U. The raft-associated protein MAL is required for maintenance of proper axon--glia interactions in the central nervous system. J Cell Biol. 2004;166:731–42. doi: 10.1083/jcb.200406092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeger TF, Bartlett B, Coskran TM, Culp JS, James LC, Krull DL, Lanfear J, Ryan AM, Schmidt CJ, Strick CA, Varghese AH, Williams RD, Wylie PG, Menniti FS. Immunohistochemical localization of PDE10A in the rat brain. Brain Res. 2003;985:113–26. doi: 10.1016/s0006-8993(03)02754-9. [DOI] [PubMed] [Google Scholar]
- Shorter J, Watson R, Giannakou M-E, Clarke M, Warren G, Barr FA. GRASP55, a second mammalian GRASP protein involved in the stacking of Golgi cisternae in a cell-free system. The EMBO Journal. 1999;18:4949–60. doi: 10.1093/emboj/18.18.4949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St John PA, Kell WM, Mazzetta JS, Lange GD, Barker JL. Analysis and Isolation of Embryonic Mammalian Neurons by Fluorescence-Activated Cell Sorting. The Journal of Neuroscience. 1986;6:1492–512. doi: 10.1523/JNEUROSCI.06-05-01492.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JR, Sharp JW, Kumari VG, Wilson M, Payne JA. The neuron-specific K-Cl cotransporter, KCC2. Antibody development and initial characterization of the protein. J Biol Chem. 1999;274:12656–64. doi: 10.1074/jbc.274.18.12656. [DOI] [PubMed] [Google Scholar]
- Wolf ME, Kapatos G. Flow cytometric analysis and isolation of permeabilized dopamine nerve terminals from rat striatum. The Journal of Neuroscience. 1989a;9:106–14. doi: 10.1523/JNEUROSCI.09-01-00106.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf ME, Kapatos G. Flow cytometric analysis of rat striatal nerve terminals. The Journal of Neuroscience. 1989b;9:94–105. doi: 10.1523/JNEUROSCI.09-01-00094.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf ME, Kapatos G. Stimulation of D2 dopamine receptors decreases intracellular calcium levels in rat anterior pituitary cells but not striatal synaptosomes: A flow cytometric study using Indo-1. Synapse. 1989c;4:353–70. doi: 10.1002/syn.890040411. [DOI] [PubMed] [Google Scholar]
- Xie Z, Adamowicz WO, Eldred WD, Jakowski AB, Kleiman RJ, Morton DG, Stephenson DT, Strick CA, Williams RD, Menniti FS. Cellular and subcellular localization of PDE10A, a striatum-enriched phosphodiesterase. Neuroscience. 2006;139:597–607. doi: 10.1016/j.neuroscience.2005.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.