

Abstract
Retinal pathologies common to human eye diseases, including abnormal retinal pigment epithelial (RPE) cells, drusen-like accumulation, photoreceptor atrophy, and choroidal neovascularization, have been reported in the Ccl2/Cx3cr1-deficient mouse. The Ccl2 gene encodes the pro-inflammatory chemokine CCL2 (MCP-1), which is responsible for chemotactic recruitment of monocyte-derived macrophages to sites of inflammation. The Cx3cr1 gene encodes the fractalkine receptor, CX3CR1, and is required for accumulation of monocytes and microglia recruited via CCL2. Chemokine-mediated inflammation is implicated in retinal degenerative diseases such as diabetic retinopathy, age-related macular degeneration, retinitis pigmentosa, and uveoretinitis, and proper chemokine signaling from the RPE, Müller glia, and astrocytes is necessary to regulate leukocyte trafficking. Therefore, this mouse, possessing aberrant chemokine signaling coupled with retinal degenerative pathologies, presents an ideal opportunity to investigate the effect of altered signaling on retinal homeostasis and photoreceptor degeneration. Since this mouse is a recent development, more data covering the onset, location, and progression rate of pathologies is needed. In the present study we establish these parameters and show two photoreceptor cell death processes. Our observations of decreased glutamine synthetase and increased glial fibrillary acidic protein suggest that Müller cells respond very early within regions where lesions are forming. Finally, we demonstrate that retinal angiomatous proliferation contributes to pathological angiogenesis in this Ccl2/Cx3cr1-deficient mouse.
Keywords: Ccl2, Cx3cr1, rodent model, confocal scanning laser ophthalmoscopy, Optical coherence tomography, 3D OCT, photoreceptor degeneration, reactive gliosis, Müller cell
1. Introduction
Recently, a double knock-out Ccl2−/−/Cx3cr1−/− mouse was created in which one chemokine and one chemokine-receptor gene, Ccl2 and Cx3cr1, were eliminated and retinal pathologies common to human eye diseases, including abnormal RPE, drusen-like accumulation, photoreceptor atrophy, and choroidal neovascularization, were reported (Tuo et al., 2007; Ramkumar et al., 2010). Detailed information on the genetic background and the generation of this Ccl2−/−/Cx3cr1−/− mouse has been previously described (Chan et al., 2008), and an analysis of the Ccl2−/−/Cx3cr1−/− mouse model of AMD is available (Raoul et al., 2010). The Ccl2 gene encodes the pro-inflammatory chemokine CCL2, also known as MCP-1, which is responsible for chemotactic recruitment of monocyte-derived macrophages to sites of inflammation (Huang et al., 2001). The Cx3cr1 gene encodes the fractalkine receptor, CX3CR1, and is required for accumulation of monocytes recruited via CCL2 (Tacke et al., 2007). In retina, CCL2 and CX3CR1 are expressed by retinal pigment epithelium (RPE), Müller glial cells, and microglia (Lee et al., 2010; Carter and Dick., 2004; Chan et al., 2005).
Chemokine signaling mediates a broad range of functions during inflammation and immunity and is essential for leukocyte trafficking (Ransohoff, et al., 2007). Immune-mediated inflammation and compromise of the blood-retina-barrier (BRB) are implicated in retinal degenerative diseases such as diabetic retinopathy, age-related macular degeneration, and uveoretinitis (Crane & Liversidge, 2008). CX3CR1 signaling alters retinal microglia dynamics (Liang et al., 2009) and regulates microglial neurotoxicity (Cardona et al., 2006). In this Ccl2 and Cx3cr1 double deficient mouse, microglial accumulation has been reported (Ross et al., 2008; Tuo et al., 2007), concomitant with drusen-like accumulation, RPE alterations, and choroidal neovascularization (Tuo et al., 2007).
CCL2- and CX3CR1-mediated signaling are particularly important in monocyte trafficking across the BRB (Crane & Liversidge, 2008). Unlike peripheral tissue, which only requires extravasation of circulating leukocytes from the vascular endothelium, leukocyte infiltration into retina requires passage of the BRB. Proper secondary chemokine signaling from cells constituting the BRB, specifically RPE and vascular endothelial cells, is necessary to regulate the passage of extravasated leukocytes into retinal tissue. Impaired signaling increases the pro-inflammatory cytokines, tumor necrosis factor alpha (TNFα) and interleukin 1-beta (IL-1β), that stimulate CCL2 synthesis by RPE (Kerkar et al, 2006; Lukiw et al., 2003; Crane & Liversidge, 2008). These pro-inflammatory cytokines also lead to upregulation of VEGF, the major pathogenic factor in retinopathy of prematurity, diabetic retinopathy, and age-related macular degeneration (Gerhardt, 2008; Grisanti & Tatar, 2008). Müller glial cells, which are closely associated with maintenance of the BRB (Tout et al., 1993), have been identified as the source of VEGF responsible for neovascularization and leakage in diabetic and oxygen-induced retinopathy (Bai et al., 2009; Wang et al., 2010).
Therefore, this Ccl2−/−/Cx3cr1−/− mouse, possessing aberrant chemokine signaling, coupled with retinal degenerative pathologies, presents an ideal avenue to investigate chemokine signaling, BRB integrity, and, ultimately, retinal degeneration. Since the Ccl2−/−/Cx3cr1−/− mouse is a recent development, more data covering the onset, location, and progression rate of pathologies is needed. In the present study we assessed the onset, location, and progression of retinal pathologies using in vivo Spectral Domain-optical coherence tomography (SD-OCT) imaging techniques coupled with histological and immunolabeling methods. These results establish a foundation for subsequent investigations of neuroprotective signaling and retinal degenerative mechanisms.
2. Materials and Methods
All animal experiments conformed to the Association for Research in Vision and Ophthalmology statement for the Use of Animals in Ophthalmic and Vision Research and were approved by the Institutional Animal Care and Use Committee for the Louisiana State University Health Sciences Center (LSUHSC), New Orleans. Ccl2−/−/Cx3cr1−/− breeder mice, constructed on a C57BL/6 background (Chan et al., 2008), were sent from the National Eye Institute and bred in the LSUHSC animal colony on a 12 h:12 h light-dark cycle (0600 h ON:1800 h OFF) at an average illumination of 20 lux (sawdust bedding level at cage center on ventilation rack). Animals were fed normal mouse chow and supplied with water ad libitum. C57BL/6 male mice (Charles River Laboratories, Wilmington, MA) were used as age-matched controls. Conventional techniques were employed for histology, immunohistochemistry, and western blots. In brief, eyes were collected and prepared for histology and immuno-localization, or the retinas were homogenized. All mice used for SD-OCT and fundus photography were anesthetized with ketamine (200 mg/kg) and xylazine (10 mg/kg), and pupils dilated with tropicamide (1%; Akorn, Inc., Buffalo Grove, IL). Non-refractive contact lenses (Veterinary Speciality Products, Shropshire, Great Britain) were used to protect the corneas from desiccation and improve image quality. High-resolution angiography (HRA) and SD-OCT images were obtained using a Spectralis HRA+OCT® imaging system (Heidelberg Engineering, Heidelberg, Germany), utilizing Spectralis software, version 4.0.
2.1 Fundus Imaging
The HRA system utilizes a confocal scanning laser ophthalmoscope (cSLO) to obtain retinal angiograms and reflectance images. Retinal reflectance images were obtained using an 820 nm laser diode for near infrared (NIR); 488 nm for red-free (RF), and 488 nm with a 500 nm long pass emission filter for auto-fluorescence (AF) and fluorescein angiography. Fluorescein injections were administered intraperitoneally (0.02 mL of 25% fluorescein; Altaire Pharmaceuticals, Inc., Aquebogue, NY) and timing started immediately thereafter. Early (40–60 s) and late (>600 s) phase images were taken and fluorescein leakage was evaluated by an ophthalmologist specialized in fundus imaging.
2.2 Spectral Domain - Optical Coherence Tomography and En Face Image Processing
We utilized dual beam SD-OCT and cSLO of the Spectralis HRA+OCT® imaging system to simultaneously acquire a series of transectional retinal images and an RF reference image for each eye. Spectral illumination was from 488, 790, and 820 nm lasers with an additional 870 nm superluminescent diode. A real time eye tracker coupled the cSLO and SD-OCT scanners to position and stabilized the OCT scan on the retina. Signal quality was greater than 20 db, scan speed was 40,000 A-scans per second, and over 25 frames were averaged per B-scan to increase signal-to-noise ratio. Axial resolution was 7 m/pixel optically and 3.5 μm/pixel digitally. An average corneal curvature value of 0.672 mm was used to produce a lateral optical resolution of 0.6635 μm/pixel (Knott et al., 2010).
Previously, we found a very high correlation between outer retinal thickness assessed by OCT and histologic data (R2 = 0.9042) (Knott et al., 2010). To accomplish this, we created an ImageJ plug-in that interpreted binary raw files exported by the Spectralis software, which enabled analysis of thickness measurements from along the entire retinal image. Consequently, we have utilized this procedure to obtain dimensional values from our OCT images.
Sequential B-scans were captured of retinal areas containing lesions to produce a 3D OCT data set. En face translations (Supplementary data, Fig 1) were created using ImageJ by re-slicing the data orthogonal to both the A- and B-scans. The resulting en face images present OCT data in a view angle analogous to that of cSLO.
2.3 Histology and Immunolocalization
Conventional methods were employed to produce plastic sections for light microscopy. Mice were sacrificed by CO2 inhalation and cervical dislocation. Following removal of eyes, corneas were slit and globes placed in fixative (2% glutaraldehyde and 2% paraformaldehyde in 0.1 M sodium cacodylate) overnight at 4° C. Each eye was then hemi- sectioned along the vertical meridian through the optic nerve, and the cornea notched near the superior margin for section orientation. After rinsing in buffer, tissue was post-fixed for 1 h in sodium cacodylate-buffered 1% OsO4, rinsed again, dehydrated through an ethanol series to acetone, and embedded in an Epon/Araldite epoxy mixture. Sections, 1 μm-thick, were obtained, stained with toluidine blue, viewed by light microscopy, and digitized for analysis.
Immunolocalization was performed on 20 μm-thick cryosections. For orientation purposes, a dot was placed on the superior edge of the cornea with a fine point permanent marker. Eyes were treated as above using 4% fresh paraformaldehyde in phosphate buffered saline, followed by cryoprotection in 15% and then 30% sucrose for 8 h and 24 h, respectively. Eyes were oriented and embedded in OCT Compound (Tissue-Tek, Sakura Finetek; Torrance, CA) just prior to sectioning. Sections were placed on glass microscope slides on a 32° C slide warmer for 30 minutes. Sections were rinsed, post-fixed with 100% cold methanol, and permeablized with 1% triton-X 100, treated with 95° C citric acid for epitope retrieval, blocked 1 h in 2% donkey serum, and placed in primary antibody for 48 h at 4° C. This was followed by incubation with secondary antibody and nuclear staining for 1 h at room temperature. Sections were then washed, coverslipped, and imaged. Primary antibodies were rabbit polyclonal anti-GFAP at 1:1000 dilution (AB5804, Millipore; Billerica, MA) and mouse monoclonal anti-Glutamine Synthetase at 1 μg/mL (MAB 302, Millipore). Fluorescent detection was by secondary antibodies that recognized the host animal of each primary antibody (0.4 μg/mL) (Invitrogen, Carlsbad, CA): AlexaFluor® 633 goat anti-rabbit IgG (A-21070) or AlexaFluor® 568 goat anti-rabbit IgG (A-11011), and AlexaFluor® 488 goat anti-mouse IgG (A-11001). Nuclei were stained with 10 μg/mL Hoechst 33258 (H3569, Invitrogen). Sections were imaged on a Zeiss LSM-510 Meta laser confocal microscope with either a 20x air or a 40x oil-immersion objective (Zeiss Plan-NEOFLUAR 40x/1.3 Oil DIC). For three-dimensional reconstructions, image stacks were acquired using the 40x oil-immersion objective. Optical slice thickness for all fluorophores was 0.9 μm. Image resolution was set to 0.45 μm per pixel and the z-interval was set to 0.45 μm to ensure cubic voxel dimensions. Visualization of fluorescent signal was achieved as follows (excitation; emission; pinhole Ø): Hoechst 33258 (405nm; 420–490nm; 1.27 Airy), AlexaFluor® 488 (488 nm; 505–550 nm; 1.00 Airy), AlexaFluor® 568 (543 nm; 560–615 nm; 0.94 Airy), AlexaFluor® 633 (633 nm; long pass 650 nm; 0.87 Airy).
2.4 Western Blot Analysis
Retinas were homogenized at 4 °C in RIPA buffer, containing broad-spectrum protease inhibitors. Protein was quantified by Lowry assay and 20 μg per sample was electrophoresed using an XCell SureLock® Mini-Cell (Invitrogen) in a NuPAGE® Novex 12% Bis-Tris Gel (Invitrogen). Proteins were transferred to 0.2 μm PVDF membranes using an iBlot® Dry Blotting System (Invitrogen) and blocked in 5% blotting grade non-fat dry milk (170-6404XTU, Bio-Rad; Hercules, CA) for 1 h at room temperature. To probe GFAP and actin, polyclonal rabbit anti-GFAP [1:1000] (see above) and mouse anti-actin [1:2000] (A5441, Sigma-Aldrich; St. Louis, MO) primary antibodies were used. Bound primary antibody was detected by HRP-linked chicken anti-rabbit IgG and chicken anti-mouse IgG secondary antibody at 0.02 μg/mL and was developed with the ECL-Plus Western-Blot analysis system according to the manufacturer’s instructions (RPN2132, Amersham, Arlington Heights, IL). Chemiluminescence was detected using a Fujifilm LAS-3000 digital scanner. GFAP and actin bands were quantified at 50 and 43 kDa, respectively, using Fujifilm Multigauge software.
3. Results
Clinically, red-free and autofluorescent fundus examinations provide information about drusen, lipofuscin, and RPE atrophy. Therefore, to initially explore the features of retinal degeneration in the Ccl2−/−/Cx3cr1−/− mouse, we used both red-free and autofluorescence cSLO funduscopy. Numerous irregular-shaped bright regions were observed on 55-degree red-free cSLO fundus images of three-month-old mutant mice (Fig 1A). Similar-shaped areas of increased autofluorescence with granular hyperfluorescent spots were apparent by 55-degree autofluorescent cSLO (Fig 1B). Colocalization of the red-free and autofluorescent features demonstrates that both imaging modalities detect the same abnormalities (Fig 1C). To determine if these areas were representative of drusen, it was necessary to establish the retinal layer within which they occurred. The only in vivo method currently available to achieve this in mouse, is SD-OCT. Corrections of eye-motion artifacts, however, are necessary to attain quality SD-OCT results of mouse retina. The autofluorescent cSLO was insufficient for the correction algorithms employed by the Spectralis HRA+OCT system; therefore, we used red-free cSLO (Fig 1D) in combination with 3D SD-OCT (Fig 1D–F).
Figure 1.

Lesions in outer nuclear layer of en face OCT co-localize with lesions in red-free and autofluorescent cSLO fundus exams. Widefield (55°) cSLO images were taken during a fundus exam of a 3-month-old DKO mouse (A–C). Irregular shaped lesions with uniform intensity are visible in a red-free cSLO image (A). An autofluorescent fundus image reveals similar irregular shapes as in A but with granular intensity patterns (B). Fundus images from A an B were registered and superimposed as red and green colors, respectively (C). Lesions observed in the red-free cSLO image colocalize with granular type lesions seen in the autofluorescent cSLO image (C). Red-free image obtained simultaneously during 3D SD-OCT (D) showing a 30° cSLO view which corresponds to the white box outline in A–C. Hyper-reflective lesions are visible in the outer nuclear layer of a single SD-OCT B-scan (E). The position of the B-scan, relative to the cSLO, is indicated by the redline (A–D, F). The blue line indicates retinal depth of the en face transform shown in F. Crossbar on blue line and white dashed line are registered with red crossbars (A-D, F) and indicates the center of a lesion in the outer nuclear layer. A single en face plane (F) transformed from the 3D SD-OCT data (blue box, A–D) through the outer nuclear layer (blue line, E) showing large irregular shaped lesions with a granular-like hyper-reflectivity similar in appearance to lesions seen in fundus autofluorescence. Contrast adjusted red-free image cropped from white boxed area in A (G). Contrast adjusted autofluorescent image cropped from white boxed area in B (H). Inverted and contrast adjusted en face OCT from F superimposed on the red-free cSLO image from D (I). Color merge of G, H and en face portion of I demonstrates that lesions observed in red-free and autofluorescent cSLO images colocalize with hyper-reflective lesions in en face OCT at the depth of the outer nuclear layer (J). Scalebar colors (A–C, G–J) represent the color of the respective image in the merged final (C, J). White box outline (A–C) indicates image area of D, G, and H. Blue box (AD) denotes region of image F. Red line (A–D, F) denotes SD-OCT B-scan position of E.
The retinal cross section illustrated by the B-scan (Fig 1E) revealed focal hyper-reflective columns distributed laterally throughout the retina. These columns traversed the ONL from the OPL to the OLM, the inner/outer segment junction, or the RPE. The en face transform of the 3D OCT data (Fig 1F), at the level of the outer nuclear layer (Fig 1E, blue line), showed irregular-shaped patterns of hyper-reflectivity with granular texture (Fig 1F) that were similar to the pattern observed by fundus autofluorescence (Fig 1B). Since 3D OCT simultaneously captures a 30-degree cSLO image (Fig 1D) for registration and eye-motion-artifact corrections, it was possible to identify the exact position of specific OCT hyper-reflective columns within the red-free cSLO image (Fig 1D and F, red crosshairs; Fig 1E, blue crosshair). This registration demonstrated that OCT abnormalities within the ONL coincided with the irregular-shaped bright regions observed in red-free funduscopy. We aligned and overlaid the 55-degree red-free and autofluorescence images with the en face OCT image (Fig 1G–J) and found that all three imaging modalities colocalized (Fig 1J), verifying that the dense regions observed by each imaging method were one and the same.
To determine if these dense regions observed by fundus imaging and OCT in the Ccl2−/−/Cx3cr1−/− mouse were also present in control C57BL/6 mice, we similarly examined three-, five, and 18-month-old eyes by autofluorescence and 3D-OCT. No autofluorescent regions were found at these ages (data not shown).
3.1 Initial lesion formation and progression
Histological sections of the Ccl2−/−/Cx3cr1−/− retinas were examined to determine the structure of the ONL densities and establish a time course for the development of these lesions. Initially, isolated columns of ONL nuclei (2–3 nuclei wide) are displaced through the OLM toward the RPE, accompanied by thin regions of edema at the ONL–inner segment interface (Fig 2A, arrow). Displacement continues toward the RPE cell layer with concomitant shortening of inner segments (Fig 2B). These lesions remain as discrete units approximately three nuclei wide, despite formation of new nearby lesions (Fig 2C). Distally, cellular debris appear as lesion development progresses. As ONL nuclei move toward the RPE layer, gaps form in the OPL, within which several morphologically distinct types of nuclei appear. Inner and outer segments shorten at the leading edge of the nuclear mass. In regions between these lesions, outer segment tips vessiculate, shorten, detach, and appear along the apical surface of the RPE. Cellular fragmentation increases as each packet of nuclei advances (Fig 2D), and photoreceptor inner and outer segments are lost. Swelling and vessiculation also disrupt the OPL, forming wedge-like protrusions into the ONL. Outer segment damage in non-lesioned areas increases. Lesions in close proximity to others fuse, forming large complex lesions that contain several distinct types of nuclei (Fig 2E). Complete degeneration of the OPL at the proximal edge of the lesion results in a nucleus-free region at the center of each lesion. Lesion progression halts as the most distal nuclei contact the RPE apical surface. Large complex lesions accumulate as adjacent lesion columns continue to form (Fig 2F). Finally, long thick cellular processes originating from the inner retina penetrate these complex lesions.
Figure 2.
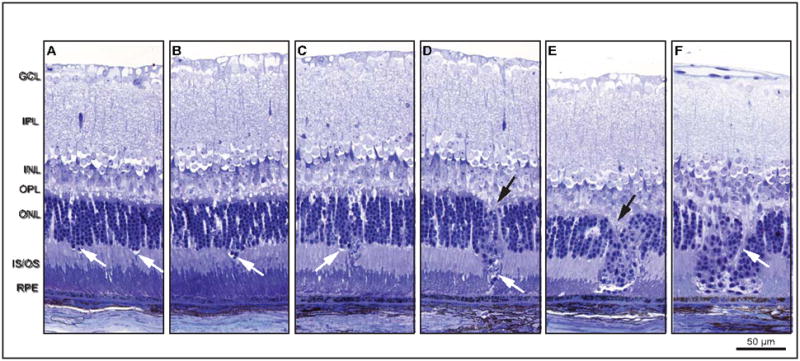
An assemblage of histologic micrographs illustrates lesion formation and progression. Lesions originate near the outer limiting membrane, followed by loss of inner and outer segments and movement of nuclei toward the retinal pigment epithelium. There is initial displacement of a column of ONL nuclei (2–3 nuclei wide) through the OLM toward the RPE (A), which is accompanied by a thin region of edema near the OLM (arrows). Outer and inner segment lengths appear relatively normal. Immediately distal to the lesion column, however, inner segments begin shortening. The OPL appears undisturbed. Displacement continues toward RPE with concomitant inner segment length reduction (B). Lesions retain discrete identity despite subsequent formation of adjacent lesions (C). The older lesion on the right contains many cellular fragments in the distal tip. Movement of ONL cell bodies distallytoward the RPE produces a gap in the OPL where several morphologically distinct nuclei are apparent (D, top arrow). Distally, inner segments are absent and outer segments contact the tip of the lesion (D, bottom arrow). Outer segments are shortened and vessiculated at their tips. As lesions progress to the level of the connecting cilium, cellular fragmentation increases and photoreceptor segments are lost. Note the newly developing lesion immediately to the left. Swelling and vessiculation disrupt the OPL and forms a wedge-like protrusion into the ONL (E, arrow). Lesions eventually fuse into large lesion complexes. Here, three lesions have fused into a single lesion complex with extensive cell debris along the right edge. Nuclear morphology within the lesion column is mixed, suggesting the presence of different cell types such as photoreceptors and immune cells (macrophage, neutrophil, and microglia). Finally, the OPL within the lesion site becomes disorganized (F). Swelling occurs at lesion front and a single row of nuclei becomes aligned along the RPE layer apical surface. Ultimately, processes originating from inner retina breach the large lesion complexes. (F, arrow).
Since lesions observed by cSLO, OCT, and histology colocalize, we next investigated the effects of age and retinal location upon these lesions using SD-OCT and cSLO autofluorescence. cSLO autofluorescence of two-, four-, and eight-month-old retinas showed the distribution of lesions. Lesions predominate throughout the inferior retina, but only occur sparsely in the superior retina, where they are confined to the inferior-most regions (Fig 3A–C). The extent of the inferior retinal lesions exceeded our limit of detection. However, the range of occurrence expands outward from the central inferior retina in a nasal and temporal direction (Fig 3D–F), with the youngest lesions located peripherally. Hyper-fluorescence increases as lesions age, except in areas of severe retinal degeneration, where autofluorescence dramatically decreases as cells and reflective cellular material are lost (Fig 3F, arrow). Only occasional lesions appear in OCT B-scans of superior retina at two months of age (Fig 3G), whereas inferior retina possesses numerous lesions (Fig 3G, arrows). Examinations at four months of age reveal an increase in the frequency of inferior retina lesions while superior retina remains relatively unaffected (Fig 3H, arrows). Severe degeneration of inferior retina can be seen by eight months (Fig 3I, arrow), whereas superior retina remains normal throughout the ages examined.
Figure 3.
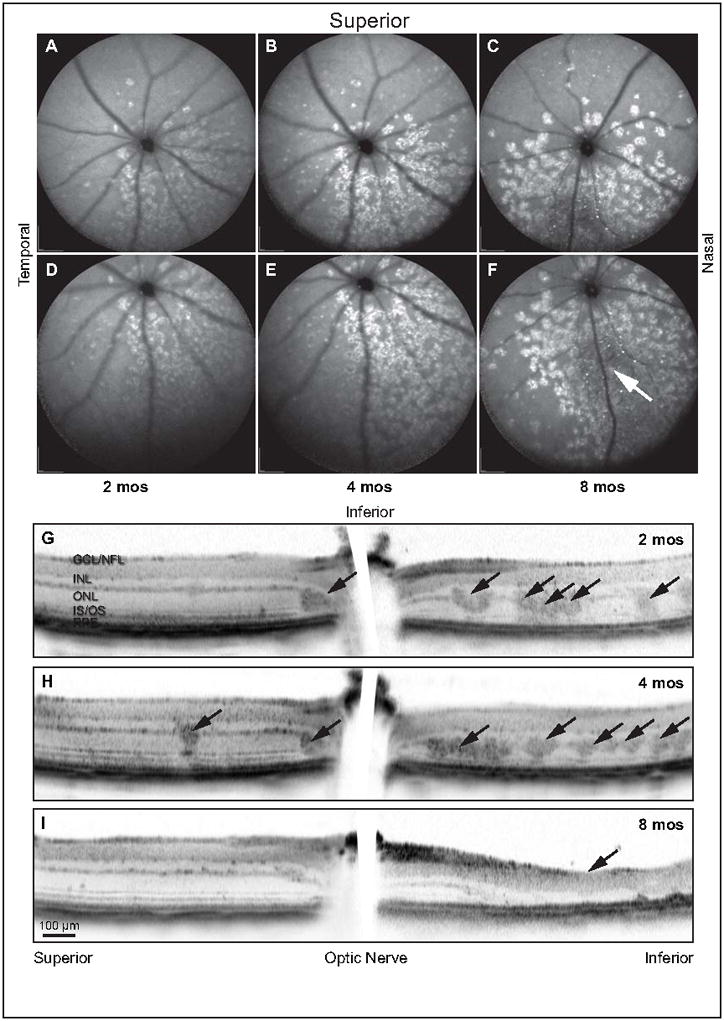
Lesions form, initially, in the inferior hemisphere, increase with age, and ultimately result in degeneration of outer retina. The central fundus from Ccl2−/−/Cx3cr1−/− mice ranging 2 – 8 months of age were examined by autofluorescent cSLO. Lesions are principally confined to the inferior retina with only sporadic occurrence in superior retina (A–C). Lesion frequency increases with age from 2 – 8 months until virtually the entire inferior hemisphere is affected. Inferior autofluorescent fundus images of the same retinas (D–F) demonstrate that lesions extend to the far periphery of the inferior hemisphere. Loss of lesions is apparent in the central inferior portion of the 8-month-old mouse (F, arrow) and correlates to the region of severe retinal degeneration. Ccl2−/−/Cx3cr1−/− retinas imaged by SD-OCT along the vertical meridian passing through the optic nerve localized lesions within the photoreceptor layer (G–I). Occasional lesions are evident in the superior retina, but numerous lesions are apparent in the inferior retina at 2 (G, arrows) and 4 (H, arrows) months of age. By 8 months, lesions are replaced by severe degeneration of outer retina in the inferior hemisphere (I, arrow). This region of sever photoreceptor loss corresponds to the hypofluorescent region indicated in F (arrow).
3.2 Long term lesion progression
Evaluation of histologic sections over an even greater age span corroborates OCT findings and further details retinal degeneration occurring after four months of age (Fig 4). Superior retina exhibits mild degeneration with age (Fig 4A–F). Disruption of photoreceptor outer segments in small regions is apparent at 1.5 months (Fig 4B, arrows). By three months of age, some outer segments appear vessiculated and the number of photoreceptor nuclei begins to decrease (Fig 4C). Outer segments are shortened at four months with focal edema present in the outer segment layer (Fig 4D, arrows). Edema is apparent throughout the entire outer segment layer by 16 months (Fig 4E, arrow), yet, ONL thickness, by nuclei count, is only moderately decreased. The density of the distal OPL, along the base of photoreceptors, is also reduced, and there is evidence of cellular swelling in both the OPL and the ONL. At 27 months of age, both inner and outer segments are disorganized (Fig 4F). Nuclear density of the ONL is significantly lower with focal regions exhibiting lateral expansion, despite only a moderate reduction in the nuclear thickness of ONL from 1 month of age. Large expanses of outer segments have degenerated (Fig 4F, arrow).
Figure 4.

Histologic sections corroborate SD-OCT observations. Focal lesions appear in the inferior hemisphere, increase with age, and result in degeneration of outer retina, thus confirming SD-OCT observations. Superior retina degenerates mildly with age. At 1 month, superior retina appears normal (A). Outer segments begin retracting at 1.5 months (B, arrows) and vessiculate by 3 months (C). Numerous focal edemas and outer segment shortening occurs by 4 months of age (D, arrows). At 16 months, the entire outer segment layer of superior retina exhibits severe edema (E, arrow). Photoreceptor inner segments become disorganized, outer segments are greatly reduced (F, arrow), and the ONL begins to thin by 27 months. In contrast, inferior retina develops large lesion complexes at 1 and 1.5 months of age (G, H, white circle). Inferior retinal lesions swell (see thicknessmarker) and become highly disorganized, with many constituent cells degenerating at 3 months (I). Edematous regions form near the RPE (arrow). At 4 months of age, only vestigial outer segments remain J, arrow) and little distinction between lesion and non-lesion regions exists. Outer segments and OPL are completely lost by 16 months and ONL is severely degenerated (K). Remarkably, 1–2 rows of outer nuclei are still present at 27 months in the inferior retina (L).
In contrast, inferior retina contains advanced complex lesions as early as one month (Fig 4G, arrows). At 1.5 months, focal lesions begin to fuse with adjacent lesions to form large complexes (Fig 4H, circled region). Retinal regions between these complexes exhibit focal edemas and reduction of outer segment length similar to that observed within the three-month-old superior retina. As inferior retinal lesions age, modest degeneration of photoreceptor nuclei occurs accompanied by enlarged somas, resulting in a temporary increase in ONL thickness (Fig 4I, vertical bar). Degeneration of the ONL continues and, in regions containing older lesions, thickness begins to decrease. Edema occurs in the subretinal space (Fig 4I, arrow) in areas associated with lesions. In the retinal regions between lesions, the ONL thickness is decreased approximately 40%, inner segment length is reduced, outer segments are severely shortened and vessiculated, and OPL is deteriorated with numerous sites of edema. By four months, the degree of inferior retinal degeneration in the most advanced lesions is severe (Fig 4J). Only vestigial outer segments remain (Fig 4J, arrow) and ONL is thoroughly disorganized. Some identifiable ONL, 1–3 nuclei thick, is still evident at 16 months of age but outer segments and OPL are completely lost (Fig 4K). Remarkably, 1–2 rows of outer nuclei are still present in the inferior retina of a 27-month-old Ccl2−/−/Cx3cr1−/− mouse (Fig 4L, arrow).
3.3 Lesion quantification by OCT
To quantitatively assess lesions, we evaluated thickness profiles of OCT B-scans using our “Open Heyex Raw” plug-in for ImageJ (Knott et al., 2010). Total retinal thickness represents the distance from the ILM to Bruch’s membrane, inner retinal thickness corresponds to the distance from the ILM to the base of photoreceptors, and non-lesion outer retinal thickness signifies the distance between Bruch’s membrane and the base of normal OCT reflectance for the ONL (the RPE plus photoreceptors). Lesion profiles were calculated using the three measured thickness profiles. Total retinal thickness in mouse is ordinarily composed of roughly equal parts inner and outer retina, and despite the occasional presence of lesions; this inner–outer proportion is unchanged in two-month-old superior retinas (Supplemental data, Fig 2A). Decline in outer retinal thickness, however, is readily apparent in inferior retina. Moreover, lesions constitute a significant fraction of the calculated outer retinal thickness. By four months of age, inferior retina exhibits severe degeneration of outer retinal thickness beyond 500 μm from optic nerve (Supplemental data, Fig 2B), yet superior retina remains unaffected. Normal outer retinal thickness persists in superior retina through eight months but degeneration of outer retinal thickness continues in inferior retina (Supplemental data, Fig 2C). Notably, lesions contribute minimally to the outer retinal thickness. Retinal degeneration in the ages assessed was primarily confined to outer retina. Minor alterations of inner retinal thickness become apparent at four months and are amplified by eight months. Integrated area under the curves for lesion and non-lesion outer retina reveals little impact on superior retina (Supplemental data, Fig 2D); lesion area is minor and outer retinal area remains reasonably stable with age. Inferior retina contains the majority of lesions (Supplemental data, Fig 2E). The fraction of outer retinal area composed of lesion is 40–50% at two months. As inferior outer retinal area decreases with age, the majority of reduction comes from the lesion fraction until all constituent cells have degenerated. By eight months of age, lesions are no longer discernible in OCT B-scans, leaving only the degenerating non-lesioned outer retina.
3.4 Neovascularization
A low rate of spontaneous choroidal neovascularization has previously been reported in the Ccl2−/−/Cx3cr1−/− mouse (Tuo et al., 2007). Accordingly, we investigated leakage in the Ccl2−/−/Cx3cr1−/− mouse using fundus fluorescein angiography and 3D OCT. Angiographic leakage was detected in approximately 30% of mice older than seven months of age (Fig 5A, asterisk). Leakage was only observed in the inferior retina and occurred in focal regions of autofluorescence. Simultaneous acquisition of 3D OCT permitted the inspection of leakage areas by OCT B-scans (Fig 5B). At the site of leakage, a low-reflectance column extends through the OPL, OLM, and photoreceptor inner/outer segments, terminating where RPE is perturbed (Fig 5B, arrow). Histologic inspection of retinas with similar pathologies reveals localized RPE cell loss, accompanied by unidentified cells associated with retinal capillaries that extend toward Bruch’s membrane (Fig 5C–F, arrows). Pigmented cells surround these unidentified cells (Fig 5D–F). These retinal capillaries ramify into larger vessels that contact Bruch’s membrane and extend parallel to its surface (Fig 5F, arrow). Disturbed choriocapillary beds were noted adjacent to these angiogenic regions, but, in this study, no discontinuity of Bruch’s membrane was observed at any stage of degeneration.
Figure 5.
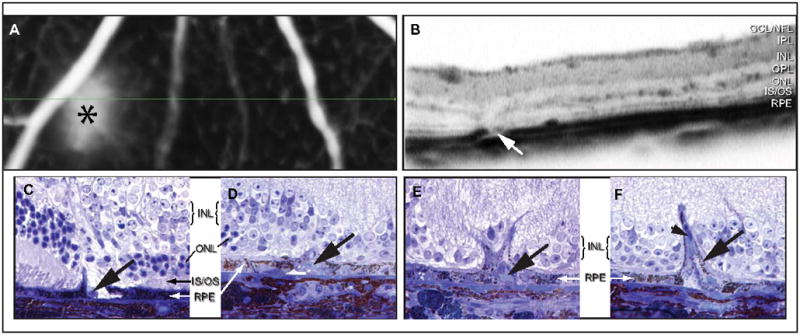
Retinal angiomatous proliferation and leakage is evident in areas of retinal degeneration of Ccl2−/−/Cx3cr1−/− mice. High-resolution fluorescein angiography reveals leakage in a 17-month-old Ccl2−/−/Cx3cr1−/− mouse (A, asterix). Simultaneous SD-OCT (B) identifies descending vasculature and RPE disruption (arrow) localized to the site of leakage. Histologic inspection of retinas with similar pathologies reveals focal RPE erosion and the presence of unknown cell types (C, arrow). RPE enclose these foreign cells (D, arrow) and create a barrier that encompasses retinal capillary outgrowths (E, arrow). In this study, no discontinuity of Bruch’s membrane was observed at any stage of this process. Retinal capillaries ultimately ramify into larger vessels that contact Bruch’s membrane and run parallel to its surface (F, arrow).
3.5 Müller cell response
Lesions originate as, and retain, a columnar format with distinct boundaries, suggesting abrupt OLM disruption. We therefore asked if Müller cells were undergoing changes during lesion formation. Cryosections of three-month-old Ccl2−/−/Cx3cr1−/− retinas were used due to the presence of numerous focal lesions in inferior retina and limited abnormalities in superior retina. Immunohistochemical localization of glutamine synthetase, a marker of Müller cell health, occurred in the OPL and at the distal and proximal ends of Müller cells at the OLM and ILM in superior retina (Fig 6A, C). Typical Müller cell processes were also evident. This labeling pattern was also apparent in non-lesion areas of inferior retina; however, labeling at the OLM and OPL and in the Müller cell processes was absent within retinal lesions (Fig 6B, D, arrows). Age-matched control mouse (C57BL/6) retinas revealed no difference in superior and inferior retinal labeling (Fig 6E–H). However, there was a difference when compared with superior retinas of the mutant animals. Whereas the mutant retinas demonstrated strong labeling at the OLM, the ILM and the OPL, control retinas only showed strong labeling at the ILM.
Figure 6.

Müller cell involvement is associated with Ccl2−/−/Cx3cr1−/− lesions by a sharp reduction of glutamine synthetase (GS), a marker of healthy Müller cells. Retinas from 3-month-old Ccl2−/−/Cx3cr1−/− mice were sectioned and labeled for GS (green) and Hoechst (blue) (A–D). For clarity, GS labeling is represented monochromatically (A, B). The superior hemisphere (A, C) exhibits normal GS patterning with strong label at the OLM, OPL, and ILM. Additionally, vertical striations archetypical of Müller cell processes can be seen. In comparison to superior hemisphere, inferior hemisphere (B, D) revealed abnormal GS labeling. The OLM and OPL bands of the GS label were absent in lesion columns (arrows). The absence of GS at the region of the OLM coincided with distally displaced nuclei characteristic of the lesions. A similar comparison was made with C57BL/6 age-matched control mice. GS labeling appeared similar in both the superior (E, G) and inferior (F, H) retina. However, there was reduced labeling in the OLM and OPL within both retinal halves. As viewed from the RPE, a 3D reconstruction of the outer nuclear layer from a non-lesioned superior (I) and lesioned inferior retina (J) illustrates the disruption of the OLM (red boxed plane).
Three-dimensional reconstructions of glutamine synthetase labeling in a section of the retina show that photoreceptor nuclei move distally toward RPE within the lesion sites. As viewed from the RPE, glutamine synthetase labeling at the plane of the OLM is continuous in the superior retina (Fig 6I, red box, green label). Conversely, at the OLM within a lesion site, inferior retinas exhibit large voids in glutamine synthetase label through which nuclei have moved toward the RPE (Fig 6J, blue label).
Reduction of glutamine synthetase and precise loss of label with column-like precision suggests a lesion-associated impairment of Müller cells. Therefore, we sought clarification of Müller cell involvement using the reactive gliosis marker, glial fibrillary acidic protein (GFAP). Lysates of three- and six-month-old Ccl2−/−/Cx3cr1−/− retinas were compared with 3.5-month-old C57BL/6 retinas by western blot (Fig 7A). At three months, prior to significant retinal degeneration, yet when lesions are profuse, GFAP expression was 3.5 fold higher than in controls (Fig 7B). Expression decreased slightly by 6 months of age, but was still over three fold higher than controls. These results clearly identify increased Müller cell reactivity very early in lesion formation. Consequently, we employed immunofluorescence to localize the increased GFAP expression. To enhance probability of detecting increased GFAP early in lesion formation, we examined 1.5-month-old Ccl2−/−/Cx3cr1−/− retinas. Within lesions of inferior retina, increased GFAP label was evident throughout the ONL and within Müller cell end-feet (Fig 7E, F). Increased GFAP label was also noted in a column in superior retina with a bright region located in the ONL (Fig 7C, D). Importantly, this discrete labeling appeared at the site of a newly forming lesion.
Figure 7.

Elevated GFAP expression and focal increases indicate an early Müller cell response as lesion onset commences in Ccl2−/−/Cx3cr1−/− mice. Whole retinal lysates from Ccl2−/−/Cx3cr1−/− mice immuno-blotted with GFAP and β-Actin (A) demonstrate a significant increase in GFAP expression at 3 and 6 months of age as compared to 3.5 month old wild type controls; bar graph (B). Immunofluorescence of GFAP in superior (C, D) and inferior (E, F) retina demonstrates that GFAP immunoreactivity is apparent within the inner processes of Müller cells in regions within, and immediately surrounding focal lesions of inner retina (E, F). More importantly, focal increases of GFAP very early in lesion development suggest Müller cell sensitivity to signals initiating photoreceptor degeneration (C, D, white arrows). C and E are monochromatic images from D and F, respectively.
4. Discussion
The goal of this study was to address in vivo the onset, location, and progression of retinal pathologies in the Ccl2−/−/Cx3cr1−/− mouse. We demonstrate that granular hyperfluorescent areas within the fundus colocalize with abnormal SD-OCT reflectance in the ONL, and correspond to lesions observed by histology.
These lesions are not typical aging features and are not swollen autofluorescent subretinal macrophages as described in aging Ccl2-knockout mice (Luhmann et al., 2009). Lesions are apparent as early as one month of age, beginning in the ONL as discrete columns of nuclei approximately three photoreceptors wide. These photoreceptors rapidly lose their inner and outer segments as their nuclei migrate toward the RPE cell layer. Lateral fusion with adjacent lesions creates large complexes which superficially resemble retinal rosettes in that they form an abnormal mass surrounded by healthy retina. However, rosette formation has been characterized as a focal abnormal arrangement of photoreceptors that occurs within the first several weeks after retina formation in some mutant mouse models, e.g., the neural retina leucine zipper-deficient (Nrl) mouse (Wenzel et al., 2007). These in-pockets of apparently healthy photoreceptors within a normal-appearing retina, which are created when small regions of the retina become detached from the RPE and fold into wrinkles, result in the inner and outer segments realigning radially toward the central space. Rosette formation also occurs as a result of pathological events occurring within the adult human retina, as noted in some Retinitis pigmentosa patients (Tulvatana et al., 1999). In retinas where rosettes form, it remains unclear whether formation is a result of neuronal “overmigration”, incorrect guidance, or inadequate focal adhesion assembly in a developmental system (Sandström et al., 2011), or from changes in the RPE/outer segment complex that promotes local detachment and reorganization of a healthy photoreceptor layer. Regardless, the large abnormal complexes that form in this Ccl2−/−/Cx3cr1−/− mouse retina occur following the loss of outer and inner segments.
Whereas lesion formation initiates in the central inferior retina of the Ccl2−/−/Cx3cr1−/− mouse, subsequent lesions appear peripherally, gradually occurring in more inferior, nasal, and temporal regions. Discrete lesion columns vanish as the component cells degenerate, with the oldest lesions “disappearing” by eight months. Thus, a timeline of lesion maturity exists from the periphery to the center of each retina. Also, within the inferior retina in the regions between lesions, the ONL is gradually reduced as photoreceptors degenerate. This is first noticeable by three months and slowly progresses until, at about two years of age, a single layer of nuclei remain. This suggests there are two photoreceptor cell death processes occurring, an initial lesion-based degeneration and a second, slow, general decline. It is important to note that our temporal description of photoreceptor degeneration is based on static images obtained from time-ordered histological sections, and it is from these images that we have attempted to assemble a plausible sequence.
An explanation for the spatio-temporal nature of lesion distribution is unavailable. However, bright light can induce stress in photoreceptor cells, which may subsequently undergo apoptotic degeneration (Gordon et al., 2002; Noell, 1980; Tanito et al., 2008; Williams and Howell, 1983). Because daily room light originates above these mice, the inferior hemisphere of the retina receives more photons, which could preferentially stress the photoreceptors in this region. The loss of the CCL2 chemokine gene and the gene for the CX3CR1 chemokine receptor in this mouse affects the ability of photoreceptors to respond to stress. Consequently, with the accumulation of stress factors and the inability to ameliorate the resulting inflammatory response, cells in the inferior retina could initiate degenerative mechanisms. Also, the intensity of overhead light impinging on the retina decreases gradually across the equator and into the superior retina, suggesting that peripheral lesions may occur later because a longer stress interval would be necessary to accrue sufficient damage. While the intensity of available light is low within our mouse colony, it has been demonstrated that photoreceptor degeneration in mouse rhodopsin mutants can be accelerated by only modest light (Krebs et al., 2009; Wang et al., 1997). Finally, it is possible that the accumulation of stress-induced factors may activate microglia, which could trigger the development of lesions. Aggregation of microglia within the lesions of Ccl2−/−/Cx3cr1−/− mice was previously reported (Ross et al., 2008; Tuo et al., 2007), and light was demonstrated to be a key triggering event in the microglial accumulation-associated retinal degeneration of Cx3cr1-deficient mice (Combadiere et al., 2007).
In addition to the formation of lesions and the degeneration of photoreceptors, the synthesis of new blood vessels is also a characteristic feature in the retina of the Ccl2−/−/Cx3cr1−/− mouse. Beyond seven months of age, neovascularization and leakage occur within the subretinal space. Abnormal retinal neovascularization, which exhibits leakage and, at late stages, may or may not anastomotize with choroidal vessels, is defined as retinal angiomatous proliferation (RAP) (Yannuzzi et al., 2001). Also, in a study on exudative age-related macular degeneration, RAP was observed in 15.1% of the cases and accounted for 30% of vascularized pigment epithelial detachments (Cohen et al., 2007). The integrity of Bruch’s membrane, in this study, implies RAP-like neovascularization of retinal capillaries without anastomosis with choroidal vessels. Therefore, in addition to previous reports of choroidal neovascularization (Chan et al., 2008), retinal angiomatous proliferation contributes to pathological angiogenesis in the Ccl2−/−/Cx3cr1−/− mouse.
The columnar form of lesions with distinct boundaries suggests abrupt local loss of adhesion molecules between Müller cells and photoreceptors at the level of the OLM. Aberrant tight junction and adherens junction protein expression has been shown to be a characteristic of the Rho−/− mouse autosomal recessive Retinitis pigmentosa model (Campbell et al., 2006), and chemokines are involved in the maintenance of these cell-to-cell junctions. Moreover, Müller cells can exhibit early focal expression of chemokines upon light-induced retinal stress (Rutar et al., 2011). Thus, Müller cell involvement can be implied. Lesion-associated decreases in glutamine synthetase, most notably in the region of the OLM, and significant increases in GFAP expression support this impression. Like glutamine synthetase, GFAP exhibits lesion-associated localization, and increased GFAP is localized to some pre-lesioned areas of retina. Together, these results demonstrate that Müller cells are responsive to the signals generated during lesion formation in the Ccl2−/−/Cx3cr1−/− mouse. Mice deficient in GFAP and vimentin attenuate the reactive responses of retinal glial cells and leukocyte infiltration (Nakazawa et al., 2007). Perturbed chemokine signaling impairs Muller cell regulation of leukocyte trafficking, and while Muller cells contribute to the formation and maintenance of the blood-retina barrier (Tout et al., 1993) and the OLM, these cells may respond to stress by disregulation of the zonula adherens junctional complexes that form cellular adhesions. Subsequent loss of OLM integrity may account for the formation of lesions and focal displacement of photoreceptor cell bodies toward the RPE. Similar displacement occurs when mice are treated with the glial toxin, DL-alpha-aminoadipic acid (AAA), a transient disruptor of the OLM (West et al., 2008). Retinal images taken at 72 hours post-AAA treatment, demonstrating loss of the OLM and disrupted retinal lamination, are remarkably similar to those of early lesions obtained in this study.
In summary, we have established the locality, onset, and progression of photoreceptor cell loss. The hyperfluorescent regions detected in the fundus correspond to focal lesions observed histologically within the photoreceptor layer. Photoreceptor degeneration occurs within the inferior retina and consists of two cell death processes. The first involves the formation of focal lesions, initially appearing at about one month of age, each of which is composed of a small number of photoreceptors. These lose their outer and inner segments, followed by migration of photoreceptor cell bodies into the subretinal space. Adjacent lesions fuse as degeneration progresses. As photoreceptors die the lesions “disappear”. This degeneration sequence is concluded by eight months of age. The second cell death process involves a slow decline in photoreceptor nuclei (ONL) within the regions between the focal lesions. This becomes apparent by three months of age and slowly continues throughout the life of the mouse. In addition, we have observed retinal angiogenesis, during which capillaries originating within the inner retina extend toward the subretinal space, contacting Bruch’s membrane. Finally, we have shown that Müller glial cells associated with focal lesions exhibit GS-negative and GFAP-positive stress-related changes within their distal processes. We suggest that the onset of focal lesion formation may result as a consequence of OLM disruption, as many cell-to-cell adhesion molecules are regulated by chemokine signaling, some of which originate within the Müller glial cells. Also, light may exacerbate the degenerative process occurring within the inferior retina. Since alterations in chemokine signaling can induce retinal changes which exhibit characteristics common with age-related macular degeneration and Retinitis pigmentosa, modulation of chemokines may provide a new approach for the regulation of retinal inflammatory responses that precede photoreceptor degeneration.
Supplementary Material
Figure S1. Schematic 3D SD-OCT data illustrates composition of the en face transform. SD-OCT measures reflectance along its optical axis and presents the intensity and axial depth of the reflectance as a column of pixels referred to as the A-scan (blue outlined vertical column of black and white rectangles). A-scans are collected at a rate of 40,000 per second along a designated path and the combination of these A-scans creates a composite known as the B-scan (gray planes). Sequential B-scan captures produce a 3D SD-OCT data set that we analyze in ImageJ using our “Open Heyex Raw” plugin. Re-slicing the 3D data set orthogonal to both the A-scan and B-scan presents OCT data in a view angle analogous to the cSLO view, termed en face OCT (green plane).
Figure S2. Retinal thickness assessment by OCT B-scans quantifies lesion contribution and degeneration in 2-, 4-, and 8-month-old Ccl2−/−/Cx3cr1−/− mouse retinas. Normally, total retinal thickness is composed of approximately equal parts inner and outer retina. Outer retinal thickness (top edge of red shaded regions) was calculated from the difference between total (solid black lines) and inner (dashed black lines) retinal thickness measurements. Healthy outer retina was also measured (blue shaded regions). The difference between calculated and healthy outer retinal thicknesses represents lesion areas (red shaded regions). At 2 months, despite some lesion area, the inner–outer thickness composition remains normal in superior retina while inferior exhibits significant lesion area and reduced outer retinal thickness (A). By 4 months, superior retina is unchanged but severe degeneration of outer retina is evident beyond 500 μm inferior to the optic nerve (B). Normal superior outer retinal thickness persists at 8 months, while severe degeneration continues in inferior retina (C). Notably, little lesion area is discernible and inner retina is the predominate source of total retinal thickness. At this point, inner retina is no longer stable across the profile but shows signs of alterations and degeneration as the inferior distance to optic nerve increases. Integrated outer retinal area reveals minor lesion area and little change over time on superior retina with age (D). In the inferior retina, lesions compose 40–50% of outer retina at 2 and 3 months (E). Lesion contribution declines with age becoming insubstantial by 8 months.
Research Highlights.
En face transformations of 3D SD-OCT identify cSLO abnormalities (red-free and autofluorescence) as the structural lesions observed by histology in the Ccl2/Cx3cr1-deficient mouse
These lesions, detectable by one month, are located predominately in the inferior retina
These lesions are distinct from the normal age-related degenerative features
Müller cell reactive gliosis is initiated prior to lesion on-set
RAP-like neovascularization originates within the retina
Acknowledgments
This research was supported by grants from the National Institutes of Health, NCRR (P20 RR016816) and NEI (R01 EY005121), the American Health Assistance Foundation (M2010091), and the Foundation Fighting Blindness (TA-NP-0808-0463-LSUNO).
Footnotes
Disclosure: Y Zhou, None; KG Sheets, None; EJ Knott, None; CE Regan, Jr, None; J Tuo, None; C-C Chan, None; WC Gordon, None; NG Bazan, None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bai Y, Ma JX, Guo J, Wang J, Zhu M, Chen Y, Le YZ. Müller cell-derived VEGF is a significant contributor to retinal neovascularization. J Pathol. 2009;219:446–454. doi: 10.1002/path.2611. [DOI] [PubMed] [Google Scholar]
- Campbell M, Humphries M, Kennan A, Kenna P, Humphries P, Brankin B. Aberrant retinal tight junction and adherens junction protein expression in an animal model of autosomal recessive Retinitis pigmentosa: The Rho(−/−) mouse. Exp Eye Res. 2006;83:484–492. doi: 10.1016/j.exer.2006.01.032. [DOI] [PubMed] [Google Scholar]
- Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, Huang D, Kidd G, Dombrowski S, Dutta R, Lee JC, Cook DN, Jung S, Lira SA, Littman DR, Ransohoff RM. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci. 2006;9:917–924. doi: 10.1038/nn1715. [DOI] [PubMed] [Google Scholar]
- Carter DA, Dick AD. CD200 maintains microglial potential to migrate in adult human retinal explant model. Curr Eye Res. 2004;28:427–436. doi: 10.1080/02713680490503778. [DOI] [PubMed] [Google Scholar]
- Chan CC, Tuo J, Bojanowski CM, Csaky KG, Green WR. Detection of CX3CR1 single nucleotide polymorphism and expression on archived eyes with age-related macular degeneration. Histol Histopathol. 2005;20:857–863. doi: 10.14670/hh-20.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CC, Ross RJ, Shen D, Ding X, Majumdar Z, Bojanowski CM, Zhou M, Salem N, Jr, Bonner R, Tuo J. Ccl2/Cx3cr1-deficient mice: An animal model for age-related macular degeneration. Ophthalmic Res. 2008;40:124–128. doi: 10.1159/000119862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen SY, Creuzot-Garcher C, Darmon J, Desmettre T, Korobelnik JF, Levrat F, Quentel G, Paliès S, Sanchez A, Solesse de Gendre A, Schluep H, Weber M, Delcourt C. Types of choroidal neovascularization in newly diagnosed exudative age-related macular degeneration. Br J Ophthalmol. 2007;91:1173–1176. doi: 10.1136/bjo.2007.115501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combadiere C, Feumi C, Raoul W, Keller N, Rodero M, Pezard A, Lavalette S, Houssier M, Jonet L, Picard E, Debre P, Sirinyan M, Deterre P, Ferroukhi T, Cohen SY, Chauvaud D, Jeanny JC, Chemtob S, Behar-Cohen F, Sennlaub F. CX3CR1-dependent subretinal microglia cell accumulation is associated with cardinal features of age-related macular degeneration. J Clin Invest. 2007;117:2920–2928. doi: 10.1172/JCI31692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane IJ, Liversidge J. Mechanisms of leukocyte migration across the blood-retina barrier. Semin Immunopathol. 2008;30:165–177. doi: 10.1007/s00281-008-0106-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhardt H. VEGF and endothelial guidance in angiogenic sprouting. Organogenesis. 2008;4:241–246. doi: 10.4161/org.4.4.7414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon WC, Casey DM, Lukiw WJ, Bazan NG. DNA damage and repair in light-induced photoreceptor degeneration. Invest Ophthalmol Vis Sci. 2002;43:3511–3521. [PubMed] [Google Scholar]
- Grisanti S, Tatar O. The role of vascular endothelial growth factor and other endogenous interplayers in age-related macular degeneration. Prog Retin Eye Res. 2008;27:372–390. doi: 10.1016/j.preteyeres.2008.05.002. [DOI] [PubMed] [Google Scholar]
- Huang DR, Wang J, Kivisakk P, Rollins BJ, Ransohoff RM. Absence of monocyte chemoattractant protein 1 in mice leads to decreased local macrophage recruitment and antigen-specific T helper cell type 1 immune response in experimental autoimmune encephalomyelitis. J Exp Med. 2001;193:713–726. doi: 10.1084/jem.193.6.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerkar S, Williams M, Blocksom JM, Wilson RF, Tyburski JG, Steffes CP. TNF-alpha and IL-1 beta increase pericyte/endothelial cell co-culture permeability. J Surg Res. 2006;132:40–45. doi: 10.1016/j.jss.2005.06.033. [DOI] [PubMed] [Google Scholar]
- Knott EJ, Sheets KG, Zhou Y, Gordon WC, Bazan NG. Spatial correlation of mouse photoreceptor-RPE thickness between SD-OCT and histology. Exp Eye Res Exp Eye Res. 2010;92:155–160. doi: 10.1016/j.exer.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs MP, White DA, Kaushal S. Biphasic photoreceptor degeneration induced by light in a T17M rhodopsin mouse model of cone bystander damage. Invest Ophthalmol Vis Sci. 2009;50:2956–2965. doi: 10.1167/iovs.08-3116. [DOI] [PubMed] [Google Scholar]
- Lee S, Varvel NH, Konerth ME, Xu G, Cardona AE, Ransohoff RM, Lamb BT. CX3CR1 deficiency alters microglial activation and reduces beta-amyloid deposition in two Alzheimer’s disease mouse models. Am J Pathol. 2010;177:2549–2562. doi: 10.2353/ajpath.2010.100265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang KJ, Lee JE, Wang YD, Ma W, Fontainhas AM, Fariss RN, Wong WT. Regulation of dynamic behavior of retinal microglia by CX3CR1 signaling. Invest Ophthalmol Vis Sci. 2009;50:4444–51. doi: 10.1167/iovs.08-3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luhmann UF, Robbie S, Munro PM, Barker SE, Duran Y, Luong V, Fitzke FW, Bainbridge JW, Ali RR, MacLaren RE. The drusen-like phenotype in aging Ccl2-knockout mice is caused by an accelerated accumulation of swollen autofluorescent subretinal macrophages. Invest Ophthalmol Vis Sci. 2009;50:5934–5943. doi: 10.1167/iovs.09-3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukiw WJ, Ottlecz A, Lambrou G, Grueninger M, Finley J, Thompson HW, Bazan NG. Coordinate activation of HIF-1 and NF-kappa B DNA binding and COX-2 and VEGF expression in retinal cells by hypoxia. Invest Ophthalmol Vis Sci. 2003;44:4163–4170. doi: 10.1167/iovs.02-0655. [DOI] [PubMed] [Google Scholar]
- Nakazawa T, Takeda M, Lewis GP, Cho KS, Jiao J, Wilhelmsson U, Fisher SK, Pekny M, Chen DF, Miller JW. Attenuated glial reactions and photoreceptor degeneration after retinal detachment in mice deficient in glial fibrillary acidic protein and vimentin. Invest Ophthalmol Vis Sci. 2007;48:2760–2768. doi: 10.1167/iovs.06-1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noell WK. There are different kinds of retinal light damage in the rat. In: Williams TP, Baker BN, editors. The effects of constant light on visual processes. Plenum Press; New York: 1980. pp. 3–28. [Google Scholar]
- Ramkumar HL, Zhang J, Chan CC. Retinal ultrastructure of murine models of dry age-related macular degeneration (AMD) Prog Retin Eye Res. 2010;29:169–190. doi: 10.1016/j.preteyeres.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raoul W, Auvynet C, Camelo S, Guillonneau X, Feumi C, Combadière C, Sennlaub F. CCL2/CCR2 and CX3CL1/CX3CR1 chemokine axes and their possible involvement in age-related macular degeneration. J Neuroinflammation. 2010;7:87–93. doi: 10.1186/1742-2094-7-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransohoff RM, Liu L, Cardona AE. Chemokines and chemokine receptors: multipurpose players in neuroinflammation. Int Rev Neurobiol. 2007;82:187–204. doi: 10.1016/S0074-7742(07)82010-1. [DOI] [PubMed] [Google Scholar]
- Ross RJ, Zhou M, Shen D, Fariss RN, Ding X, Bojanowski CM, Tuo J, Chan C-C. Immuological protein expression profile in Ccl2/Cx3cr1-deficient mice with lesions similar to age-related macular degeneration. Exp Eye Res. 2008;86:675–683. doi: 10.1016/j.exer.2008.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutar M, Natoli R, Valter K, Provis JM. Early focal expression of the chemokine Ccl2 by Müller cells during exposure to damage-inducing bright continuous light. Invest Ophthal Vis Sci. 2011;52:2379–2388. doi: 10.1167/iovs.10-6010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandström J, Heiduschka P, Beck SC, Philippar U, Seeliger MW, Schraermeyer U, Nordheim A. Degeneration of the mouse retina upon dysregulated activity of serum response factor. Mol Vis. 2011;17:1110–1127. [PMC free article] [PubMed] [Google Scholar]
- Tacke F, Alvarez D, Kaplan TJ, Jakubzick C, Spanbroek R, Llodra J, Garin A, Liu J, Mack M, van Rooijen N, Lira SA, Habenicht AJ, Randolph GJ. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J Clin Invest. 2007;117:185–194. doi: 10.1172/JCI28549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanito M, Kaidzu S, Ohira A, Anderson RE. Topography of retinal damage in light-exposed albino rats. Exp Eye Res. 2008;87:292–5. doi: 10.1016/j.exer.2008.06.002. [DOI] [PubMed] [Google Scholar]
- Tout S, Chan-Ling T, Holländer H, Stone J. The role of Müller cells in the formation of the blood-retinal barrier. Neurosci. 1993;55:291–301. doi: 10.1016/0306-4522(93)90473-s. [DOI] [PubMed] [Google Scholar]
- Tulvatana W, Adamian M, Berson EL, Dryja TP. Photoreceptor rosettes in autosomal dominant retinitis pigmentosa with reduced penetrance. Arch Ophthalmol. 1999;117:399–402. doi: 10.1001/archopht.117.3.399. [DOI] [PubMed] [Google Scholar]
- Tuo J, Bojanowski CM, Zhou M, Shen D, Ross RJ, Rosenberg KI, Cameron DJ, Yin C, Kowalak JA, Zhuang Z, Zhang K, Chan C-C. Murine Ccl2/Cx3cr1 deficiency results in retinal lesions mimicking human age-related macular degeneration. Invest Ophthalmol Vis Sci. 2007;48:3827–3836. doi: 10.1167/iovs.07-0051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Lam TT, Tso MO, Naash MI. Expression of a mutant opsin gene increases the susceptibility of the retina to light damage. Vis Neurosci. 1997;14:55–62. doi: 10.1017/s0952523800008750. [DOI] [PubMed] [Google Scholar]
- Wang J, Xu X, Elliott MH, Zhu M, Le YZ. Müller cell-derived VEGF is essential for diabetes-induced retinal inflammation and vascular leakage. Diabetes. 2010;59:2297–305. doi: 10.2337/db09-1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel A, von Lintig J, Oberhauser V, Tanimoto N, Grimm C, Seeliger MW. RPE65 is essential for the function of cone photoreceptors in NRL-deficient mice. Invest Ophthalmol Vis Sci. 2007;48:534–42. doi: 10.1167/iovs.06-0652. [DOI] [PubMed] [Google Scholar]
- West EL, Pearson RA, Tschernutter M, Sowden JC, MacLaren RE, Ali RR. Pharmacological disruption of the outer limiting membrane leads to increased retinal integration of transplanted photoreceptor precursors. Exp Eye Res. 2008;86:601–611. doi: 10.1016/j.exer.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams TP, Howell WL. Action spectrum of retinal light-damage in albino rats. Invest Ophthalmol Vis Sci. 1983;24:285–287. [PubMed] [Google Scholar]
- Yannuzzi LA, Negrão S, Iida T, Carvalho C, Rodriguez-Coleman H, Slakter J, Freund KB, Sorenson J, Orlock D, Borodoker N. Retinal angiomatous proliferation in agelated macular degeneration. Retina. 2001;21:416–434. doi: 10.1097/00006982-200110000-00003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Schematic 3D SD-OCT data illustrates composition of the en face transform. SD-OCT measures reflectance along its optical axis and presents the intensity and axial depth of the reflectance as a column of pixels referred to as the A-scan (blue outlined vertical column of black and white rectangles). A-scans are collected at a rate of 40,000 per second along a designated path and the combination of these A-scans creates a composite known as the B-scan (gray planes). Sequential B-scan captures produce a 3D SD-OCT data set that we analyze in ImageJ using our “Open Heyex Raw” plugin. Re-slicing the 3D data set orthogonal to both the A-scan and B-scan presents OCT data in a view angle analogous to the cSLO view, termed en face OCT (green plane).
Figure S2. Retinal thickness assessment by OCT B-scans quantifies lesion contribution and degeneration in 2-, 4-, and 8-month-old Ccl2−/−/Cx3cr1−/− mouse retinas. Normally, total retinal thickness is composed of approximately equal parts inner and outer retina. Outer retinal thickness (top edge of red shaded regions) was calculated from the difference between total (solid black lines) and inner (dashed black lines) retinal thickness measurements. Healthy outer retina was also measured (blue shaded regions). The difference between calculated and healthy outer retinal thicknesses represents lesion areas (red shaded regions). At 2 months, despite some lesion area, the inner–outer thickness composition remains normal in superior retina while inferior exhibits significant lesion area and reduced outer retinal thickness (A). By 4 months, superior retina is unchanged but severe degeneration of outer retina is evident beyond 500 μm inferior to the optic nerve (B). Normal superior outer retinal thickness persists at 8 months, while severe degeneration continues in inferior retina (C). Notably, little lesion area is discernible and inner retina is the predominate source of total retinal thickness. At this point, inner retina is no longer stable across the profile but shows signs of alterations and degeneration as the inferior distance to optic nerve increases. Integrated outer retinal area reveals minor lesion area and little change over time on superior retina with age (D). In the inferior retina, lesions compose 40–50% of outer retina at 2 and 3 months (E). Lesion contribution declines with age becoming insubstantial by 8 months.