

Abstract
The mechanistic basis of the high toxicity to lung macrophages of coarse PM from the California wildfires of 2008 was examined in cell culture experiments with mouse macrophages. Wildfire PM directly killed macrophages very rapidly in cell culture at relatively low doses. The wildfire coarse PM are about four times more toxic to macrophages on an equal weight basis than the same sized PM collected from normal ambient air (no wildfires) from the same region and season. There was a good correlation between the extent of cytotoxicity and the amount of oxidative stress observed at a given dose of wildfire PM in vitro. Our data implicate NF-kB signaling in the response of macrophages to wildfire PM, and suggest that most, if not all, of the cytotoxicity of wildfire PM to lung macrophages is the result of oxidative stress. The relative ratio of toxicity and of expression of biomarkers of oxidant stress between wildfire PM and “normal” PM collected from ambient air is consistent with our previous results in mice in vivo, also suggesting that most, if not all, of the cytotoxicity of wildfire PM to lung macrophages is the result of oxidative stress. Our findings from this and earlier studies suggest that the active components of coarse PM from the wildfire are heat-labile organic compounds. While we can not rule out a minor role for endotoxin in coarse PM preparations from the collected wildfire PM in our observed results both in vitro and in vivo, based on experiments using the inhibitor Polymyxin B most of the oxidant stress and pro-inflammatory activity observed was not due to endotoxin.
Keywords: Air Pollution, Coarse PM, Oxidative stress, cytotoxicity
INTRODUCTION
Respirable particulate matter (PM) is one of the air pollutants regulated under the National Ambient Air Quality Standards by the United States Environmental Protection Agency. Current standards focus on the concentration and size of the PM in the atmosphere. Source attribution of PM and the chemical composition of PM in the various size classes and from different sources is an area of active investigation in many laboratories worldwide. In addition to the major anthropogenic sources of PM, especially automobile emissions and combustion of fossil fuels for power generation, there are significant natural sources of PM. One such natural source worldwide is wildfires, which can give rise to high levels of PM emissions from products of incomplete combustion of trees, grasses, and other organic sources. The PM emissions from wildfires can travel long distances and their components can be subject to photochemical oxidation, especially in the sunny and arid conditions characteristic of California and much of the western United States in the late spring, summer, and early autumn months when most of these wildfires occur. In 2008, California was profoundly affected by a series of large wildfires that occurred over a large part of the forested areas of the state (Wegesser et al., 2009). The PM from this wildfire episode, especially the coarse PM fraction (PM10-2.5), was unusually toxic in mouse bioassays as compared to PM collected at different times from the same geographical area in the absence of wildfire activity (Wegesser et al., 2009, 2010).
We have studied the basis of this enhanced toxicity in wildfire-derived PM and have demonstrated that intratracheally-instilled wildfire PM decreases the number of macrophages recovered from mice by lung lavage at PM doses that realistically approximate total doses that might have been inhaled by populations exposed to the emissions from the wildfires of 2008 (Wegesser et al., 2009). Based upon experiments that directly compared the response of mouse lungs to instillation of coarse PM from the wildfire episode to the response of mouse lungs to comparable sized PM collected from ambient air from the same area during the same season the previous year (when there was no significant wildfire activity), we were able to conclude that the wildfire PM was 3- to 4-times more toxic to mouse lungs at a dose of 50 ug than was the “normal” PM from the region (Wegesser et al., 2009).
The concept of wood smoke or wildfire-derived PM being able to specifically kill lung macrophages has been suggested previously. Toxicological and epidemiological health effects of wood smoke were reviewed by Naeher et al. (2007), who concluded that many of the reported effects could be the consequence of wood smoke-specific toxicity to lung macrophages. A suggested mechanism to explain these toxic effects of the inhaled wood smoke-derived PM was oxidative stress arising from interactions of PM components with lung macrophages. More generally, Li et al. (2003) have linked oxidant stress induced by exposure to particulate air pollutants to lung disease, as have many other research groups. A plausible mechanism linking oxidative stress to redox cycling by semiquinones derived from polycyclic organic compounds in combustion-derived PM was suggested by Pryor and colleagues (2001).
There is an extensive literature published on the effects of various kinds of PM on cultured cells of lung origin that has attempted to explore mechanistic aspects of cell injury, cell repair, and oxidative stress induced by PM exposure, and the mechanistic basis for the acute inflammatory response of the lung to exposure to PM. Many of these studies have focused on the response of either cultured airway epithelial cells or pulmonary alveolar macrophages to PM exposure in vitro. For example, Chirino et al. (2010) recently reported that PM10 collected from ambient air in Mexico City induced oxidative damage and biomarkers of oxidative stress in A549 cells (a human lung epithelial cell line). Among the biomarkers was a 55% decrease in GSH levels and decreases in several antioxidant enzymes. There were no observed changes in cell viability. These authors used doses of 100 ug of PM to 1,000,000 cells in these experiments. Danielson et al. (2011) reported that cultured lung epithelial and monocytic cells produced high levels of free radicals upon exposure to wood smoke-derived PM.
The current study was designed to examine the mechanisms of the unusual toxicity to lung macrophages of the coarse PM from the wildfire of 2008 that we observed in our mouse bioassays (Wegesser et al., 2009) using cell culture experiments with the RAW 264.7 established cell line of mouse macrophages. Our studies were designed to answer the following three questions. (1) Does exposure to coarse PM from the 2008 California wildfires directly kill macrophages in vitro at doses that are reasonable approximations to the doses that we used for our in vivo experiments in mice? (2) Is the relative ratio of toxicity and the expression of biomarkers of oxidant stress for cultured macrophages exposed to wildfire PM and “normal” PM consistent with our previous results in vivo in mice? (3) What is the mechanism by which wildfire PM kills macrophages? Can we infer what critical intracellular signaling pathways are responsible for the greater response of macrophages to wildfire PM as compared to “normal” PM?
MATERIALS AND METHODS
Particle collection and sources
Particulate matter (PM) used in these experiments was collected with a high-volume air sampler equipped with a cascade impactor as previously described (Wegesser and Last, 2008). Coarse PM (10.2-2.1 μm MMAD) samples were collected from the cascade impactor substrate, scraped into Eppendorf tubes, weighed, and stored either dry or in suspension at 1 or 2 mg/mL in water or in phoshpate-buffered saline at −20°C until used. Ambient air coarse particles collected one year later from the same region with the same sampler operated under the same conditions and flow rate was used as a control. The wildfire PM sample was collected in June of 2008 from a rural area in the San Joaquin Valley near the town of Escalon (Wegesser et al., 2009). The control (“Fresno”) sample was collected in the city of Fresno, to the southwest of Escalon in the San Joaquin Valley, in July of 2009. This collection site has been described elsewhere (den Hartigh et al., 2010). The chemical composition of the Fresno PM is dominated by automotive emissions (den Hartigh et al., 2010).
Cell Culture
Murine macrophage RAW 264.7 cells (American Type Culture Collection, Manassas, Va), an established cell line, were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% Fetal Bovine Serum at 37°C in a 5% CO2 humidified chamber. All experiments were conducted with cells between passages 6–10 to sustain uniformity of the cells throughout the course of the project, and n=3 replicate wells on each plate for all of the doses or time points tested in all of the reported experiments.
Treatment of cultured macrophages
RAW 264.7 cells were grown in semi-suspension on 100mm non-tissue culture treated petri dishes. Once cells reached approximately 80% of the total cell number attainable under these culture conditions, they were counted (adherent cells + non-adherent cells in the medium) and diluted to 5 × 105 cells in a volume of 1 mL of medium containing Penicillin plus Streptomycin and Normocin™ (100μg/mL), which was put into each well of a 12-well tissue culture plate. After 30 minutes of incubation at 37°C to allow the cells to attach to the well, for time course experiments either 10μg of PM (1mg/ml), or 10μl of sterile Phosphate Buffered Saline (PBS) was added to each well. The cells were incubated for the indicated times between 30 minutes and 24 hours, with samples taken at 30 minutes, 1 hour, 2 hours, 4 hours, 6 hours, and/or 24 hours as indicated in the figure legends. For dose-response experiments cells were incubated for 24 hours with the indicated amount of wildfire or Fresno PM added. At the time of collection cells were scraped from the culture well into the culture medium with a plastic policeman and the combined adherent and non-adherent cells were assessed for viability using a Trypan Blue exclusion assay (Bratt et al., 2009).
Determination of NF-kB activation
RAW-Blue™ cells (InvivoGen, San Diego, CA), a cell line derived from RAW 264.7 macrophages containing an NF-κB-driven reporter gene (Bruschi et al., 2010), were used to determine activation of NF-κB by measurement of the expression of secreted embryonic alkaline phosphatase (SEAP). In the presence of specific agonists (in this case PM10-2.1), these cells are induced to activate signaling pathways that lead to NF-κB activation. Once activated these cells secrete alkaline phosphatase, which is detectable and measurable by the use of QUANTI-Blue™ (InvivoGen, San Diego, CA) SEAP detection medium (Bruschi et al., 2010). To detect NF-κB activation, RAW-Blue™ cells were grown in semi-suspension in 100mm non-tissue culture treated petri dishes until they reached approximately 80% confluence, then the cells were counted and resuspended in Growth Medium (DMEM, 4.5 g/l glucose, 10% heat-inactivated fetal bovine serum, 100μg/ml Normocin™, 2mM L-glutamine). Twenty μl of PM10-2.1 at various doses (prepared by dilution into PBS of 1–2mg/ml stock solutions of frozen PM) was added to each well of a flat-bottomed 96-well plate. Endotoxin-free water, PBS, and DMSO (0.1ng) were tested as negative controls. 100,000 cells in a total volume of 1 mL of medium were added per well and incubated at 37°C for 24hrs. The following day, QUANTI-Blue™ was added to the supernatants prepared from the induced RAW-Blue™ cells. The plate was finally incubated for 30 minutes at 37°C and the alkaline phosphatase levels were determined using a spectrophotometer at a wavelength of 650nm.
Polymyxin B experiments
We determined what, if any, role was being played by lipopolysaccharide (LPS), the soluble fraction of endotoxin arising from the cell walls of Gram-negative bacteria, in the responses of RAW 264.7 macrophages to the wildfire PM. To do this, we treated RAW-Blue™ cells with polymyxin B, an antibiotic that specifically binds LPS and prevents its binding to TLR4 receptors on the macrophages, using the methods described by Shoenfelt et al. (2009). Briefly, Polymyxin B was added to the PM suspension at a concentration of 10 ug/mL and the resulting mixture was pre-incubated for 30 minutes with the LPS-binding agent prior to addition of PM to the cells during their 24 hours of incubation with the particles.
Experimental design
Preliminary range finding experiments were performed with both wildfire and ambient air PM to determine the linear portion of the dose-response curves over a wide range of doses. Subsequent experiments were designed to compare the response to different PM preparations from the slopes of the linear portion of the dose-response curve for that particular PM source. Thus, the actual PM doses used for our comparative studies of wildfire and ambient air PM differed, as may be noted by examination of the scales in each of the figures.
Statistical analysis of data
Prism 4.0 (GraphPad Software, San Diego, CA) was used for data analysis. Values are expressed as mean values ±SE (standard error). Data were analyzed by ANOVA, followed by post-test with Tukey’s honestly significant difference test to correct for multiple comparisons. Linear regression analysis was performed using a 95% confidence interval. Differences between experimental groups were considered significant if p values were found to be <0.05.
RESULTS
Toxicity of wildfire PM to cultured macrophage cells
Dose-response curves for cell killing in vitro by different PM preparations
We exposed RAW 264.7 macrophages to various doses of either the wildfire PM or ambient air (Fresno) PM for 24 hours and evaluated the number of live and dead cells present as a percentage of the total cells counted. We found a good dose-response relationship for both the wildfire and ambient air PM (Figures 1A and 1B). A linear dose response curve described the results, with r2 values of 0.44 and 0.73, respectively (Figure 2). Linear regression analysis gave a slope for the wildfire PM of 1.87± 0.67 versus a slope for the ambient air PM of 0.37± 0.09 for the ambient air PM. The difference between these slopes was significant (P = 0.03). The ratio of the slopes, 1.87/0.37 = 5.05, is a measure of the relative toxicity of the two PM preparations. Thus, the wildfire PM is about 5 times more toxic to the macrophages in vitro than is the PM preparation collected from normal ambient air from the same region one year previously.
Figure 1. Dose-response relationships for RAW 264.7 macrophage killing by wildfire and ambient air PM.
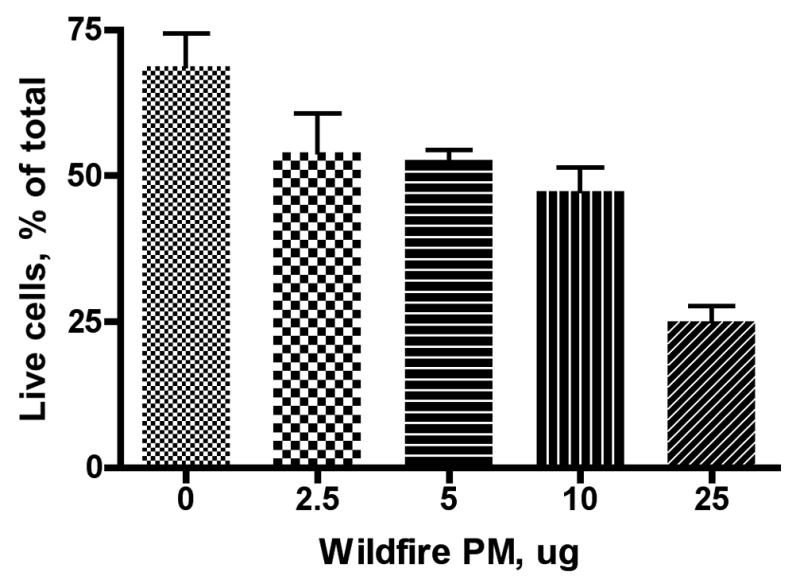

Cells (5 × 105 cells/mL) were seeded into 12-well tissue culture plates. After 30 minutes cells were treated with the indicated dose of either wildfire PM or PM collected from ambient air during a time when there were no regional wildfires or 10μl of sterile Phosphate Buffered Saline (PBS), then incubated for 24 hours. Results are presented as mean values ± SE. Three replicate wells were used for each condition tested (i.e. n=3) in this and subsequent figures. Cells were suspended by scraping and assessed for viability using Trypan Blue. A: Wildfire PM; B: Ambient air PM collected near Fresno, CA in the absence of regional wildfires. Note difference in scale on the X-axis (dose).
Figure 2. Linear regression analysis of dose-response relationships for RAW 264.7 macrophage killing by wildfire and ambient air PM.
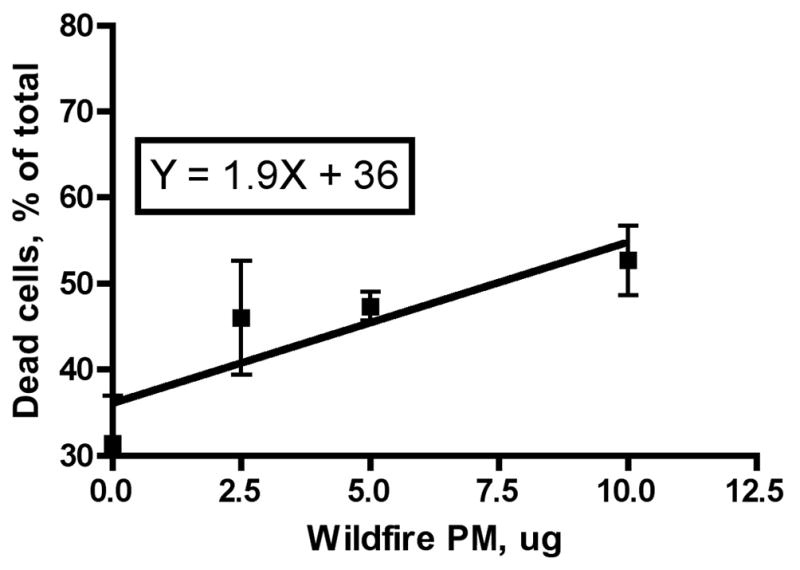
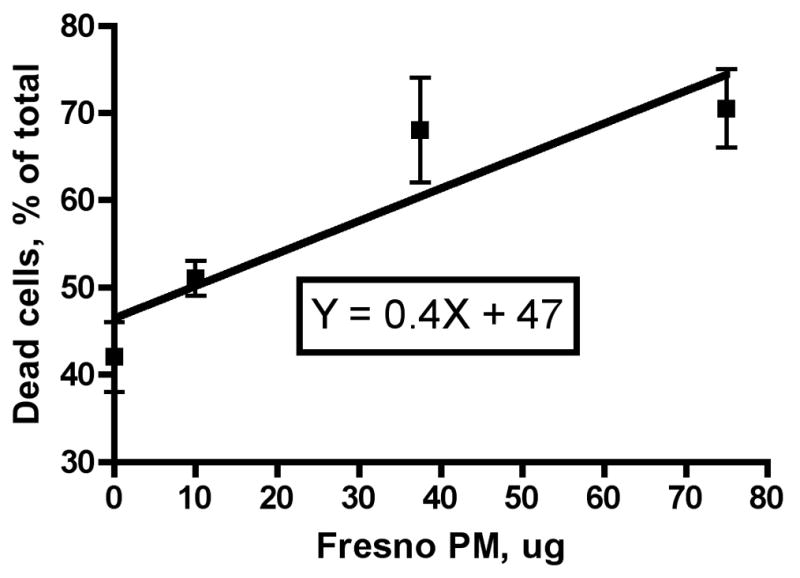
The individual results from the experiments described in Figure 1 were used to analyze the data by linear regression analysis. A: Wildfire PM, B: Ambient air PM collected near Fresno, CA in the absence of regional wildfires. Note difference in scale on the X-axis (dose).
Time course of wildfire PM toxicity in vitro
Cells were plated at 0 time, allowed 30 minutes to attach to the plastic substratum, then incubated with either PM or PBS (controls) for time periods from 0.5–24 hours. We observed rapid killing of the cells by the PM preparations; maximum cell death occurred within 0.5 hours of exposure to the PM (Figure 3). Presumably, proliferation of surviving cells caused the percentage of live cells to increase at the subsequent time points analyzed. At 24 hours after addition of PM the relative numbers of dead cells present were the same in both control and wildfire PM-treated cultures, so the PM-treated cells must have proliferated faster (the ratio of the slopes of the lines, −2.1/−1.4, suggests that the PM-treated cells were replicating 50% faster than the controls) after the initial phagocytosis of PM and death of the cells in these experiments.
Figure 3. Time course of RAW 264.7 macrophage killing by wildfire PM.
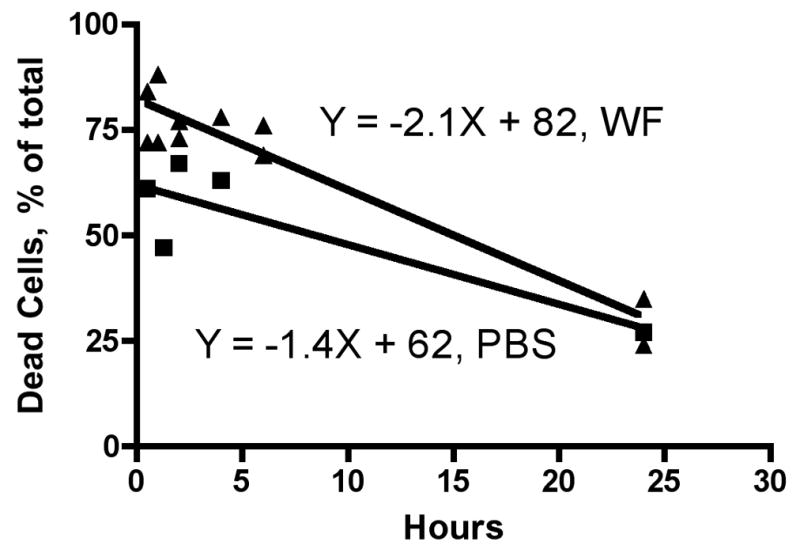
Cells were grown to approximately 80% confluence in semi-suspension in 100mm petri dishes, counted, and diluted to 5 × 105 cells/mL into 12-well tissue culture plates with Penicillin/Streptomycin and Normocin™ (100μg/ml). After 30 minutes cells were treated with either 10μg of PM (WF, 1mg/ml), or 10μl of sterile Phosphate Buffered Saline (PBS), then incubated for the indicated times. Cells were suspended by scraping and assessed for viability using Trypan Blue.
Mechanism of wildfire PM-induced macrophage cell death
QUANTI-Blue™ experiments to quantify NF-kB activation
Dose-response curves were run for wildfire PM and for PM isolated from ambient air the previous year using the same sampler and cascade impactor from a nearby area during a time of no wildfire activity. Comparison of the slopes of the dose-response curves obtained by linear regression analysis, as shown in Figure 4, clearly demonstrates that the wildfire PM more strongly activates NF-kB than does the ambient air PM (P<0.0001). The slopes of the different lines obtained with the two sources of PM were 0.23 ± 0.03 (r2=0.83) and 0.056 ± 0.002 (r2=0.96) for the wildfire PM and the ambient air PM, respectively. The ratio of these slopes is 0.23/0.056 = 4.3.
Figure 4. Stimulation of expression of an NF-kB reporter gene by wildfire PM or normal ambient air-derived PM as a function of PM dose.
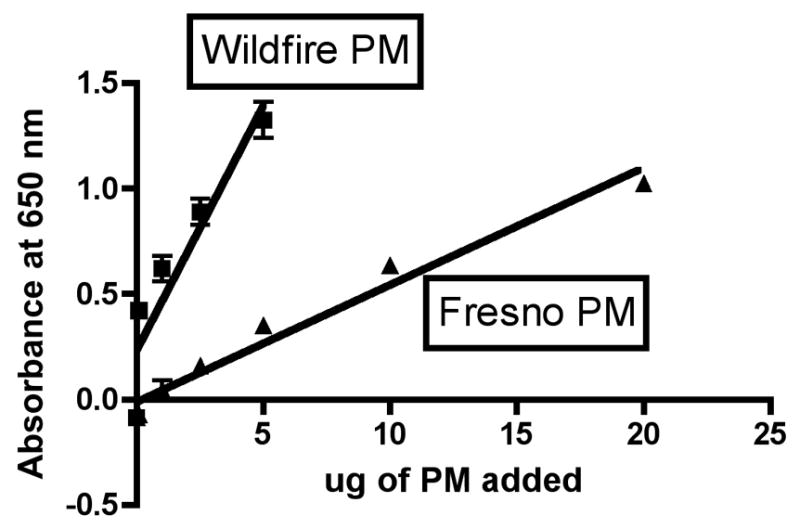
20μl of wildfire PM10-2.1 at the indicated doses was added to each well of a flat-bottom 96-well plate, including endotoxin-free water, PBS and DMSO (0.1ng) as matched controls. 100,000 RAW-Blue™ cells were added per well and incubated at 37°C for 24hrs. The following day, QUANTI-Blue™ was added to the supernatant after removal of cells by centrifugation. The plate was incubated for 30 minutes and alkaline phosphatase concentration was determined by measurement of absorbance at 650nm.
The same linear regression analysis allows us to ask another question: Is the slope of the dose response curve significantly different from zero? For both the wildfire PM and the ambient air PM collected when no wildfires were occurring, the answer is yes, with a P value <0.0001 for both PM types. Thus, we can conclude that the ambient air PM also causes oxidative stress and stimulates an NF-kB-driven reporter gene in this macrophage cell line, albeit more weakly than wildfire PM at a comparable dose. These results are also correlated with the dose-response curves observed for cell death with these same PM preparations (Figures 1A and 1B), where linear regression analysis also indicated slopes significantly different from zero (P = 0.0194 and 0.0066 for wildfire and Fresno PM, respectively). Thus, the amount of stimulation of an NF-kB-driven reporter gene and the extent of macrophage cell death as a function of PM dose are correlated in this series of experiments.
Is endotoxin the active component in wildfire PM?
LPS is known to act by binding to TLR4 receptors on the surface of macrophages and thereby turning on a signaling system (via MyD88) that results in activation of NF-kB. To determine whether endotoxin (as LPS) was playing a role in the activation of NF-kB in the QuantiBlue cells treated with wildfire PM, we incubated the wildfire PM with Polymyxin B to extract and complex any LPS present in the particles before assay with the QuantiBlue cells. The results of this set of experiments are shown in Figure 5. The slope of the dose-response curve for the RAW 264.7 macrophages with no added Polymyxin B was 0.08 ± 0.00 (r2= 0.93), while the comparable slope with Polymyxin B pretreatment was 0.06 ± 0.00 (r2=0.95). These slopes were significantly different (P = 0.0068); however, the maximum possible relative contribution from LPS seems to be about 0.08/0.06 = 133% of the difference in activity between wildfire PM ± Polymyxin B. The relative contribution from LPS seems to be no more than about 15% of the difference in toxicity observed between the wildfire PM and other sources of coarse PM isolated from ambient air. For example, in this experiment we also performed a dose-response analysis with the Fresno PM analyzed above in parallel with the wildfire PM. Thus the same batch of cells, the same reagents, and the same 36-well plate were used for both sources of PM ± Polymyxin B. There was no significant difference between the slopes of the lines obtained by linear regression analysis of the dose-response curves for Fresno PM ± Polymyxin B (P = 0.1862). We found a slope for the Fresno PM with no added Polymyxin B of 0.025 ± 0.003 (r2 = 0.85). The ratio of the slopes for wildfire/Fresno PM was 0.08/0.025 = 3.2, so we can conclude that there was 320% additional NF-kB activation activity in the wildfire PM as compared to the Fresno PM.
Figure 5. Effect of Polymyxin B on wildfire induced expression of NF-kB by cultured RAW 264.7 macrophages.
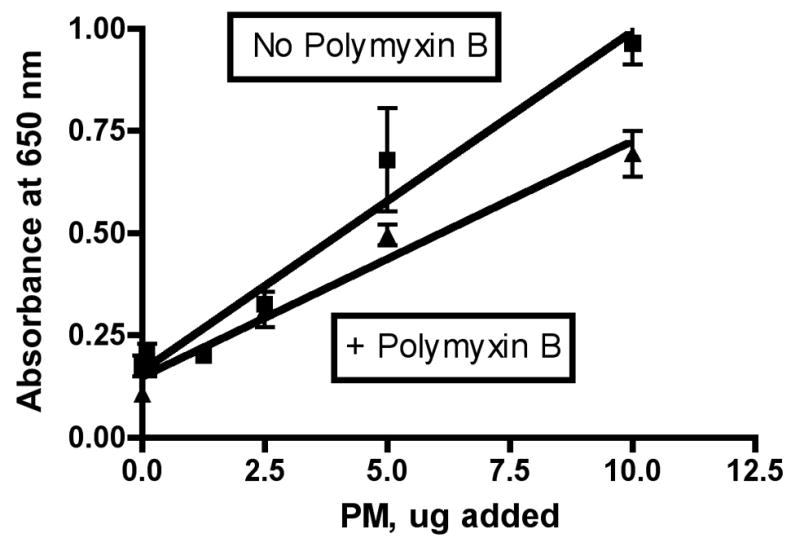
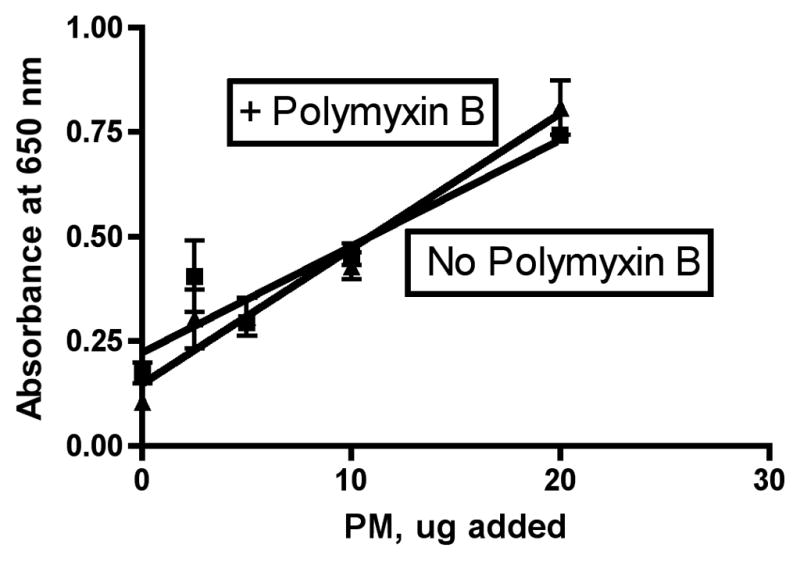
Experiments were performed as described in the legend to Figure 4. Polymyxin B was added to the PM suspension at a concentration of 10 ug/mL and the resulting mixture was pre-incubated for 30 minutes with the antibiotic (LPS binding agent) prior to addition to the cells where indicated. A, wildfire PM; B, Fresno PM.
DISCUSSION
We asked three questions at the start of this study. Our current results allow us to directly address the answers to all of these questions.
(1) Does exposure to coarse PM from the 2008 California wildfires directly kill cultured macrophages at doses that are reasonable approximations to the doses that we used for our previously published in vivo experiments in mice (Wegesser and Last, 2009)?
Our current studies have clearly demonstrated the rapid and effective killing of macrophages in vitro by wildfire PM (Figs. 1 and 2). This is an important new result insofar as it establishes that the wildfire PM itself is adequate and sufficient to kill macrophages in cell culture. In vivo experiments do not allow us to rule out the putative contribution of other resident lung or inflammatory cells or of agents like cytokines, chemokines, or oxidants produced by lung cells or inflammatory cells other than macrophages in the mechanism of toxicity.
There are at least three other studies we have found in the literature that report the effects of coarse PM from wood or wildfire smoke on cultured RAW 264.7 mouse macrophages. Kubatova et al. (2006) observed a depletion of reduced cellular glutathione (GSH) of 58% in 180,000 cells (initial plating density) exposed to the combined aqueous extracts of the equivalent dose to 100 ug/mL of PM from combustion of wood in a fireplace stove; in the same study, no significant cytotoxicity (as measured by LDH release) was observed at PM equivalent doses below 200 ug of PM. Leonard et al. (2007) reported that there was no significant increase of H2O2 production or lipid peroxidation in 5,000,000 or 50,000,000 RAW 264.7 cells, respectively, at a dose of 100 ug/mL of coarse PM collected from wildfire smoke. Significant increases of both parameters were observed with equivalent doses of fine or ultrafine PM in the same experiments, suggesting the biological relevance of the reported negative observations with coarse PM. Jalava et al. (2006) found significantly increased cytotoxicity (decreased viability by MTT test) to 50,000 cells per well of RAW 264.7 macrophages exposed to 15–300 ug of coarse wildfire PM, as compared to a reagent blank or in comparison to PM isolated from normal ambient air. In the same studies the coarse PM induced apoptotic cell death in 4–7% of the cells, with no difference between wildfire and ambient air PM reported.
It is difficult to answer the question of comparative toxicity in vivo and in vitro without having to make several assumptions because the mode and medium of exposure is so different in vitro and in vivo. For example, let us assume that the appropriate exposure metric for such a comparison is weight of PM/macrophage cell exposed. This is an easy number to determine for the cell culture experiments where we exposed 500,000 cells per tissue culture well to known doses of wildfire PM. This is not an easy number to determine for the intratracheal instillation studies. One simple way to try to do this is to assume that the population of macrophages being exposed to the instilled PM is the same population that we can freely lavage from the lungs. The average number of macrophages we recover from a normal mouse after lung lavage is about 125,000–150,000 cells. From the data in Figure 2B of Wegesser and Last (2009), we can see that we get significantly increased killing of macrophages in vivo at a dose of 25 ug/125,000 cells. This would scale to a dose of 125,000/500,000 × 25 ug, or ~6 ug in vitro. As shown in Figure 1A, our results are consistent with the wildfire PM being able to kill macrophages at comparable doses of PM to this amount (~6 ug) in vitro.
We also calculated the potential 24-hour dose of wildfire PM a mouse would have received at the point of collection of our wildfire samples, based on the typical ventilation rate of a mouse, the expected fractional deposition of coarse PM in the lung, and the observed peak concentration of coarse PM of 381 ug/M3 during our collection site of the wildfire PM (Wegesser et al., 2009). That concentration would have given a total dose to a hypothetical mouse at the site of about 20 ug of PM.
(2) Is the relative ratio of toxicity between wildfire PM and “normal” PM to cultured macrophages consistent with our previous results in mice?
The answer to this question is yes. In our previous studies we reported that the number of pulmonary macrophages in the lung lavage fluid prepared from mice instilled with coarse PM from “normal” ambient air was about 3- to 4-fold higher than the number of macrophages found in lavage fluid from mice instilled with the same dose (50 ug) of wildfire PM (Figure 2B, Wegesser et al., 2009). The actual number of macrophages counted in the lavage fluid in this in vivo experiment, which may be hard to see from the cited figure, were as follows: 50 ug of wildfire PM = 70,604 ± 15,067 (N=6); 50 ug of PM from ambient air collected the previous year = 224,845 ± 49,437 (N = 6); ratio (comparative toxicity) = 3.2. We concluded in our earlier paper, “wildfire PM10–2.1 may be especially toxic to pulmonary alveolar macrophages or that these wildfire PM10–2.1 create a condition in which macrophages are difficult to extract from the lungs by lavage, perhaps because of enhanced adherence to alveolar surfaces.” Based upon our results in the present study, that the wildfire PM are 4-fold more toxic to cultured pulmonary macrophages in vitro than are comparable PM from “normal” ambient air, we can now conclude that the wildfire PM are indeed more toxic than PM normally present in the ambient air in this region, rather than the macrophages being inherently stickier after exposure to wildfire PM. It is striking that we see the same ratio, about 4, for the difference between the wildfire PM and the ambient air PM in terms of inflammatory activity in vivo, macrophage killing in vivo, macrophage killing in vitro, and stimulation of an NF-kB-driven reporter gene in vitro. However, we would not interpret this finding to be indicative that all of the observed effects in vivo are solely attributable to outcomes that are macrophage-based.
While the apparent consistency between our previous results in vivo and the current results in vitro could have occurred by chance, these results strongly support the hypothesis that the higher toxicity in mice of wildfire PM than “normal” PM collected from ambient air the previous year from the same region on about the same date is related to the striking ability of the wildfire PM to kill pulmonary macrophages at lower doses than “normal” PM. The property of increased oxidative stress and enhanced toxicity to macrophages of wildfire PM as compared to ambient air PM at the same dose may have clinical significance. There are reports in the epidemiological literature of increased exacerbations of asthma and an increase in emergency room visits during other California wildfire episodes, as compared to the same hospitals on similar dates without wildfire activity, including a large series of wildfires in Southern California in October of 2003 (Delfino et al., 2009; Kunzli et al., 2006; Viswathan et al., 2006), the severe wildfire of 1999 on the Hoopa Indian Reservation in Humboldt County in northwest California (Lee et al., 2009), a series of wildfires in 1987 in Northern California (Duclos et al., 1990), and an urban wildfire in 1991 in the Oakland Hills between Oakland and Berkeley (Shusterman et al., 1993).
We might also ask whether the doses of PM we used in the current cell culture experiments bear any reasonable relationship to doses of PM that a mouse might inhale in a whole body exposure chamber under reasonable conditions of inhalation exposure. If we assume a mouse inhales a minute volume that is approximately ten times less than a rat with fifteen times its body weight, we can calculate that the mouse inhales about 50 ug/hour when exposed to a concentration of coal fly ash of about 4.2 mg/M3 (Raabe et al., 1981). The concentrations we used for the current group of in vitro experiments, 2.5–75 ug of PM, are certainly comparable given the assumptions made. We can also attempt to compare the dose of PM that we used in these experiments with doses of PM previously used by other investigators for studying PM toxicity in vitro. This turns out to be surprisingly difficult due to the various dose metrics used in different studies and the lack of essential information to allow for ready inter-conversion of different dose metrics such as particle mass, mass per unit of surface area, particle concentration, etc. However, we can conclude that our experiments are clearly being performed at the low end of the typical dose ranges used by investigators exposing cultured cells to PM10-2.5 in vitro in the previous literature, as would be predicted with wildfire PM that is significantly more toxic than PM isolated from normal ambient air that is not influenced by emissions from wildfires.
It is noteworthy that the death of the macrophages in lungs of mice exposed to wildfire PM is probably not instantaneous, as adequate time seems to exist for signals to be released that result in recruitment of large numbers of neutrophils to the lungs of exposed mice (Wegesser et al., 2009). In this respect the wildfire PM seem to elicit similar results as intratracheally instilled quartz particles in mice, which also efficiently kill pulmonary macrophages while causing a major influx of neutrophils as a component of the lung’s inflammatory response (Adamson and Bowden, 1984).
(3) What is the mechanism by which wildfire PM kills macrophages? Can we infer what critical intracellular signaling pathways are responsible for the greater response of macrophages to wildfire PM as compared to “normal” PM at comparable doses?
The answer to the question of the mechanism of macrophage cytotoxicity upon exposure to wildfire PM seems to be oxidant stress to the macrophage cells in the airways, and this answer seems to be unequivocal. We have previously reported depletion of total antioxidants in the supernatant of the lung lavage fluid obtained from mice instilled intratracheally with wildfire PM (Wegesser et al., 2010). In the current study we have demonstrated activation of NF-kB in macrophages exposed to the same wildfire PM in vitro. We have shown (Fig. 5) that Polymyxin B, which binds lipopolysaccharide (LPS) the soluble active component of endotoxin and prevents its binding to TLR4 receptors, that (at least most of the) activation of NF-kB by wildfire PM10-2.1 is not due to endotoxin. However, previous studies with PM10-2.5 isolated from ambient air have suggested that the effects of coarse PM may be mediated by recognition of the particles by TLR4 receptors on the macrophages independent of any endotoxin effect (Shoenfelt et al., 2009), and we have no reason to rule out the possibility that this suggested mechanism for the initial interaction of particles with macrophage TLR4 receptors could account for the effects of the wildfire PM in our studies.
The concept that PM causes oxidative stress to airway epithelial cells and to pulmonary macrophages has been suggested previously. For example, Jalava et al. (2006) reported cytotoxicity to cultured RAW 264.7 macrophages exposed to 15–300 ug/mL of coarse PM prepared from wildfire smoke. Danielsen et al. (2011) reported that PM from wood smoke (and ambient air) caused oxidative stress, DNA damage, and inflammatory cytokine production in cultured epithelial and monocyte cell lines. What is novel in the current studies is the concept that the oxidative stress produced by wildfire PM is causally linked to the killing of macrophages at concentrations of PM that might reasonably occur in polluted air. Much of the existing literature links oxidative stress to inflammation (review: Mazzoli-Rocha et al., 2010). Oxidant stress-induced signaling pathways include NF-kB and other calcium ion-regulated pathways and protein kinase (e.g., MAPK) cascades, and AP-1 activation (Mazzoli-Rocha et al., 2010; Donaldson et al., 2003; Xia et al., 2007). The ultimate result of activation of these pathways on the cell would be complex, but would include activation of transcription of various genes including antioxidant enzymes, potential intracellular stress responses, and apoptotic cell death.
Based upon the existing literature, we can say with some confidence that activation of the NF-kB pathway is associated with induction of apoptosis. Therefore in preliminary experiments we examined whether wildfire PM killed macrophages in vivo by inducing apoptosis, presumably secondary to induction of oxidative stress in these target cells. Preliminary experiments using TUNEL staining with tissue sections taken from lavaged lungs of mice instilled intratracheally with wildfire PM10-2.1 6 or 24 hours previously showed the presence of apoptotic cells in cytospin preparations of lavaged cells (data not shown). These results are consistent with other studies of the effects of PM on cultured lung cells, both macrophages and epithelial cells (Li et al., 2003; Mazzoli-Rocha et al., 2010; Soberanes et al., 2009; Hetland et al., 2004; Chin et al., 1998; Obot et al., 2002).
What is the active component(s) in wildfire PM?
From the combined results of our previous studies and the current results we can summarize the following attributes of the active components of the wildfire PM. (1) The most active agent(s) are heat labile (130°C for 24 hours), and therefore are probably organic compounds. Due to the relatively small amount of wildfire PM we were able to collect, detailed chemical analysis of these particles has of necessity been very limited (Wegesser etal., 2010). (2) The relative contribution of endotoxin to the inflammatory or oxidant activity of wildfire PM, if any, is a minor (or negligible) component of the total activity present. This result stands in contrast to the findings of Guastadisegni et al. (2010), and suggests that differences in ambient air sample collection or handling techniques, especially relative humidity during collection of the PM samples, may be a factor in reported levels of endotoxin in coarse PM collected from various locations around the world. Guastadisegni et al. collected samples in The Netherlands, Germany, Sweden, and Italy, all from areas in which ambient relative humidity would be high, while our samples were collected in the Central Valley of California in late June, where relative humidity would be very low. (3) The inflammatory activity of the wildfire PM is associated with oxidant stress, which appears to be correlated with triggering the NF-kB signaling pathway in target cells. (4) The active agent(s) can, and do, kill alveolar macrophages at very low doses of PM in these experiments, suggesting the possibility that these results are relevant to environmental exposures. It is also worth noting that coarse PM (PM10-2.5) seem to be able to induce inflammation in cultured cells as strongly as fine PM (PM2.5-1) on an equal mass basis (Schwarze et al., 2007).
Highlights.
Coarse PM from wildfires is more cytotoxic to macrophages than “normal” coarse PM from ambient air at equivalent doses by mass.
Exposure of cultured macrophages containing a reporter gene to wildfire coarse PM demonstrates activation of the NF–kB pathway.
These studies demonstrate that oxidative stress is the major contributing mechanism to macrophage killing by wildfire coarse PM in vitro and in vivo.
Acknowledgments
We thank Dr. Renee Solis for her gift of the RAW 264.7 cells used in these studies. We also thank Dr. Angela Linderholm for her patient advice and guidance while we learned the proper methodology to grow these cells in our own laboratory. Dr. Teresa Wegesser collected the wildfire PM samples and Professor Kent Pinkerton generously supplied the Fresno PM preparation we used for these experiments. The research was supported in part by NIH grants UL1RR02416 and HL-076415. Dr. Jennifer Bratt and Keisha Williams were supported in part by NIH Training Grants ES-07058 and HL-07013, respectively, during these studies.
Footnotes
Conflict of Interest Statement: The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adamson IY, Bowden DH. Role of polymorphonuclear leukocytes in silica-induced pulmonary fibrosis. Am J Pathol. 1984;117:37–43. [PMC free article] [PubMed] [Google Scholar]
- Bratt JM, Franzi LM, Linderholm AL, Last MS, Kenyon NJ, Last JA. Arginase enzymes in isolated airways from normal and nitric oxide synthase 2-knockout mice exposed to ovalbumin. Toxicol Appl Pharmacol. 2009;234:273–280. doi: 10.1016/j.taap.2008.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruschi M, Pirri G, Giuliani A, Nicoletto SF, Baster I, Scorciapino MA, Casu M, Rinaldi AC. Synthesis, characterization, antimicrobial activity and LPS-interaction properties of SB041, a novel dendrimeric peptide with antimicrobial properties. Peptides. 2010;31:1459–1467. doi: 10.1016/j.peptides.2010.04.022. [DOI] [PubMed] [Google Scholar]
- Chin BY, Choi ME, Burdick MD, Strieter RM, Risby TH, Choi AM. Induction of apoptosis by particulate matter: role of TNF-alpha and MAPK. Am J Physiol. 1998;275:L942–949. doi: 10.1152/ajplung.1998.275.5.L942. [DOI] [PubMed] [Google Scholar]
- Chirino YI, Sánchez-Péreza Y, Osornio-Vargas AR, Morales-Bárcenasa R, Gutiérrez-Ruíz MC, Segura-García Y, Rosas I, Pedraza-Chaverrid J, García-Cuellar CM. PM10 impairs the antioxidant defense system and exacerbates oxidative stress driven cell death. Toxicol Lett. 2010;193:209–216. doi: 10.1016/j.toxlet.2010.01.009. [DOI] [PubMed] [Google Scholar]
- Danielsen PH, Møller P, Jensen KA, Sharma AK, Wallin H, Bossi R, Autrup H, Mølhave L, Ravanat JL, Bried JJ, de Kok TM, Loft S. Oxidative Stress, DNA Damage, and Inflammation Induced by Ambient Air and Wood Smoke Particulate Matter in Human A549 and THP-1 Cell Lines. Chem Res Toxicol. 2011;24:168–184. doi: 10.1021/tx100407m. [DOI] [PubMed] [Google Scholar]
- Delfino RJ, Brummel S, Wu J, Stern H, Ostro B, Lipsett M, Winer A, Street DH, Zhang L, Tjoa T, Gillen DL. The relationship of respiratory and cardiovascular hospital admissions to the southern California wildfires of 2003. Occup Environ Med. 2009;66:189–197. doi: 10.1136/oem.2008.041376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Hartigh LJ, Lamé MW, Ham W, Kleeman MJ, Tablin F, Wilson DW. Endotoxin and polycyclic aromatic hydrocarbons in ambient fine particulate matter from Fresno, California initiate human monocyte inflammatory responses mediated by reactive oxygen species. Toxicol In Vitro. 2010;24:1993–2002. doi: 10.1016/j.tiv.2010.08.017. [DOI] [PubMed] [Google Scholar]
- Donaldson K, Stone V, Borm PJA, Jimenez LA, Gilmour PS, Schins RPF, Knaapen ADM, Rahman I, Faux SP, Brown DM, Macnee W. Oxidative stress and calcium signaling in the adverse effects of environmental particles (PM10) Free Radical Biol Med. 2003;34:1369–1382. doi: 10.1016/s0891-5849(03)00150-3. [DOI] [PubMed] [Google Scholar]
- Duclos P, Sanderson LM, Lipsett M. The 1987 forest fire disaster in California: assessment of emergency room visits. Arch Environ Health. 1990;45:53–58. doi: 10.1080/00039896.1990.9935925. [DOI] [PubMed] [Google Scholar]
- Guastadisegni C, Kelly FJ, Cassee FR, Gerlofs-Nijland ME, Janssen NAH, Pozzi R, Brunekreef B, Sandström T, Mudway I. Determinants of the Proinflammatory Action of Ambient Particulate Matter in Immortalized Murine Macrophages. Environ Health Perspect. 2010;118:1728–1734. doi: 10.1289/ehp.1002105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetland RB, Cassee FR, Refsnes M, Schwarze PE, Låg M, Boere AJF, Dybing E. Release of inflammatory cytokines, cell toxicity and apoptosis in epithelial lung cells after exposure to ambient air particles of different size fractions. Toxicol in Vitro. 2004;18:203–212. doi: 10.1016/s0887-2333(03)00142-5. [DOI] [PubMed] [Google Scholar]
- Jalava PI, Salonen RO, Hälinen AI, Penttinen P, Pennanen AS, Sillanpää M, Sandell E, Hillamo R, Hirvonen MR. In vitro inflammatory and cytotoxic effects of size-segregated particulate samples collected during long-range transport of wildfire smoke to Helsinki. Toxicol Appl Pharmacol. 2006;215:341–353. doi: 10.1016/j.taap.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Kubatova A, Dronen LC, Picklo MJ, Hawthorne SB. Midpolarity and Nonpolar Wood Smoke Particulate Matter Fractions Deplete Glutathione in RAW 264.7 Macrophages. Chem Res Toxicol. 2006;19:255–261. doi: 10.1021/tx050172f. [DOI] [PubMed] [Google Scholar]
- Künzli N, Avol E, Wu J, Gauderman WJ, Rappaport E, Millstein J, Bennion J, McConnell R, Gilliland FD, Berhane K, Lurmann F, Winer A, Peters JM. Health effects of the 2003 Southern California wildfires on children. Am J Respir Crit Care Med. 2006;174:1221–1228. doi: 10.1164/rccm.200604-519OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TS, Falter K, Meyer P, Mott J, Gwynn C. Risk factors associated with clinic visits during the 1999 forest fires near the Hoopa Valley Indian Reservation, California, USA. Int J Environ Health Res. 2009;19:315–327. doi: 10.1080/09603120802712750. [DOI] [PubMed] [Google Scholar]
- Leonard SS, Castranova V, Chena BT, Schwegler-Berry D, Hoover M, Piacitelli C, Gaughan DM. Particle size-dependent radical generation from wildland fire smoke. Toxicology. 2007;236:103–113. doi: 10.1016/j.tox.2007.04.008. [DOI] [PubMed] [Google Scholar]
- Li N, Hao M, Phalen RF, Hinds WC, Nel AE. Particulate air pollutants and asthma: a paradigm for the role of oxidative stress in PM-induced adverse health effects. Clin Immunol. 2003;109:250–265. doi: 10.1016/j.clim.2003.08.006. [DOI] [PubMed] [Google Scholar]
- Mazzoli-Rocha F, Fernandes S, Einicker-Lamas M, Zin WA. Roles of oxidative stress in signaling and inflammation induced by particulate matter. Cell Biol Toxicol. 2010;26:481–498. doi: 10.1007/s10565-010-9158-2. [DOI] [PubMed] [Google Scholar]
- Naeher LP, Brauer M, Lipsett M, Zelikoff JT, Simpson CD, Koenig JQ, Smith KR. Woodsmoke Health Effects: A Review. Inhalat Toxicol. 2007;19:67–106. doi: 10.1080/08958370600985875. [DOI] [PubMed] [Google Scholar]
- Obot CJ, Morandi MT, Beebe TP, Hamilton RF, Holian A. Surface components of airborne particulate matter induce macrophage apoptosis through scavenger receptors. Toxicol Appl Pharmacol. 2002;184:98–106. [PubMed] [Google Scholar]
- Raabe OG, Tyler WS, Last JA, Schwartz LW, Lollini LO, Fisher GL, Wilson FD, Dungworth DL. Studies of the chronic inhalation of coal fly ash by rats. Ann Occup Hyg. 1981;26:189–211. [PubMed] [Google Scholar]
- Schwarze PE, Øvrevik J, Hetland RB, Becher R, Cassee FR, Lag M, Løvik ME, Dybing E, Refsnes M. Importance of Size and Composition of Particles for Effects on Cells In Vitro. Inhalat Toxicol. 2007;19(Suppl 1):17–22. doi: 10.1080/08958370701490445. [DOI] [PubMed] [Google Scholar]
- Sehlstedt M, Dove R, Boman C, Pagels J, Swietlicki E, Löndahl J, Westerholm R, Bosson J, Barath S, Behndig AF, Pourazar J, Sandström T, Mudway IS, Blomberg A. Antioxidant airway responses following experimental exposure to wood smoke in man. Particle Fibre Toxicol. 2010;7:21. doi: 10.1186/1743-8977-7-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoenfelt J, Mitkus RJ, Zeisler R, Spatz RO, Powell J, Fenton MJ, Squibb KA, Medvedev AE. Involvement of TLR2 and TLR4 in inflammatory immune responses induced by fine and coarse ambient air particulate matter. J Leukoc Biol. 2009;86:303–312. doi: 10.1189/jlb.1008587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shusterman D, Kaplan JZ, Canabarro C. Immediate health effects of an urban wildfire. West J Med. 1993;158:133–138. [PMC free article] [PubMed] [Google Scholar]
- Soberanes S, Urich D, Baker CM, Burgess Z, Chiarella SE, Bell EL, Ghio AJ, De Vizcaya-Ruiz A, Liu J, Ridge KM, Kamp DW, Chandel NS, Schumacker PT, Mutlu GM, Budinger GR. Mitochondrial complex III-generated oxidants activate ASK1 and JNK to induce alveolar epithelial cell death following exposure to particulate matter air pollution. J Biol Chem. 2009;284:2176–2186. doi: 10.1074/jbc.M808844200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squadrito GL, Cueto R, Dellinger B, Pryor WA. Quinoid redox cycling as a mechanism for sustained free radical generation by inhaled airborne particulate matter. Free Radical Biol Med. 2001;31:1132–1138. doi: 10.1016/s0891-5849(01)00703-1. [DOI] [PubMed] [Google Scholar]
- Viswanathan S, Eria L, Diunugala N, Johnson J, McClean C. An analysis of effects of San Diego wildfire on ambient air quality. J Air Waste Manag Assoc. 2006;56:56–67. doi: 10.1080/10473289.2006.10464439. [DOI] [PubMed] [Google Scholar]
- Wegesser TC, Franzi LM, Mitloehner FM, Eiguren-Fernandez A, Last JA. Lung Antioxidant and Cytokine Responses to Coarse and Fine Particulate Matter from the Great California Wildfires of 2008. Inhalat Toxicol. 2010;22:561–570. doi: 10.3109/08958370903571849. [DOI] [PubMed] [Google Scholar]
- Wegesser TC, Last JA. Lung Response to Coarse PM: Bioassay in Mice. Toxicol Appl Pharmacol. 2008;230:159–166. doi: 10.1016/j.taap.2008.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegesser TC, Pinkerton KE, Last JA. California Wildfires of 2008: Coarse and Fine Particulate Matter Toxicity. Environ Health Perspect. 2009;117:893–897. doi: 10.1289/ehp.0800166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia T, Kovochich M, Brant J, Hotze M, Sempf J, Oberley T, Sioutas C, Yeh JI, Wiesner MR, Nel AE. Comparison of the Abilities of Ambient and Manufactured Nanoparticles To Induce Cellular Toxicity According to an Oxidative Stress Paradigm. Nano Lett. 2006;6:1794–1807. doi: 10.1021/nl061025k. [DOI] [PubMed] [Google Scholar]