

Abstract
Over-consumption of ethanol (EtOH) represents a major health problem. This study was to test the cytotoxicity of EtOH in cardiac stem cells or myoblasts, and the potential protective effect of apolipoprotein-J (ApoJ), a stress-responding, chaperone-like protein in high density lipoprotein, on EtOH-injured cardiac myoblasts. In culture, EtOH-exposed canine fetal myoblasts underwent apoptosis in a concentration- and time-dependent manner. Transduction of ApoJ by cDNA transfection markedly reduced EtOH-induced apoptosis in the cells. ApoJ expression also restored partially the mitochondrial membrane potential and prevented the release of cytochrome-c from mitochondria into cytoplasma. Thus, ApoJ serves as a cytoprotective protein that protects cardiac stem cells against cytotoxicity of EtOH.
Keywords: Apoptosis, stem cells, ethanol, cytochrome-c, mitochondria, lipoprotein, heart
Introduction
Alcohol abuse results in more than 20% of hospital admissions in the USA. In a complex process, ethanol (EtOH)-induced toxicity causes widespread cell damage [1; 2] and death via either apoptosis or necrosis [3; 4]. The cytotoxic actions of EtOH are complicated, and perhaps mediated, in part, through oxidative stress and altered calcium homeostasis [5]. As a storehouse for intracellular calcium, a source of reactive oxygen species, and a sensor of oxidative stress [6; 7], mitochondria play a key role in regulation of apoptosis under a variety of pathological conditions, including ischemia, hypoxia, and myocardial infarction [8; 9; 10].
Resulting from the asymmetric distribution of protons and other ions across the membranes, the electrochemical potential across mitochondrial membrane, Δ Ψ, is known to be highly sensitive to apoptotic stimulation. As a index of mitochondrial function in living cells, Δ Ψ can be measured with an indicator dye, e.g., rhodamine 123 (Rh123), which fluoresces in direct proportion to Δ Ψ [11]. Decreased Δ Ψ occurs in cells undergoing apoptosis induced by oxidative agents, such as oxidized low density lipoprotein (oxLDL) [12] and hydrogen peroxide [13]. Injured mitochondria can release cytochrome-c into the cytoplasm when cells are treated with proapoptotic stimuli [14; 15]. On entry into the cytosol, cytochrome-c activates the apoptosome containing the caspase-activating protein Apaf-1 and caspase-9, and subsequently the caspase cascade that induce apoptosis [16]. EtOH exposure can induce apoptosis of human neural stem cells. The mechanisms by which EtOH induces myogenic stem cell apoptosis are not well defined; therefore, no therapeutic approach is available for preventing EtOH-induced stem cell toxicity.
Apolipoprotein-J (ApoJ) is a multifunctional glycoprotein widely existing in tissues and body fluids. ApoJ-deficient mice exhibits an increased susceptibility to autoimmune myocarditis [17]. Increased expression of ApoJ confers resistance to apoptosis induced by heat shock and oxidative stress [18; 19]. As a component of high-density lipoprotein (HDL) [20; 21], ApoJ protects endothelial cells against apoptosis by inhibiting the dissipation of mitochondrial potential and the release of cytochrome-c into the cytoplasm [22]. This study examined the effects of ApoJ on EtOH-induced mitochondrial dysfunction, the release of cytochrome-c, and apoptosis in canine fetal myoblasts.
Materials and methods
Fetal myoblast cell cultures
Canine fetal myoblast cell cultures were prepared from fetal hearts. Briefly, myoblasts were separated by digesting fetal heart tissues in 10 mL of 0.25% trypsin on a shaking water bath for 5 min at 37°C. After dissociation, cells were counted in a hemacytometer and plated in 25 cm2 tissue culture flask (Becton Dickinson) at a density of 106 cells/flask. IMDM (Gibco) supplemented with 15% fetal bovine serum, 100 U/ml penicillin, and 100 ug/ml streptomycin was used for culture. The cells were incubated at 5% CO2/95% room air for 10–14 days before use.
Apo J cDNA transfection
Human ApoJ cDNA (hApoJ, accession # BC19588) was purchased from ResGen IMAGE consortium cDNA clones (IMAGE:4915444). Briefly, cDNA coding for human ApoJ was isolated from pCMV-sport 6 plasmids by using Hind III and EcoRV. The 1.7 kb fragment containing the full length human ApoJ cDNA was ligated into the mammalian expression vector pcDNA3 (Invitrogen), and transfected into canine fetal myoblast cells by using Lipofectamine2000 (Invitrogen). Transfected cells were divided 48 hours later and maintained in complete medium containing 300 μg/ml G418. After attaining confluent growth, the cells were subjected to experimental assays.
Determination of ApoJ protein expression by Western blot analysis
ApoJ–transfected and mock myoblast cells were lysed in 50mM Tris-HCl, pH 8.0, 150mM NaCl, 1% Igepal CA-630 (NP-40), 0.5% sodium deoxycholate, 1% sodium dodecyl sulfate and a mixture of protease inhibitors (1mM phenylmethylsulfonyl fluoride, 1 μg/ml aprotinin, and 1 μg/ml leupeptin). Protein extracts were prepared by sonicating the lysed cells 3 times for 5 seconds. Cell debris was removed by centrifugation at 13,000 rpm for 20 min at 4°C. An equal volume of 2X SDS reducing gel-loading buffer was then added, and the samples were boiled for 5 minutes. The protein concentration in the cell extracts was quantified via a spectrophotometer with the use of the BCA protein assay kit (Pierce) before adding the loading buffer. Polyclonal antibody against ApoJ was obtained from Santa Crutz Biotechnology. Immunopositive bands were visualized by enhanced chemiluminescence (ECL, Amersham). Blots were stripped and probed again with monoclonal anti-β-actin antibody (Sigma) as a control for equal loading.
Quantification of fragmented DNA in cell death by ELISA
DNA fragmentation in cell death was analyzed using a cell death detection ELISA kit (Roche) according to the manufacturer's instructions. Briefly, canine fetal myoblast cells were exposed to 4 different concentrations of EtOH (25 mM, 50 mM, 75 mM, and 100 mM) for 3 different time periods (16, 24, and 48 hours) to determine the effect of EtOH on apoptosis. After EtOH exposure, cells were lysed in 100μl of lysis buffer and centrifuged for 10 min at 1500 rpm Triplicate 20 μl samples of supernatant were placed into the streptavidin-coated microtest plates for analysis. DNA fragmentation was quantified by measuring absorbance at 405 nm with a reference wavelength at 492 nm.
Determination of Mitochondrial Δ Ψ
To determine the effects of EtOH on Δ Ψ, fetal myoblast cells were exposed to EtOH at the doses up to 100 mM for 3 different time periods (4, 8, and 16 hours). After the experimental treatment, the cells were washed with PBS (pH 7.2, 1 mL) 3 times, then incubated for 15 min with 5 μM Rh123 (Molecular Probes) in 1 mL of PBS. After 15 min the cells were washed 3 times with PBS, and 1 mL of PBS was then added to the plates. Cells were removed from the culture plates with a scraper and placed into cytometer tubes. The indicator fluorescence was measured (excitation/emission wavelengths 480/530 nm) with a fluorescence plate reader. The emission values were expressed as the mean fluorescent peak height of the samples normalized to a percentage of the control value.
Cytochrome-C Measurement
The cytosolic level of cytochrome-c was determined by using an ELISA (R&D Systems, Minneapolis, MN, USA). Briefly, after EtOH exposure, ApoJ – transfected and non-transfected fetal myoblast cell cultures were washed twice with PBS, and cell membrane permeabilized in PBS with 0.5% Triton X-100. The cell lysates were centrifuged at 14,000 rpm for 15 min at 4 °C. Cytosolic proteins were determined using the Bradford assay, and the same amounts of proteins from each sample loaded into 96-well microplates with immobilized anti-cytochrome-c. After incubation for 2 hours at room temperature, the plates were washed 3 times with washing buffer. A substrate solution containing tetramethylbenzidine and hydrogen peroxide was added to each well, and the plates were incubated for 30 minutes at room temperature. Then, a stop solution containing hydrochloric acid was added to terminate the reaction. Absorbance was determined by using a microplate reader at 450/575 nm dual wavelengths. The data were expressed as the mean optical density of the samples normalized to a percentage of the control value.
Statistics
Data are presented as mean ± SEM. Statistical analysis was performed by using ANOVA or Student's t test when appropriate. A p value < 0.05 was considered significant. All experiments were performed at least 3 times.
Results
EtOH exposure induces apoptotic cell death in canine fetal myoblasts
In culture, canine fetal myoblasts exposed to EtOH underwent apoptotic death in a manner dependent upon incubation time and concentrations of EtOH. Analysis of histone-associated DNA fragments showed that incublation of the cells with 100 mM EtOH caused a concentration- and time-dependent DNA fragmentation and cell loss in the cultures over the 48 h interval (Fig. 1). Significant cell death occurred in the cultures exposed to EtOH at 100 mM for 48 hours. The untreated myoblasts showed little changes in DNA fragmentation and morphology when cultured under the same conditions excerpt for EtOH treatment.
Fig. 1.
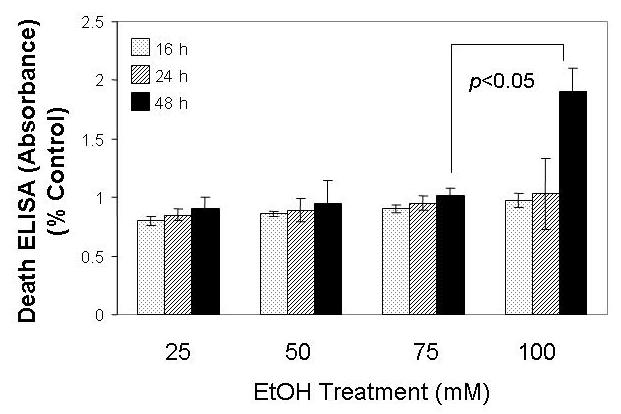
EtOH induced fetal myoblast cell apoptosis. The non-transfected fetal myoblast cells were exposed to the specified concentration of EtOH for 16 h, 24 h and 48 h, and cells were assessed for apoptosis by analyzing histone-associated DNA fragments.
ApoJ expression confers resistance to EtOH induced apoptosis of canine fetal myoblasts
To determine whether ApoJ exerts a protective effect on cardiovascular cells against a toxic agent, such as EtOH, ApoJ expression was transduced in canine fetal myoblasts by stable cDNA transfection. Western blot analysis with anti-human ApoJ antibodies confirmed a marked increase in ApoJ expression in the transfected myoblasts but not mock transfected cells (Fig. 2 A). To determine the effect of ApoJ in EtOH-induced apoptosis, 100 mM EtOH was added into the cells overexpressing ApoJ. The death ELISA showed that cells with stable expression of ApoJ had significantly lower levels of histone-associated DNA fragments than the mock control cells when exposed to 100 mM EtOH for 48 h than did non-transfected cells (Fig 2B).
Fig.2.
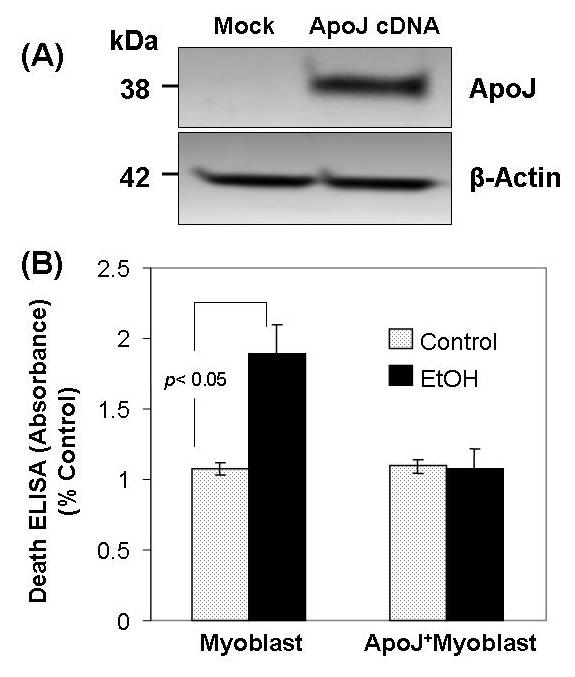
Apo J expression transduced by cDNA transfection prevents EtOH - induced apoptosis in fetal myoblast cells. Fig. 2 A. Immunoblot analysis showing ApoJ expression in fetal myoblasts transfected with or without ApoJ. Total cellular proteins extracted from fetal myoblasts and from fetal myoblasts transfected with ApoJ were subjected to 10% SDS-PAGE, then transferred to polyvinylidene fluoride membranes, blotted with anti-Apo J and anti-β-actin antibody, and detected by enhanced chemiluminescence. Anti-β-actin antibody served as a control for equal loading. Fig. 2 B. Apoptosis of ApoJ – transfected or mock control canine myoblasts exposed to EtOH. Both fetal myoblasts transfected with or without ApoJ were exposed to 100 mM EtOH for 48 h, and apoptosis was analyzed for histone-associated DNA fragments.
ApoJ expression partially restores the mitochondrial membrane potential (Δ Ψ) in canine myoblasts exposed to EtOH
Mitochondrial dysfunction characterizes apoptosis induced by cytotoxic substances. Analysis of the mitochondrial membrane potential Δ Ψ was performed as a functional measurement of mitochondrial function in the fetal myoblasts. Canine fetal myoblasts exposed to EtOH showed a decrease in Δ Ψ in a concentration- and time-dependent manner (Fig. 3 A). At the cytotoxic dose (100 mM) EtOH decreased Δ Ψ at 16 h, but not at 4 h and 8 h (Fig. 3 B). ApoJ expression by transduction with cDNA protected fetal myoblast cells against the cytotoxicity of EtOH on Δ Ψ; the EtOH - induced decrease in Δ Ψ at 16 h was significantly less in ApoJ - transfected cells than in non-transfected stem cells (Fig. 3 B).
Fig. 3.
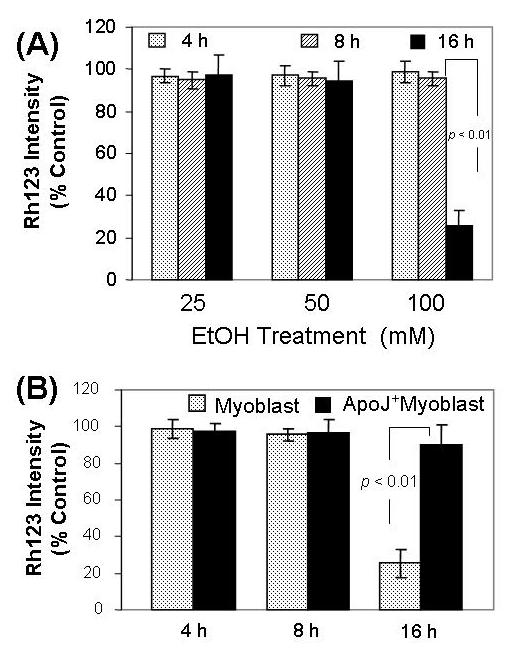
ApoJ expression restores the mitochondrial membrane potential in fetal myoblasts exposed to EtOH. Fig.3A. Effect of EtOH on mitochondrial membrane potential in fetal myoblast cells. The non - transfected fetal myoblast cells were exposed to 25 mM, 50 mM and 100 mM EtOH for 4 h, 8 h and 16 h, then loaded with 1 μM rhodamine 123 (Rh123). After washing, the cells were analyzed for fluorescence at 480/530 nm (excitation/emission) in a fluorescence plate reader. Fig 3B. Effects of ApoJ on EtOH – induced mitochondrial dysfunction. Both fetal myoblasts transfected with or without ApoJ were exposed to 100 mM EtOH for 4 h, 8 h and 16 h, then loaded with 1 μM rhodamine 123 (Rh123). After washing, the cells were analyzed for fluorescence at 480/530 nm (excitation/emission) in a fluorescence plate reader.
ApoJ expression reduces the release of cytochrome-c from mitochondria into the cytoplasma
Concurrently with the changes in the mitochondrial membrane protential, mitochondrial cytochrome-c may be released into the cytoplasma due to a partial leakage of mitochondrial membrane triggered by cytotoxic agents. Canine fetal myoblasts exposed to 100 mM EtOH showed increased cytosolic levels of cytochrome-c after 24 h of exposure but not after 4 h, 8 h and 16 h (Fig. 4). Expression of ApoJ transduced by cDNA transfection attenuated this EtOH-induced increase in the cytosolic levels of cytochrome-c as shown by the significant smaller change seen in EtOH - treated transfected cells as compared with non-transfected cells (Fig. 4).
Fig. 4.
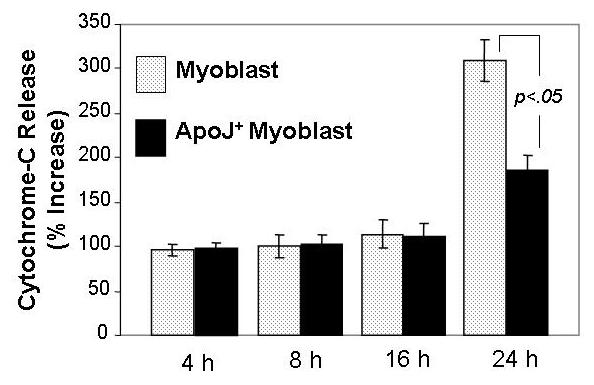
Effects of EtOH on cytochrome-c release in ApoJ-transfected or mock control fetal myoblasts. Both fetal myoblasts transfected with or without ApoJ were exposed to 100 mM EtOH for 4h, 8 h, 16 h and 24 h, and the cytosolic fractions were obtained by centrifugation. Cytochrome-c levels were assayed by ELISA, with absorbance determined using a microplate reader at 450/575 nm (excitation/emission) dual wavelengths. The results were expressed as a percentage of the untreated control ± SEM of at least three plates per group in three replicate experiments.
Discussion
This study provides the first experimental evidence showing that ApoJ attenuates EtOH-induced apoptosis in fetal myoblast cells. Furthermore, the studies of mitochondrial function demonstrates that this cytoprotection against cytotoxic doses of EtOH conferred by ApoJ may be mediated, at least partially, through stabilization of Δ Ψ and the retention of mitochondrial cytochrome-c. Others have reported similarly that Apo J attenuates apoptosis induced by heat shock and oxidative stress [18]. ApoJ is a heat shock protein-like stress-responding protein, and a component of HDL known to be cytoprotective. Our findings appear to be consistent with recent reports on the anti-apoptotic effects of ApoJ in non-stem cell cells. Others have shown that HDL inhibits apoptosis induced by ox-LDL in vascular cells [23; 24] , prevents apoptosis of endothelial progenitor cells through the inhibition of caspase-3 activity [25], and blocks apoptosis of endothelial cells by inhibiting dissipation of mitochondrial potential and release of cytochrome-c into the cytoplasm [22].
Our data showed exposure to EtOH for 16 h decreased Δ Ψ without affecting cytochrome-c release (measured 24 h after exposure to EtOH) or toxicity (measured 48 h after exposure to EtOH). These data suggest that the EtOH - induced decrease in Δ Ψ by itself is not sufficient to induce toxicity. However, the finding that EtOH affects Δ Ψ only at cytotoxic concentrations (100 mM) and not at non-cytotoxic concentrations (25 and 50 mM) indicates that EtOH – induced mitochondrial dysfunction contributes to the cytotoxicity towards myoblasts.
Our observation shows that the EtOH-induced decrease in Δ Ψ (16 h) occurred before the release of cytochrome-c (24 h), suggesting that EtOH might have attacked the mitochondria and increased the permeability of the mitochondrial membrane. This EtOH-induced cytochrome-c release may be a consequence of the opening of the mitochondrial permeability transition (MPT) channel. The finding is consistent with other reports that impaired mitochondria can release cytochrome-c into the cytoplasm, where it can bind to Apaf-1 and activate the caspase networks that induce apoptosis [26; 27; 28]. Furthermore, the finding that only cytotoxic concentrations of EtOH could induce cytochrome-c release and the fact that cytochrome-c release (24 h) occurred before cytotoxicity was induced (48 h) indicate that EtOH-induced cytotoxicity requires the release of cytochrome-c from mitochondria, which is consistent with previous findings that cytochrome-c release is necessary for apoptosis in many cell lines [14; 29; 30]. However, the experiments that directly examine the requirement for cytochrome-c in apoptosis must await the generation of conditional null mutants of cytochrome-c that eliminate the death-promoting activity but maintain the essential function of cytochrome-c in oxidative phosphorylation [15].
In conclusion, our findings show that ApoJ inhibits EtOH-induced apoptosis in fetal myoblast cells by reducing mitochondrial dysfunction. Specifically, the protective mechanism of ApoJ involves preserving the mitochondrial membrane potential and maintaining the mitochondrial retention of cytochrome-c. These results may be therapeutically useful in preventing EtOH-induced stem cell toxicity.
Acknowledgements
This work was supported partly by grants from NIH/NHLBI, Texas State and Texas Heart Institute. We thank Qun Kong and Judy Ober for their technical support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Di Gennaro C, Biggi A, Barilli AL, Fasoli E, Carra N, Novarini A, Delsignore R, Montanari A. Endothelial dysfunction and cardiovascular risk profile in long-term withdrawing alcoholics. J Hypertens. 2007;25:367–73. doi: 10.1097/HJH.0b013e328010929c. [DOI] [PubMed] [Google Scholar]
- 2.Rubert G, Minana R, Pascual M, Guerri C. Ethanol exposure during embryogenesis decreases the radial glial progenitorpool and affects the generation of neurons and astrocytes. J Neurosci Res. 2006;84:483–96. doi: 10.1002/jnr.20963. [DOI] [PubMed] [Google Scholar]
- 3.Kang K, Oh YK, Choue R, Kang SJ. Scutellariae radix extracts suppress ethanol-induced caspase-11 expression and cell death in N(2)a cells. Brain Res Mol Brain Res. 2005;142:139–45. doi: 10.1016/j.molbrainres.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 4.Bhave SV, Hoffman PL. Ethanol promotes apoptosis in cerebellar granule cells by inhibiting the trophic effect of NMDA. J Neurochem. 1997;68:578–86. doi: 10.1046/j.1471-4159.1997.68020578.x. [DOI] [PubMed] [Google Scholar]
- 5.Li Y, King MA, Meyer EM. alpha7 nicotinic receptor-mediated protection against ethanol-induced oxidative stress and cytotoxicity in PC12 cells. Brain Res. 2000;861:165–7. doi: 10.1016/s0006-8993(99)02457-9. [DOI] [PubMed] [Google Scholar]
- 6.Hyun DH, Hunt ND, Emerson SS, Hernandez JO, Mattson MP, Cabo RD. Up-regulation of plasma membrane-associated redox activities in neuronal cells lacking functional mitochondria. J Neurochem. 2007 doi: 10.1111/j.1471-4159.2006.04411.x. [DOI] [PubMed] [Google Scholar]
- 7.Melov S. Mitochondrial oxidative stress. Physiologic consequences and potential for a role in aging. Ann N Y Acad Sci. 2000;908:219–25. doi: 10.1111/j.1749-6632.2000.tb06649.x. [DOI] [PubMed] [Google Scholar]
- 8.Tsutsui H, Ide T, Kinugawa S. Mitochondrial oxidative stress, DNA damage, and heart failure. Antioxid Redox Signal. 2006;8:1737–44. doi: 10.1089/ars.2006.8.1737. [DOI] [PubMed] [Google Scholar]
- 9.Beal MF. Mitochondrial dysfunction in neurodegenerative diseases. Biochim Biophys Acta. 1998;1366:211–23. doi: 10.1016/s0005-2728(98)00114-5. [DOI] [PubMed] [Google Scholar]
- 10.Lemasters JJ, Nieminen AL, Qian T, Trost LC, Herman B. The mitochondrial permeability transition in toxic, hypoxic and reperfusion injury. Mol Cell Biochem. 1997;174:159–65. [PubMed] [Google Scholar]
- 11.Satoh T, Enokido Y, Aoshima H, Uchiyama Y, Hatanaka H. Changes in mitochondrial membrane potential during oxidative stress-induced apoptosis in PC12 cells. J Neurosci Res. 1997;50:413–20. doi: 10.1002/(SICI)1097-4547(19971101)50:3<413::AID-JNR7>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 12.Li Y, Higashi Y, Itabe H, Song YH, Du J, Delafontaine P. Insulin-like growth factor-1 receptor activation inhibits oxidized LDL-induced cytochrome-c release and apoptosis via the phosphatidylinositol 3 kinase/Akt signaling pathway. Arterioscler Thromb Vasc Biol. 2003;23:2178–84. doi: 10.1161/01.ATV.0000099788.31333.DB. [DOI] [PubMed] [Google Scholar]
- 13.Li Y, Meyer EM, Walker DW, Millard WJ, He YJ, King MA. Alpha7 nicotinic receptor activation inhibits ethanol-induced mitochondrial dysfunction, cytochrome-c release and neurotoxicity in primary rat hippocampal neuronal cultures. J Neurochem. 2002;81:853–8. doi: 10.1046/j.1471-4159.2002.00891.x. [DOI] [PubMed] [Google Scholar]
- 14.Reed JC. Cytochrome-c: can't live with it--can't live without it. Cell. 1997;91:559–62. doi: 10.1016/s0092-8674(00)80442-0. [DOI] [PubMed] [Google Scholar]
- 15.Deshmukh M, Johnson EM., Jr. Evidence of a novel event during neuronal death: development of competence-to-die in response to cytoplasmic cytochrome-c. Neuron. 1998;21:695–705. doi: 10.1016/s0896-6273(00)80587-5. [DOI] [PubMed] [Google Scholar]
- 16.Krajewski S, Krajewska M, Ellerby LM, Welsh K, Xie Z, Deveraux QL, Salvesen GS, Bredesen DE, Rosenthal RE, Fiskum G, Reed JC. Release of caspase-9 from mitochondria during neuronal apoptosis and cerebral ischemia. Proc Natl Acad Sci U S A. 1999;96:5752–7. doi: 10.1073/pnas.96.10.5752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McLaughlin L, Zhu G, Mistry M, Ley-Ebert C, Stuart WD, Florio CJ, Groen PA, Witt SA, Kimball TR, Witte DP, Harmony JA, Aronow BJ. Apolipoprotein J/clusterin limits the severity of murine autoimmune myocarditis. J Clin Invest. 2000;106:1105–13. doi: 10.1172/JCI9037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rosenberg ME, Girton R, Finkel D, Chmielewski D, Barrie A, 3rd, Witte DP, Zhu G, Bissler JJ, Harmony JA, Aronow BJ. Apolipoprotein J/clusterin prevents a progressive glomerulopathy of aging. Mol Cell Biol. 2002;22:1893–902. doi: 10.1128/MCB.22.6.1893-1902.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dumont P, Chainiaux F, Eliaers F, Petropoulou C, Remacle J, Koch-Brandt C, Gonos ES, Toussaint O. Overexpression of apolipoprotein J in human fibroblasts protects against cytotoxicity and premature senescence induced by ethanol and tertbutylhydroperoxide. Cell Stress Chaperones. 2002;7:23–35. doi: 10.1379/1466-1268(2002)007<0023:ooajih>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jones SE, Jomary C. Clusterin. Int J Biochem Cell Biol. 2002;34:427–31. doi: 10.1016/s1357-2725(01)00155-8. [DOI] [PubMed] [Google Scholar]
- 21.Wilson MR, Easterbrook-Smith SB. Clusterin is a secreted mammalian chaperone. Trends Biochem Sci. 2000;25:95–8. doi: 10.1016/s0968-0004(99)01534-0. [DOI] [PubMed] [Google Scholar]
- 22.Nofer JR, Levkau B, Wolinska I, Junker R, Fobker M, von Eckardstein A, Seedorf U, Assmann G. Suppression of endothelial cell apoptosis by high density lipoproteins (HDL) and HDL-associated lysosphingolipids. J Biol Chem. 2001;276:34480–5. doi: 10.1074/jbc.M103782200. [DOI] [PubMed] [Google Scholar]
- 23.Negre-Salvayre A, Dousset N, Ferretti G, Bacchetti T, Curatola G, Salvayre R. Antioxidant and cytoprotective properties of high-density lipoproteins in vascular cells. Free Radic Biol Med. 2006;41:1031–40. doi: 10.1016/j.freeradbiomed.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 24.Li XA, Guo L, Dressman JL, Asmis R, Smart EJ. A novel ligand-independent apoptotic pathway induced by scavenger receptor class B, type I and suppressed by endothelial nitric-oxide synthase and high density lipoprotein. J Biol Chem. 2005;280:19087–96. doi: 10.1074/jbc.M500944200. [DOI] [PubMed] [Google Scholar]
- 25.Noor R, Shuaib U, Wang CX, Todd K, Ghani U, Schwindt B, Shuaib A. High-density lipoprotein cholesterol regulates endothelial progenitor cells by increasing eNOS and preventing apoptosis. Atherosclerosis. 2006 doi: 10.1016/j.atherosclerosis.2006.06.023. [DOI] [PubMed] [Google Scholar]
- 26.Shidoji Y, Nakamura N, Moriwaki H, Muto Y. Rapid loss in the mitochondrial membrane potential during geranylgeranoic acid-induced apoptosis. Biochem Biophys Res Commun. 1997;230:58–63. doi: 10.1006/bbrc.1996.5883. [DOI] [PubMed] [Google Scholar]
- 27.Kruman I, Guo Q, Mattson MP. Calcium and reactive oxygen species mediate staurosporine-induced mitochondrial dysfunction and apoptosis in PC12 cells. J Neurosci Res. 1998;51:293–308. doi: 10.1002/(SICI)1097-4547(19980201)51:3<293::AID-JNR3>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 28.Mattson MP, Duan W. “Apoptotic” biochemical cascades in synaptic compartments: roles in adaptive plasticity and neurodegenerative disorders. J Neurosci Res. 1999;58:152–66. [PubMed] [Google Scholar]
- 29.Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng TI, Jones DP, Wang X. Prevention of apoptosis by Bcl-2: release of cytochrome-c from mitochondria blocked. Science. 1997;275:1129–32. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- 30.Garrido R, Mattson MP, Hennig B, Toborek M. Nicotine protects against arachidonic-acid-induced caspase activation, cytochrome-c release and apoptosis of cultured spinal cord neurons. J Neurochem. 2001;76:1395–403. doi: 10.1046/j.1471-4159.2001.00135.x. [DOI] [PubMed] [Google Scholar]