

Abstract
Background/Aims
Cytosolic phospholipase A2α (cPLA2α) is a rate-limiting key enzyme controlling the release of arachidonic acid (AA) substrate for the synthesis of prostaglandins and leukotrienes. This study was designed to explore the role of hepatocyte cPLA2α in Fas-mediated liver injury, in vivo.
Methods
Transgenic mice with targeted expression of cPLA2α under control of the albumin-promoter enhancer and wild-type mice were injected intraperitoneally with anti-Fas antibody Jo2 or lipopolysaccharide plus D-galactosamine and monitored for liver injury and survival at various time points.
Results
The cPLA2α Tg mice resist Fas-induced liver failure, as reflected by the lower serum transaminase levels, fewer apoptotic hepatocytes, reduced caspase activation, and reduced PARP cleavage when compared to the matched wild type mice. Inhibition of cPLA2α by its pharmacological inhibitor, pyrrolidine, enhanced Jo2-induced liver injury in both cPLA2α Tg and wild type mice. Hepatic overexpression of cPLA2α increases the expression of EGFR in the liver and the EGFR inhibitor, AG1478, exacerbated Jo2-mediated liver injury. The cPLA2α transgenic mice develop more prominent liver tissue damage than wild-type mice after LPS/D-galactosamine injection.
Conclusion
Hepatocyte cPLA2α protects against Fas-induced liver injury and this effect is mediated at least in part through upregulation of EGFR.
Keywords: Cytosolic phospholipase A2, liver, Fas, apoptosis, epidermal growth factor receptor, LPS
INTRODUCTION
Cytosolic phospholipase A2α (cPLA2α) is a rate-limiting key enzyme that releases arachidonic acid (AA) from membrane phospholipid for the synthesis of biologically active lipid mediators, including prostaglandins (PGs), leukotrienes (LTs) and platelet-activating factor (PAF). Whereas the free AA cleaved by cPLA2α is subsequently converted to PGs by cyclooxygenases (COXs) and LTs by lipoxygenases (LOXs), the lysophospholipid is converted to PAF, lysophosphatidic acid (LPA), and sphingosine-1-phosphate (S1P)(5, 12, 13, 45). These lipid products function as local hormones through binding to their cellular receptors in autocrine or paracrine fashions or serve as intracellular second messengers to mediate a variety of physiological and pathophysiological functions in different organ systems, such as inflammation, cell proliferation, and carcinogenesis(5, 12, 13, 21, 33, 45).
In the liver, the cPLA2α-derived lipid mediators are implicated in several aspects of liver pathobiology, such as hepatocyte growth, liver injury, hepatic microcirculation and hepatocarcinogenesis. For example, liver regeneration following partial hepatectomy in mice and rats is associated with PLA2 activation, COX-2 upregulation and increased prostaglandin production, and the regenerative response is suppressed by treatment with COX inhibitors(6, 7, 25, 30, 40, 42, 52, 57). PGs stimulate DNA synthesis in primary cultures of neonatal and adult rat hepatocytes(2, 38, 39, 47, 48); these effects are exerted in synergism with epidermal growth factor (EGF), in a manner consistent with a co-mitogenic stimulation(26, 38, 39). The cPLA2α-controlled prostaglandin cascade also interacts with other growth factors and cytokines (including HGF and IL-6) to coordinately regulate hepatic cell growth(1, 59). Additionally, PGs also play an important role in hepatocarcinogenesis, as exemplified by the following observations: (1) COX-2 expression is increased in human and animal HCC tissues; (2) activation of cPLA2α/COX-2/PGE2 signaling pathway enhances HCC cell growth and invasiveness; and (3) inhibition of COX-2 prevent the growth of HCC cells in vitro and in vivo(58). Moreover, the cPLA2-derived prostaglandins and PAF are also implicated in several other liver diseases, such as hepatosteatosis/steatohepatitis(19, 61), lipopolysaccharide-induced liver injury(15, 43), portal hypertension and ascites(29, 35, 53, 60). All of these findings underscore the importance of cPLA2/COX-2-controlled prostaglandin signaling in liver biology and in the pathogenesis of liver diseases.
In spite of the involvement of arachidonic acid metabolism in liver pathobiology, detailed assessment of hepatic cPLA2α function in vivo has been limited by the lack of a genetic animal model. This study was designed to examine the effect and mechanism of cPLA2α in Fas-induced liver injury in vivo. We generated transgenic mice with targeted expression of cPLA2α in the liver by using the albumin promoter-enhancer driven vector and the produced cPLA2α transgenic mice were injected with the anti-Fas antibody Jo2 to document the extent of liver injury. Our data show that the cPLA2α Tg mice resist Fas-induced liver failure, as reflected by the lower serum transaminase levels, fewer apoptotic hepatocytes, reduced caspase activation, and reduced PARP cleavage when compared to the matched wild type mice. Consistent with these observations, inhibition of cPLA2α by its pharmacological inhibitor, pyrrolidine, enhanced Jo2-induced liver injury in both cPLA2α Tg and wild type mice. Furthermore, hepatic overexpression of cPLA2α increases the expression of EGFR in the liver and the EGFR inhibitor, AG1478, exacerbated Jo2-mediated liver injury. These results suggest that cPLA2α prevents Fas-induced liver failure at least in part through upregulation of EGFR.
MATERIALS AND METHODS
Materials
Hamster monoclonal anti-mouse Fas antibody (Jo2) was purchased from PharMingen (San Diego, CA). The antibodies against cPLA2α, PARP, caspase-3, cyclin D1, Mcl-1, Bcl-xL, were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The antibodies against EGFR, phospho-PTEN (Ser380), phospho-Akt (Ser473/Thr308), Akt, JNK, phospho-JNK(Thr183/Tyr185) were purchased from Cell Signaling (Beverly, MA). The EIA kits for PGE2, PGF2α, LTB4, LTC4/D4/E4 and the antibody against GAPDH were purchased from Cayman Chemical (Ann Arbor, MI). Horseradish peroxidase-linked streptavidin and chemiluminescence detection reagents were from Amersham Pharmacia Biotech, Inc. (Piscataway, NJ). The cPLA2α inhibitor (pyrrolidine derivative (cat. 525143)) was purchased from Calbiochem (San Diego, CA). The EGFR inhibitor (AG1478) and the caspase substrates (Ac-DEVD-AFC for caspase 3, Ac-IETD-AFC for caspase 8, and Ac-LEHD-AFC for caspase 9) were obtained from EMD Bioscience (Gibbstown, NJ).
Animals
Transgenic mice with targeted expression of cPLA2α in the liver were developed by using the well-established albumin promoter-enhancer driven vector. To construct the albumin promoter-cPLA2α transgene, a 2.8 kb human cPLA2 cDNA containing the entire coding region of human cPLA2α was inserted into the first exon of the human growth hormone gene controlled by the mouse albumin enhancer/promoter(4, 55). This transgene was micro-injected into mouse zygotes (B6SJL/F1 eggs) and five transgenic lines were produced. The transgenic lines were maintained by backcrossing to the C57BL/6 wild type mice. The transgenic mice were identified by genotyping using tail DNA samples. The cPLA2α transgenic mice used in this study were derived from one transgenic line that was backcrossed to C57BL/6 wild type mice for more than five consecutive generations. The wild type C57BL/6 mice were obtained from Jackson Laboratories (Bar Harbor, MA) and the colonies were maintained at our animal facility. The cPLA2α transgenic mice used in this study were derived from one transgenic line that was backcrossed to C57Bl/6 wild type mice for 7 consecutive generations. The mice at the age of 8–10 weeks were utilized for experiments, with age- and sex-matched wild type C57BL/6 mice as controls. The animals were kept at 22°C under a 12-h light/dark cycle and received food and water ad libitum. The handling of mice and experimental procedures were conducted in accordance with experimental animal guidelines.
Experimental Protocol
Male C57BL/6 wild type mice and cPLA2α Tg mice were used for experiments at the age of 8–10 weeks. The mice were administered intraperitoneally (i.p.) with 0.5 μg/g body weight of Jo2 to induce acute fulminant hepatic failure (the reagents were dissolved in sterile nonpyrogenic saline solution). To determine the survival rate, the wild type mice (n=10) and cPLA2α Tg mice (n=10) were monitored continuously after Jo2 injection until animal death. In other experiments, the animals (n = 6) were sacrificed at specific time points to obtain blood and liver tissues. The liver tissues were rapidly excised, and the specimens were immediately cut into small fragments and subjected to standard formalin fixation and paraffin-embedding for histological evaluation and TUNEL stain. The remaining liver samples were immediately frozen in liquid nitrogen and stored at −80°C for future preparation of tissue homogenates. The blood samples were centrifuged at 3000 rpm for 15 min, and the sera were collected and stored at −80°C. Serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) activities were measured using an automatic analyzer at the Hospital Chemistry Department. For inhibitor pre-treatment experiments, mice were pre-treated intraperitoneally with vehicle (DMSO), pyrrolidine (3 mg/kg) or AG1478 (25 mg/kg) for 30 min before Jo2 administration and the animals were sacrificed at 4 hours after Jo2 administration to obtain blood and liver tissues. Separate experiments were carried out to evaluate the extent of liver injury at a late time point, in which the cPLA2α transgenic and wild type mice were treated with lower dose of Jo2 (0.25 μg/g body weight) and the animals were sacrificed 16 hours after Jo2 injection. All experimental animals used in this study were treated according to the protocol approved by the University Animal Care and Use Committee (Protocol # 0303501).
Hematoxylin and Eosin Staining
For histological analysis, liver tissue was fixed in 10% neutral-buffered formalin and embedded in paraffin. Sections of 5-μm thickness were affixed to slides, deparaffinized, and stained with hematoxylin and eosin (H&E) to determine morphologic changes.
TUNEL Stain
The extent of hepatocyte apoptosis was detected by terminal deoxynucleotidy1-transferase (TdT)-mediated deoxyuridine triphosphate-digoxigenin (dUTP) nick-end labeling (TUNEL). TUNEL-positive cells were counted by randomly selecting high-power fields (400 x) distributed over 6 independent sections. The numbers of TUNEL-positive and -negative cells were compiled and the percentages of TUNEL-positive cells were calculated.
Immunoblotting
Mouse liver tissues were homogenized on ice in a modified NP-40 lysis buffer (150 mM NaCl, 1.0 % NP-40, and 50 mM Tris, pH 8.0, 1 mM DTT) containing protease inhibitor cocktail tablets (Roche Diagnostics GmbH). The liver extracts was collected by centrifugation at the speed of 15,000g at 4°C for 15 minutes to remove cell debris and stored in aliquots at −80°C until use. The protein concentrations in the liver extracts were determined by the Bio-Rad protein assay (Bio-Rad, CA). 30 μg of liver protein was subjected to SDS-PAGE and the separated proteins were electrophoretically transferred onto the nitrocellulose membranes (BioRad, CA). Nonspecific binding was blocked with PBS-T (0.05% Tween 20 in PBS) containing 5% non-fat milk for 1 hr at room temperature. The membranes were then incubated overnight at 4°C with antibodies against cPLA2α, PARP, caspase-3, EGFR, phospho-PTEN (Ser380), phospho-Akt (Ser473/Thr308), Akt, cyclin D1, Mcl-1, Bcl-xL, JNK, phospho-JNK(Thr183/Tyr185) in PBS-T containing 1% non-fat milk at the dilutions specified by the manufacturers. Following three washes with PBS-T, the membranes were then incubated with the horseradish peroxidase-conjugated secondary antibodies at 1:10,000 dilution in PBS-T containing 1% non-fat milk for 1 hour at room temperature. The membranes were then washed 3 times with PBS-T and the protein bands were visualized with the ECL Western blotting detection system according to the manufacturer’s instructions. GAPDH was used as the loading control.
Analysis of caspase activities
After Jo2 antibody challenge, caspase-3, 8 and 9 activities were measured in liver extracts from mice pretreated with or without inhibitors. Briefly, 30 μg of liver protein was incubated with 20 μM fluorogenic substrates Ac-DEVD-AFC, Ac-IETD-AFC, and Ac-LEHD-AFC for caspase-3, caspase-8, caspase-9 activity, respectively. The fluorescence signals were monitored by a fluorometer (Tecan, GENios) at excitation wavelengths of 400 nm and emission wavelengths of 510 nm and the background signals were corrected. The caspase activities were expressed as the fold changes over the control samples (from corresponding wild type mice).
Statistical Analysis
All values were expressed as the mean + standard deviation. The statistical significance of differences between groups was analyzed with the homoscedastic Student’s t-Test, and p<0.05 was considered to be statistically significant.
RESULTS
Development of transgenic mice with targeted expression of cPLA2α in the liver
To determine the effect of hepatocyte cPLA2 in Fas-induced hepatocyte apoptosis, we developed transgenic mice with targeted expression of human cPLA2α in the liver by using the well-established albumin promoter-enhancer driven vector(4, 55). These mice were generated by microinjection of the cPLA2α transgene (complete cPLA2α cDNA cloned into the albumin promoter-driven vector) into mouse zygotes(14). The cPLA2α transgenic mice develop normally with no significant liver inflammation or histological abnormality under normal housing conditions, although cPLA2α transgenic mice show slightly higher body weight and liver weight than wild type mice (p<0.01) (Table 1). The liver tissues from the cPLA2α transgenic mice show increased levels of several biologically active lipid mediators in the cPLA2α pathway, including prostaglandin E2 (PGE2), prostaglandin F2α (PGF2α), leukotriene B4 (LTB4), cysteinyl leukotrienes (LTC4/D4/E4) and platelet activating factor (PAF), when compared to the wild type mice (Supplemental Figure S1).
Table 1.
Overexpression of cPLA2α in mice increases body weight and liver weight
| Wild type | cPLA2α Tg | |
|---|---|---|
| Body weight (g) | 22.63±0.68 | 25.82±1.23** |
| Liver weight (g) | 1.13±0.06 | 1.30±0.10* |
| Relativeliver weight (%) | 5.01±0.21 | 5.03±0.23 |
Values are expressed as mean ± SD from 8 male mice (9 weeks of age). Relative liver weight is expressed as a percentage of body weight.
p<0.01.
p<0.01 compared with wild type mice.
Hepatic overexpression of cPLA2α protects mice against Fas-induced liver failure
To investigate the effect of hepatocyte cPLA2α in Fas-induced hepatocyte apoptosis, the cPLA2α Tg mice and the age/sex-matched wild type mice (n=10) were injected intraperitoneally with a single dose of purified hamster anti-mouse Fas monoclonal antibody Jo2 (0.5 μg/g body weight) and followed for 24 hours to investigate the survival status. We found that the wild type mice exhibited early mortality than cPLA2α Tg mice after Jo2 treatment. In wild type mice group, death appeared at 5 h after Jo2 injection and mortality became apparent at 6.5 to 7.5 h and all mice died by 10 h (Fig. 1A). However, in cPLA2α Tg mice, first animal death was observed at 7 h and all animals died by 22 h. The survival status indicates that hepatic overexpression of cPLA2α prolongs the survival time up to 12 hours (Fig. 1A).
Figure 1. Hepatic overexpression of cPLA2α prevents Fas-induced liver injury.
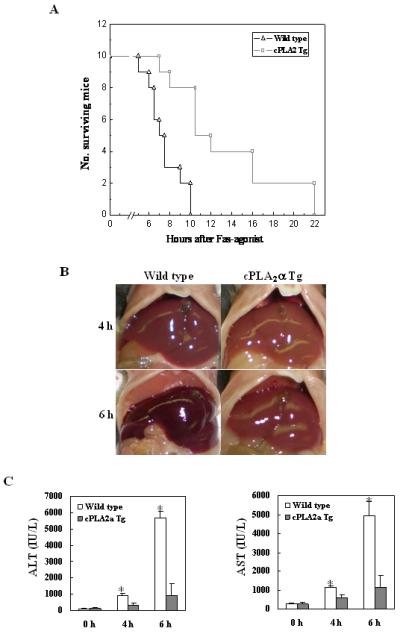
The cPLA2α Tg mice and their age/sex-matched wild type mice were injected intraperitoneally with a single dose of purified hamster anti-mouse Fas monoclonal antibody Jo2 (0.5 μg/g body weight) to induce hepatocyte apoptosis. (A) Survival curve of wild type and cPLA2α Tg mice after Jo2 injection. The cPLA2α Tg (n=10) and wild type mice (n=10) at 8-10 weeks of age received an intraperitoneal injection of Jo2 and the animals were closely monitored for activity and survival. (B) Gross photographs of liver taken 4 hours and 6 hours after Jo2 injection. The livers of wild type mice turned to dark red after Jo2 injection because of massive hepatic hemorrhage, which was observed at 4 hour and became much more prominent at 6 h. In contrast, the livers of cPLA2α Tg mice were completely normal at 4 h and became slightly red at 6 h. (C) Serum levels of ALT and AST at 4 h and 6 h after Jo2 injection. Blood samples were collected and sera were separated for transaminase analysis. The cPLA2α Tg mice show significantly lower serum ALT and AST levels than the wild type mice after Jo2 treatment. The data are expressed as mean ±SD from 6 mice (*p<0.01 vs. corresponding cPLA2α Tg mice, Student’s t test).
Based on the survival rate, additional cPLA2α Tg and wild type mice were sacrificed at specific time points (0, 4 and 6 h) after Jo2 administration to obtain blood samples and liver tissues for liver enzyme and tissue analyses. The livers of wild type mice turned to dark red after Jo2 injection because of massive hepatic hemorrhage, which was observed at 4 hour and became much more prominent at 6h. In contrast, the livers of cPLA2α Tg mice were completely normal at 4 h and became slightly red at 6 h (Fig. 1B). Accordingly, the wild type mice showed significantly higher serum alanine transaminase (ALT) and aspartate transaminase (AST) levels than cPLA2α Tg mice at both 4 and 6 h time points (p<0.01) (Fig. 1C). Histological examination of the liver tissues revealed more prominent hepatocyte apoptosis and liver damage in the wild type mice than in cPLA2α Tg mice (Fig. 2). The wild type mice showed multifocal hepatocyte apoptosis at 4 hours and massive hepatocyte apoptosis with hemorrhage at 6 hours. In contrast, the livers from the cPLA2α Tg mice showed no significant histological abnormalities at 4 h and only mild scattered apoptosis at 6 h (Fig. 2A and B). The number of TUNEL-positive hepatocytes in the wild type mice is significantly higher than in the cPLA2α Tg mice at both 4 and 6 hour time points (p<0.01) (Fig. 2C). These results indicate that hepatic overexpression of cPLA2α protects the liver from Fas-induced hepatocyte apoptosis and liver injury.
Figure 2. Hepatic overexpression of cPLA2α suppresses Fas-induced hepatocyte apoptosis and liver tissue damage.
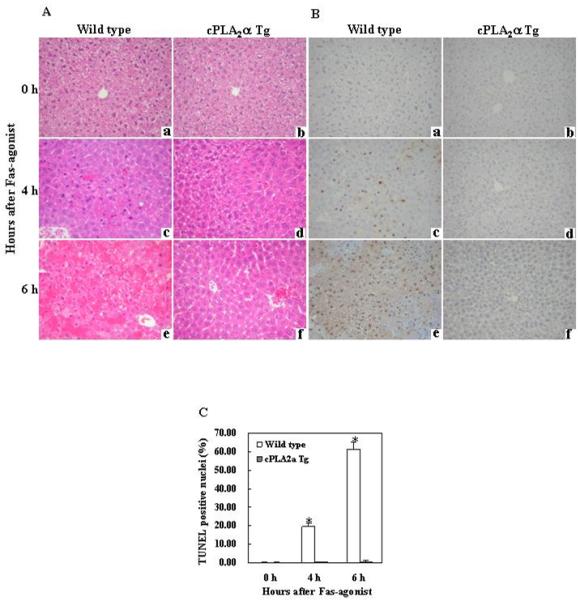
The cPLA2α Tg mice and their age/sex-matched wild type mice were administered intraperitoneally with saline or Jo2 ( 0.5 μg/g body weight ). The animals were sacrificed at 0 (a and b), 4 (c and d) and 6 (e and f) hours after injection and the liver tissues were harvested for histological evaluation. Formalin-fixed and paraffin-embedded sections (5 μm thick) were stained with hematoxylin and eosin (H&E) (A), and terminal deoxynucleotidy1 – transferase-mediated deoxyuridine triphosphate-digoxigenin nick-end labeling (TUNEL) (B) (200 × ). After Jo2 administration, the livers of the wild type mice exhibit more prominent hemorrhagic necrosis, hepatocyte apoptosis and degeneration (c and e), when compared to the livers of cPLA2α Tg mice (d and f). The number of TUNEL-positive hepatocytes in the wild type mice is significantly higher than in the cPLA2α Tg mice (*p<0.01 compared to the corresponding cPLA2α Tg mice, the data are expressed as mean ±SD from 6 mice) (C).
Hepatic overexpression of cPLA2α prevents Fas-induced caspase activation
Fas activates caspase-8 after binding to its ligand (e.g. Jo2); activated caspase-8 subsequently stimulates the caspase cascade leading to the activation of caspase-9 and caspase-3. Given the involvement of caspase 3, 8, and 9 in Fas-induced apoptosis, we next measured their activities in the cPLA2α Tg and wild type livers. As shown in Fig. 3A, although Jo2 injection significantly increased the levels of hepatic caspase 3, 8, and 9 activities in the wild type mice (p<0.01 compared to the corresponding saline treatment groups), the same concentration of Jo2 only minimally altered caspase activities in the cPLA2α Tg mice. Thus, hepatic caspase 3, 8, and 9 activities in Jo2-treated wild type mice are significantly higher than in Jo2-treated cPLA2α Tg mice.
Figure 3. Hepatic overexpression of cPLA2α protects the liver from Fas-induced apoptosis.
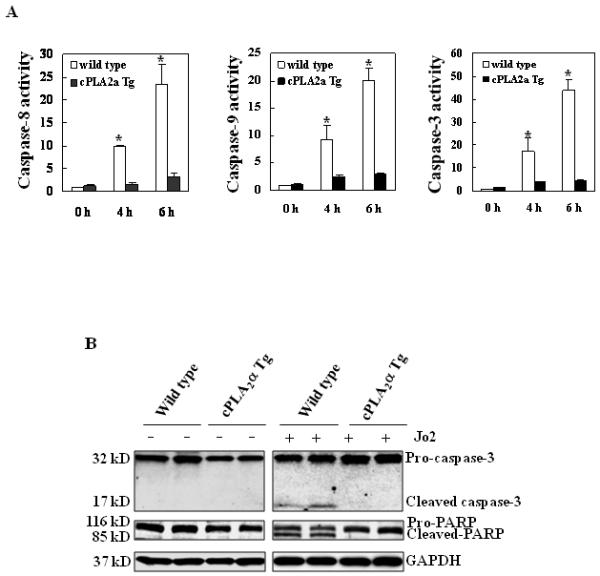
The cPLA2α Tg and their matched wild type mice were administered intraperitoneally with saline or Jo2 (0.5 μg/g body weight). The animals were sacrificed 4 hours after the injection and the liver tissues were harvested and homogenized. (A) The levels of caspases-3, 8, and 9 activities in liver homogenates. Caspase-3, 9, and 8 activities were measured by fluorometric assay with Ac-DEVD-AFC, Ac-LEHD-AFC, and Ac-IETD-AFC as the substrates, respectively. The wild type mice showed higher caspase activities than cPLA2α Tg mice after Jo2 treatment. The results are expressed as mean ±SD of fold changes over wild type livers (*p<0.01 compared to the corresponding cPLA2α Tg mice, n = 6 for each group). (B) Western blot analysis to detect caspase-3 and PARP cleavage. Liver homogenates from the cPLA2α Tg and wild type livers were subjected to SDS-PAGE and Western blot analysis to determine the protein level of proform and cleaved caspase-3 and PARP. Western blot for GAPDH was used as the loading control. Jo2 treatment for 4 hours induced capase-3 and PARP cleavage in the wild type mice, but not in the cPLA2α Tg mice.
Since caspase-3 is the primary executioner of programmed cell death and PARP is one of the main cleavage targets of caspase-3 in vivo, we next performed western blot analysis to determine the levels of cleaved caspase-3 and PARP in the liver tissues. As shown in Fig. 3B, Jo2 treatment for 4 hours induced the cleavage of caspase-3 and PARP in the wild type mice, but not in the cPLA2α Tg mice. To further examine the role of hepatic cPLA2α on Fas-induced apoptosis, additional experiments were carried out at a late time point [16 hours after injection with lower dose of Jo2 (0.25 μg/g body weight)]. At the latter experimental condition, the cPLA2α Tg mice also showed reduced liver injury, few apoptotic hepatocytes and reduced caspase-3 activity compared to the wild type mice (Fig. 4). These results all demonstrate that cPLA2α signaling prevents Fas-induced hepatocyte apoptosis.
Figure 4. Hepatic overexpression of cPLA2α protects the liver from Fas-induced apoptosis (16 hours after Jo2 injection).
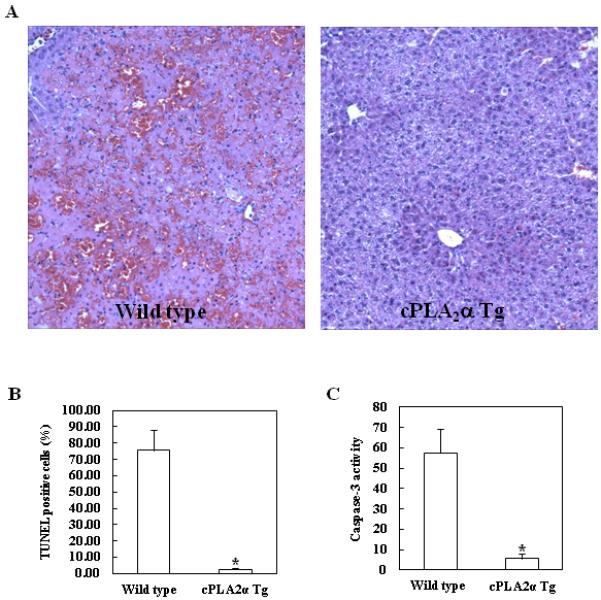
The cPLA2α Tg mice and their age/sex-matched wild type mice were intraperitoneally administered with 0.25 μg/g body weight of Jo2 and the animals were sacrificed 16h after Jo2 injection. The liver tissues were harvested for histological evaluation and caspase activity assay. (A) Formalin-fixed and paraffin-embedded sections (5 μm thick) were stained with H&E (Magnification x 100). The wild type mice showed more prominent hepatocyte apoptosis and liver injury when compared to the cPLA2α Tg mice. (B) The number of TUNEL-positive hepatocytes in the wild type mice was significantly higher than that in the cPLA2α Tg mice. The data are expressed as means ±standard deviation from six mice per group (*p<0.01). (C) The activity of caspase 3 in the wild type mice was significantly increased compared with the cPLA2α Tg mice (*p<0.01).
Inhibition of cPLA2α exacerbates Fas-induced hepatocyte apoptosis and liver injury in wild type mice
To further investigate the role of cPLA2α in Fas-induced hepatocyte apoptosis, we next employed pharmacological inhibitors of cPLA2α in Jo2-challenged wild type mice. The cPLA2α inhibitor, pyrrolidine, was intraperitoneally injected into wild type mice (3 mg/kg body weight) 30 minutes before administration of Jo2 (0.5 mg/kg body weight); the animals were sacrificed 4 hours after Jo2 injection. As shown in Fig. 5A, pretreatment with the cPLA2α inhibitor resulted in more prominent hepatocyte apoptosis in response to Jo2 challenge. The number of TUNEL-positive hepatocytes in the wild type mice pretreated with pyrrolidine is significantly higher than in the mice pretreated with vehicle control (p<0.01) (Fig. 5B). Pretreatment of wild type mice with pyrrolidine induced significantly higher serum ALT and AST levels when compared to pretreatment with vehicle control (p<0.01) (Fig. 5C). Furthermore, pretreatment of wild type mice with pyrrolidine also induced more caspase-8, 9, 3 activation in the liver than pretreatment with vehicle (p<0.01) (Fig. 5D). The cPLA2α inhibitor also enhanced Fas-induced liver injury in the cPLA2α transgenic mice (see below). These results provide pharmacological evidence for the role of cPLA2α in prevention of Fas-induced liver injury.
Figure 5. Effects of the cPLA2 inhibitor on Fas-induced hepatocyte apoptosis and liver injury in wild type mice.
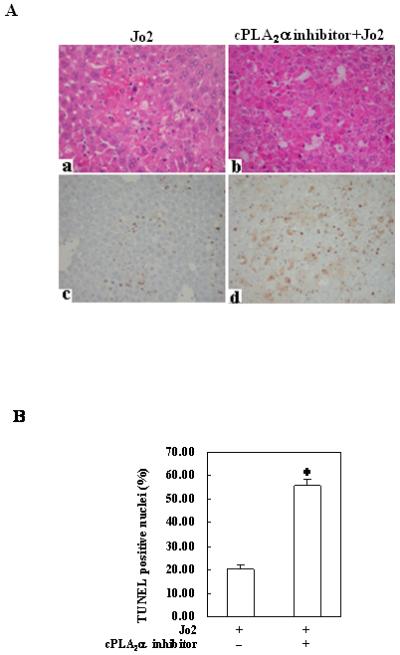
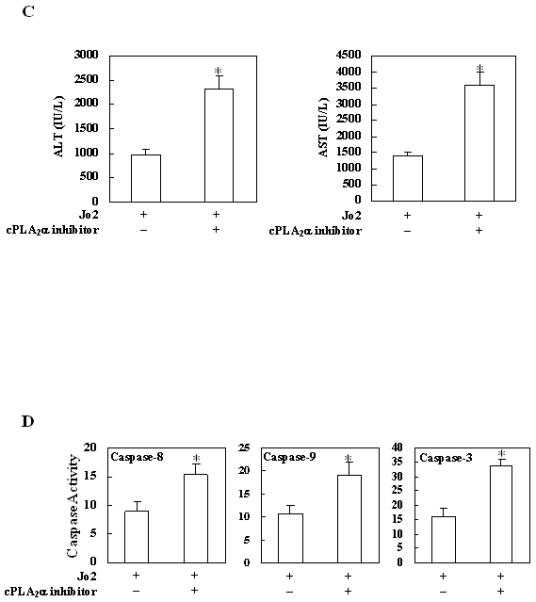
The animals were injected intraperitoneally with the cPLA2α inhibitor pyrrolidine (3 mg/kg body weight) 30 minutes before intraperitoneal administration of Jo2 (0.5 mg/kg body weight). The animals were sacrificed 4 hours after Jo2 injection and the liver tissues were harvested. (A) Representative H&E and TUNEL stains (200×) of the liver tissues from mice pretreated with or without inhibitors (all the mice received Jo2 injection). (B) Quantitation of TUNEL-positive hepatocytes in mice pretreated with or without inhibitors (*p < 0.01 compared to the corresponding wild type mice without inhibitor pretreatment, n = 6 for each group). (C) Serum transaminases. Blood samples were collected at the time of sacrifice and sera were separated for transaminase analysis. Pretreatment of wild type mice with cPLA2 inhibitor induced significantly higher serum ALT and AST levels when compared to pretreatment with vehicle control. The data are expressed as mean ±SD from 6 mice (*p<0.01 vs. corresponding mice pretreated with vehicle, Student’s t test). (D) Caspase activities. The harvested liver tissues were homogenized for subsequent caspase activity assay. Caspase-3, 9, and 8 activities were measured by fluorometric assay with Ac-DEVD-AFC, Ac-LEHD-AFC, and Ac-IETD-AFC as substrate, respectively. Pretreatment with cPLA2 inhibitor induced significantly higher caspase activities than the vehicle control. The results are expressed as mean ±SD of fold changes over wild type livers (*p<0.01 compared to the corresponding mice pretreated with vehicle, n = 6 for each group).
Hepatic overexpression of cPLA2α upregulates the expression of EGFR
To elucidate the mechanism by which cPLA2α protects against Fas-induced hepatocyte apoptosis in our system, we further performed western blot analyses to determine the levels of several molecules that are implicated in hepatocyte survival. We first examined the level of EGFR in the cPLA2α Tg and wild type mice. As shown in Fig. 6, the level of EGFR is increased in the cPLA2α Tg liver compared to wild type liver. This pattern of alteration is observed in mice with or without Jo2 treatment. Since EGFR is known to phosphorylate and activate Akt and this process is facilitated by PTEN phosphorylation, we next examined the phosphorylation level of Akt and PTEN in the cPLA2α Tg and wild type livers. As shown in Fig. 6, the phosphorylation of Akt and PTEN in the liver of cPLA2α Tg mice is higher than in the wild type mice. The levels of cyclin D1, Mcl-1 and Bcl-xL, three downstream targets of Akt, are slightly increased in the cPLA2α Tg mice than in the wild type mice (Fig. 6). These findings suggest a potential role of EGFR and related signaling in cPLA2α mediated hepatocyte survival, in vivo. The latter assertion is further supported by the findings that overexpression of cPLA2α in the liver increases EGFR mRNA (Fig. 7A) and that the cPLA2α inhibitor, pyrrolidine, inhibited hepatic expression of EGFR mRNA and protein (Fig. 7 A & B).
Figure 6. Changes of anti-apoptosis related signaling molecules in livers with altered expression of cPLA2α.
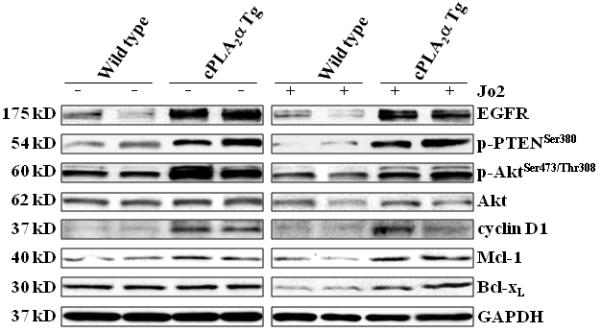
The cPLA2α Tg and wild type mice were injected intraperitoneally with saline or Jo2 (0.5 μg/g body weight). The livers were harvested 4 hours after the injection and the liver tissues were then homogenized. The obtained cellular proteins were subjected to SDS-PAGE and Western blot analysis to determine the protein levels of EGFR, phospho-PTEN, phospho-Akt, Akt, cyclin D1, Mcl-1 and Bcl-xL. Western blot for GAPDH was shown as the loading control.
Figure 7. The expression of EGFR is increased in the cPLA2α transgenic mice. The effect of the cPLA2α inhibitor (pyrrolidine).
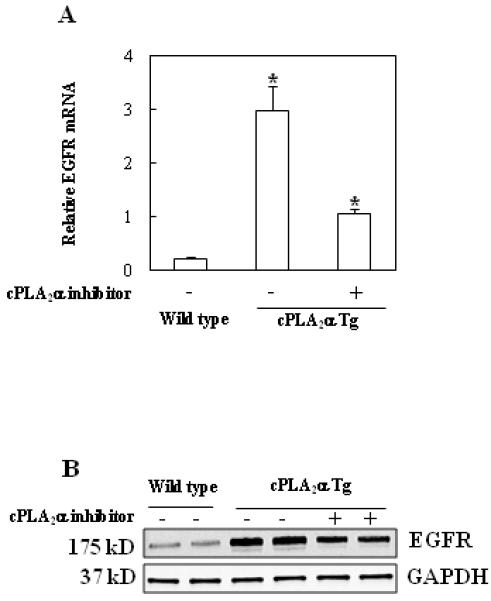
The cPLA2α Tg and wild type mice were treated intraperitoneally with vehicle (DMSO) or pyrrolidine (3 mg/kg) and animals were sacrificed at 4 hours after injection. The liver tissues were then homogenized to extract mRNA for real time quantitative PCR (qPCR) and protein for Western blotting analysis. (A) Real time quantitative PCR (qPCR) showed increased EGFR mRNA level in the cPLA2α Tg mice compared to the wild type mice. Treatment with the cPLA2α inhibitor, pyrrolidine, significantly reduced EGFR mRNA level. The data are presented as Mean + SD (n = 3, *p<0.01). (B) Western blot analysis showed reduced level of EGFR protein in mice treated with pyrrolidine.
Inhibition of cPLA2 and EGFR exacerbates Fas-induced hepatocyte apoptosis and liver injury
To further investigate the role of cPLA2 and EGFR in Fas-induced hepatocyte apoptosis, we next employed pharmacological inhibitors of cPLA2 and EGFR in the cPLA2α transgenic mice. For the cPLA2α inhibitor regimen, the cPLA2α Tg mice received intraperitoneal injection of pyrrolidine (3 mg/kg body weight) 30 minutes before administration of Jo2 (0.5 mg/kg body weight); the animals were sacrificed 4 hours after Jo2 injection. As shown in Fig. 8A, pretreatment with the cPLA2α inhibitor pyrrolidine resulted in more prominent hepatocyte apoptosis in response to Jo2 challenge. The number of TUNEL-positive hepatocytes in the cPLA2α Tg mice pretreated with pyrrolidine is significantly higher than in the mice pretreated with vehicle control (p<0.01) (Fig. 8B). Pretreatment with pyrrolidine induced significantly higher serum ALT and AST levels when compared to pretreatment with vehicle control (p<0.01) (Fig. 8C). Furthermore, the cPLA2α inhibitor also induced more caspase-8, 9, 3 activation in cPLA2α Tg mice (p<0.01) (Fig. 8D).
Figure 8. Effect of the cPLA2α inhibitor or EGFR inhibitor on Fas-induced liver injury in cPLA2α Tg mice.
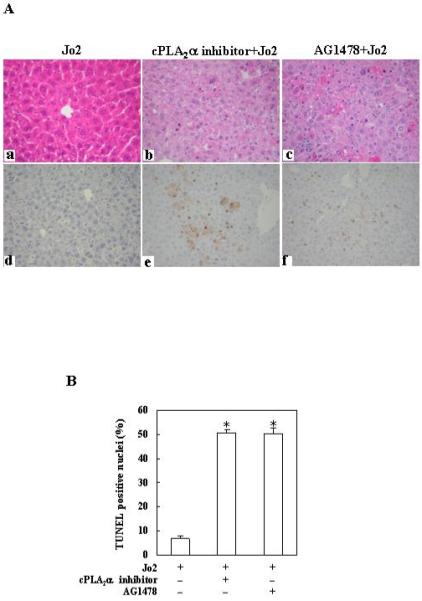
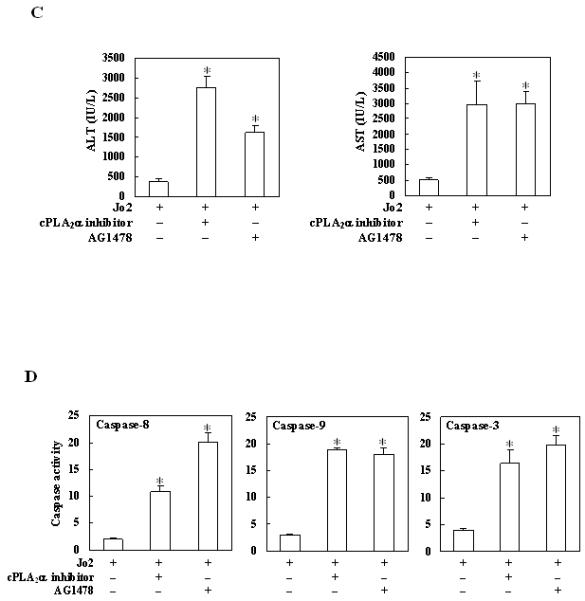
The mice were injected intraperitoneally with the cPLA2α inhibitor pyrrolidine (3 mg/kg body weight) or the EGFR inhibitor AG1478 (25 mg/kg body weight) 30 minutes before intraperitoneal administration of Jo2 (0.5 mg/kg body weight). The animals were sacrificed 4 hours after Jo2 injection and the liver tissues were harvested. (A) Representative H&E (a-c) and TUNEL stains (d-f) (200×) of the liver tissues from mice pretreated with or without inhibitors (all the mice received Jo2 injection). (B) Quantitation of TUNEL-positive hepatocytes in mice pretreated with or without inhibitors (*p < 0.01 compared to the corresponding cPLA2α Tg mice without inhibitor pretreatment, n = 6). (C) Serum transaminases. Upon sacrifice the blood samples were collected for serum transaminase analysis. Pretreatment with inhibitors induced significantly higher serum ALT and AST levels when compared to pretreatment with vehicle control. *p<0.01 compared to the corresponding cPLA2α Tg mice without inhibitor pretreatment (n = 6). (D) Caspase activities. The liver tissue homogenates were analyzed for caspase-3, 9, and 8 activities by fluorometric assay with Ac-DEVD-AFC, Ac-LEHD-AFC, and Ac-IETD-AFC as substrate, respectively. The data are expressed as mean ±SD of changes over wild type livers (n = 6 for each group). *p<0.01 compared to the corresponding cPLA2α Tg mice without inhibitor pretreatment.
For the EGFR inhibitor regimen, the cPLA2α Tg mice were pretreated with the EGFR inhibitor, AG1478 (25 mg/kg body weight), or vehicle 30 minutes before Jo2 injection. Histological examination of the liver tissues harvested 4 hours after Jo2 injection showed more prominent hepatocyte apoptosis in AG1478 pretreated mice (Fig. 8A). The number of TUNEL-positive hepatocytes in the AG1478 pretreated group is significantly higher than in the control group (p<0.01) (Fig. 8B). The mice pretreated with AG1478 showed significantly higher serum ALT and AST levels (Fig. 8C) and higher caspase-8, 9, 3 activities (Fig. 8D) (p<0.01), when compared with the mice pretreated with vehicle.
Given that JNK phosphorylation has been implicated in Fas-mediated apoptosis and that JNK might switch otherwise observed proliferative EGFR activity towards cell death in hepatic stellate cells(49), we performed further experiments to examine the status of JNK activation in our cPLA2α transgenic model. As shown in Figure 9, the levels of JNK or phospho-JNK were not altered in the cPLA2α transgenic and wild type mice (with or without Jo2 treatment). Thus, JNK activation does not appear to be involved in Jo-2-induced liver injury in the cPLA2α transgenic mice.
Figure 9. The level of JNK and phospho-p54/46-JNK in cPLA2α Tg mice and wild type mice.

The cPLA2α Tg and wild type mice were intraperitoneally administered with 0.5 μg/g or 0.25 μg/g body weight of Jo2 and sacrificed at 4h or 16h, respectively. The liver tissue proteins were subjected to SDS-PAGE and western blot analysis to determine the protein level of JNK and phospho-p54/46-JNK. GAPDH is used as the loading control.
DISCUSSION
PLA2s are key enzymes that catalyze the hydrolysis of membrane phospholipids to release bioactive lipids that play important roles in various physiological and pathological processes. Among all of the PLA2 forms, the Group IVa cPLA2α exhibits a high selectivity for liberating arachidonic acid that is subsequently metabolized by a panel of downstream enzymes for eicosanoid production. Accordingly, cPLA2α has been shown to play a pivotal role in several pathobiological conditions such as inflammation, cell growth regulation, tissue injury and carcinogenesis(5, 12, 13, 21, 33, 45). However, the role of cPLA2α in liver biology and liver diseases has not been defined. This study provides the first in vivo evidence for the role of cPLA2α in protection against Fas-induced hepatocyte apoptosis and liver injury. Our findings suggest that upregulation of EGFR represents an important mechanism for cPLA2α-mediated resistance to Fas-induced liver injury.
Our data have showed that hepatic overexpression of cPLA2α partially prevents Fas-induced liver injury (4 and 6 hours after Jo2 challenge). However, this is not a full protection, as the cPLA2α transgenic mice eventually died at later time points. It is unclear why the cPLA2α transgenic mice only confer partial protection against Fas-induced liver injury, although it is possible that this may relate to the extrahepatic systemic effects associated with Jo-2 injection, which has been documented in various previous studies(20, 41). For example, Rodriguez et al(41) reported that although intravenous administration of Jo2 did not induce fulminant hepatic destruction in AAT-bcl-2 transgenic mice in which human bcl-2 cDNA was placed under the control of hepatocyte-specific a1-antitrypsin gene promoter, these animals died. The authors suggest that the acute lethality resulted from stimulation of Fas receptors on target organs or cells other than the liver, given that Fas receptor is expressed in various extrahepatic organs/cells including lymphoid cells in the thymus and peripheral lymphoid tissues as well as the heart, lung, kidney, and small intestine, as documented in various previous studies (23, 50). Accordingly, Kakinuma et al(20) found cell apoptosis in the spleen, thymus, lymph nodes, Peyer’s patch, instestine (besides the liver) after intravenous administering of anti-Fas antibody to mice. In lieu of the previous studies, we cannot exclude the possible contribution of systemic toxicity to the lethality of cPLA2α transgenic mice after Jo2 challenge. In our model we did not observe apparent apoptosis or significant caspase activation in the liver of cPLA2α transgenic mice 6 hours after Jo2 treatment; therefore, the possible involvement of mechanisms other than apoptosis (such as necrosis) cannot be excluded.
EGFR is a key receptor tyrosine kinase in the liver which plays an important role in liver regeneration and hepatocarcinogenesis(18, 31, 34, 46). In human HCC cells, PGE2 transactivates (phosphorylates) EGFR in human HCC cells and this mechanism is important for HCC cell growth and invasion(16). In primary hepatocytes, PGE2 has also been found to enhance EGFR signaling through modulation of downstream mitogenic signaling pathways(9). The role of EGFR in hepatocyte growth is exemplified by the facts that EGFR ligands (transforming growth factor-α or EGF) enhance the growth of cultured hepatocytes in vitro(27, 28) and that conditional deletion of EGFR in mice impairs liver regeneration in vivo(34). Bilodeau and colleagues(11, 32) reported that EGF signaling pathway had antiapoptotic effects on mouse hepatocytes. However, up till now, the role of EGFR in cPLA2α-mediated hepatic actions has not been addressed. This study provides the first in vivo evidence for upregulation of EGFR in cPLA2α-mediated protection against Fas-induced hepatocyte apoptosis. The involvement of EGFR in cPLA2α-mediated protection against Fas-induced hepatocyte apoptosis is supported by the observation that AG1478, a specific EGFR tyrosine kinase inhibitor, reversed cPLA2α protection against Fas-induced liver injury (Fig. 8). Given that EGFR is able to phosphorylate and activate Akt and this process is facilitated by PTEN phosphorylation, we also examined the phosphorylation level of Akt and PTEN in the liver tissues from the cPLA2α Tg mice. Our data indicate that transgenic expression of cPLA2α increases the levels of p-Akt and p-PTEN in the livers. Additionally, the levels of three Akt downstream targets (Mcl-1, Bcl-xL and Cyclin D1) are also slightly increased in the cPLA2α transgenic livers. Taken together, these findings support a role of EGFR and related signaling in cPLA2α mediated hepatocyte survival, in vivo. Our results are consistent with the previous observations that EGF-mediated PI3-K/Akt activation protects mouse hepatocytes from Fas-induced injury(11, 24, 32), although possible involvement of other molecules cannot be excluded.
We did not observe significant JNK activation in Jo2-treated cPLA2α transgenic mice, suggesting that JNK probably is not a major factor in cPLA2α-mediated upregulation of EGFR and prevention of apoptosis. However, this interpretation has a potential pitfall, in light of the lack of significant JNK phosphorylation in Jo2-treated wild type mice in our system, which is contrast to some other studies(36, 54). The exact reason for this difference is not clear, although possible explanations include the specificity and affinity of the antibodies against JNK and p-JNK, the time of Jo2 treatment or other specific experimental conditions.
A number of previous studies have been conducted to determine the effect of cPLA2α on cell proliferation and apoptotic signaling in several nonhepatic cell types. Whereas cPLA2α has been suggested in mediating cell death by oxidant substances and certain apoptotic signals(10, 17, 37, 44, 56), this enzyme has also been shown to prevent cell death (8, 51) and even to be inactivated in apoptotic cells by proteolytic cleavage (hence not mediating cell death)(3, 22). It is possible that the prosurvival effect of cPLA2α is likely caused by the elevated formation of a metabolite derived from the action of cPLA2α on phospholipids, i.e. free AA or a lysophospholipid. This concept is consistent with the observations that when cells commit to apoptosis in response to death ligands such as Fas ligand or tumor necrosis factor-α, an early cellular event is the proteolytic cleavage of cPLA2α by caspases to prevent liberation of AA and its subsequent metabolism to eicosanoids(3, 22). On the other hand, the Group VI Ca2+-independent PLA2 (iPLA2), a PLA2 isozyme implicated in phospholipid remodeling, remains intact during apoptosis and has been suggested to facilitate apoptotic cell death process. Nevertheless, the role of PLA2α isoforms on apoptosis appears to be dependent on different cell types and specific experimental conditions. In light of the discrepancies observed from different in vitro cell culture systems, our data presented in the current study disclose an important role of hepatic cPLA2α, in vivo. Since the levels of several eicosanoids (PGE2, PGF2α, LTB4, LTC4/D4/E4) and PAF are increased in the liver tissues from the cPLA2α transgenic mice, it remains unclear which of these lipid mediators may convey the protection against Fas-induced liver injury. Given that PGE2 is produced in highest amount compared to other metabolites in cPLA2α transgenic livers and that PGE2 is known to activate EGFR in primary and transformed hepatocytes(9, 16), it is possible that PGE2 may play a key role in preventing Fas-induced liver injury. However, the potential involvement of other cPLA2α downstream lipid mediators cannot be excluded.
Our experimental findings suggest that cPLA2α is not a general cytoprotective mediator in the liver. The data presented in this study show that cPLA2α protects the liver from Fas-induced apoptosis via up-regulation of EGFR and activation of its downstream Akt signaling pathway. On the other hand, our further experiments have shown that the cPLA2α transgenic mice develop more prominent liver tissue damage than wild-type mice after LPS/D-galactosamine injection (Supplementary Figure S2 and S3). Thus, hepatic cPLA2α may mediate different responses depending on the context of liver injuries. At the present time, it remains unclear why overexpression of cPLA2α could not protect the liver from LPS/D-galactosamine-mediated liver injury. One possible explanation is that cPLA2α exhibits no significant effect on the activation of JNK which is pivotal in LPS/D-galactosamine-induced liver injury; however, it is possible that other factors might also be implicated. As EGFR activation is known to mediate anti-apoptotic effect via activation of Akt and inhibition of Bid in hepatocytes(11, 32), the role of cPLA2α in LPS/D-galactosamine-mediated liver injury is likely to be complex and warrants further investigation; however, this is beyond the scope of the current study.
Given the role of cPLA2α in liver injuries as documented in the current study, it is conceivable that this enzyme may represent a potential target for therapeutic intervention. However, when or where to inhibit or enhance cPLA2α therapeutically requires careful consideration of the underlying cause of liver injuries and the potential benefit or risk associated with the intervention.
In summary, this study provides novel evidence for hepatocyte cPLA2α in prevention of Fas-induced liver injury. We have shown that this effect is mediated at least in part through upregulation of EGFR. The availability of the cPLA2α transgenic mice will allow future studies to further define the role of cPLA2α in different animal models of liver injuries and liver diseases.
Supplementary Material
Acknowledgments
This work is partially supported by grants from the National Institutes of Health R01 grants CA106280, CA102325, CA134568 and DK077776 (to T.W.). G.L. is supported in part by the China Scholarship Council and the S&T Development Plan of Jilin Province (20090727).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
No conflicts of interest exist.
REFERENCES
- 1.Adachi T, Nakashima S, Saji S, Nakamura T, Nozawa Y. Roles of prostaglandin production and mitogen-activated protein kinase activation in hepatocyte growth factor-mediated rat hepatocyte proliferation. Hepatology. 1995;21(6):1668–1674. [PubMed] [Google Scholar]
- 2.Andreis PG, Whitfield JF, Armato U. Stimulation of DNA synthesis and mitosis of hepatocytes in primary cultures of neonatal rat liver by arachidonic acid and prostaglandins. Exp Cell Res. 1981 Aug;134(2):265–272. doi: 10.1016/0014-4827(81)90425-0. [DOI] [PubMed] [Google Scholar]
- 3.Atsumi G, Tajima M, Hadano A, Nakatani Y, Murakami M, Kudo I. Fas-induced arachidonic acid release is mediated by Ca2+-independent phospholipase A2 but not cytosolic phospholipase A2, which undergoes proteolytic inactivation. J Biol Chem. 1998;273(22):13870–13877. doi: 10.1074/jbc.273.22.13870. [DOI] [PubMed] [Google Scholar]
- 4.Bell A, Chen Q, DeFrances MC, Michalopoulos GK, Zarnegar R. The five amino acid-deleted isoform of hepatocyte growth factor promotes carcinogenesis in transgenic mice. Oncogene. 1999;18(4):887–895. doi: 10.1038/sj.onc.1202379. [DOI] [PubMed] [Google Scholar]
- 5.Burke JE, Dennis EA. Phospholipase A2 structure/function, mechanism, and signaling. J Lipid Res. 2009 Apr;50(Suppl):S237–242. doi: 10.1194/jlr.R800033-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Callery MP, Mangino MJ, Flye MW. Kupffer cell prostaglandin-E2 production is amplified during hepatic regeneration. Hepatology. 1991 Aug;14(2):368–372. [PubMed] [Google Scholar]
- 7.Casado M, Callejas NA, Rodrigo J, Zhao X, Dey SK, Bosca L, et al. Contribution of cyclooxygenase 2 to liver regeneration after partial hepatectomy. Faseb J. 2001 Sep;15(11):2016–2018. doi: 10.1096/fj.01-0158fje. [DOI] [PubMed] [Google Scholar]
- 8.Casas J, Gijon MA, Vigo AG, Crespo MS, Balsinde J, Balboa MA. Overexpression of cytosolic group IVA phospholipase A2 protects cells from Ca2+-dependent death. J Biol Chem. 2006 Mar 3;281(9):6106–6116. doi: 10.1074/jbc.M505230200. [DOI] [PubMed] [Google Scholar]
- 9.Dajani OF, Meisdalen K, Guren TK, Aasrum M, Tveteraas IH, Lilleby P, et al. Prostaglandin E2 upregulates EGF-stimulated signaling in mitogenic pathways involving Akt and ERK in hepatocytes. J Cell Physiol. 2008 Feb;214(2):371–380. doi: 10.1002/jcp.21205. [DOI] [PubMed] [Google Scholar]
- 10.Dong M, Guda K, Nambiar PR, Rezaie A, Belinsky GS, Lambeau G, et al. Inverse association between phospholipase A2 and COX-2 expression during mouse colon tumorigenesis. Carcinogenesis. 2003 Feb;24(2):307–315. doi: 10.1093/carcin/24.2.307. [DOI] [PubMed] [Google Scholar]
- 11.Ethier C, Raymond VA, Musallam L, Houle R, Bilodeau M. Antiapoptotic effect of EGF on mouse hepatocytes associated with downregulation of proapoptotic Bid protein. Am J Physiol Gastrointest Liver Physiol. 2003 Aug;285(2):G298–308. doi: 10.1152/ajpgi.00040.2003. [DOI] [PubMed] [Google Scholar]
- 12.Fitzpatrick FA, Soberman R. Regulated formation of eicosanoids. J Clin Invest. 2001;107(11):1347–1351. doi: 10.1172/JCI13241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Funk CD. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science. 2001 Nov 30;294(5548):1871–1875. doi: 10.1126/science.294.5548.1871. [DOI] [PubMed] [Google Scholar]
- 14.Han C, Bowen WC, Li G, Demetris AJ, Michalopoulos GK, Wu T. Cytosolic phospholipase A2alpha and peroxisome proliferator-activated receptor gamma signaling pathway counteracts transforming growth factor beta-mediated inhibition of primary and transformed hepatocyte growth. Hepatology. 2010 Aug;52(2):644–655. doi: 10.1002/hep.23703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Han C, Li G, Lim K, DeFrances MC, Gandhi CR, Wu T. Transgenic expression of cyclooxygenase-2 in hepatocytes accelerates endotoxin-induced acute liver failure. J Immunol. 2008 Dec 1;181(11):8027–8035. doi: 10.4049/jimmunol.181.11.8027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Han C, Michalopoulos GK, Wu T. Prostaglandin E(2) receptor EP(1) transactivates EGFR/MET receptor tyrosine kinases and enhances invasiveness in human hepatocellular carcinoma cells. J Cell Physiol. 2006 Apr;207(1):261–270. doi: 10.1002/jcp.20560. [DOI] [PubMed] [Google Scholar]
- 17.Hayakawa M, Ishida N, Takeuchi K, Shibamoto S, Hori T, Oku N, et al. Arachidonic acid-selective cytosolic phospholipase A2 is crucial in the cytotoxic action of tumor necrosis factor. J Biol Chem. 1993 May 25;268(15):11290–11295. [PubMed] [Google Scholar]
- 18.Hopfner M, Sutter AP, Huether A, Schuppan D, Zeitz M, Scherubl H. Targeting the epidermal growth factor receptor by gefitinib for treatment of hepatocellular carcinoma. J Hepatol. 2004 Dec;41(6):1008–1016. doi: 10.1016/j.jhep.2004.08.024. [DOI] [PubMed] [Google Scholar]
- 19.Ii H, Hatakeyama S, Tsutsumi K, Sato T, Akiba S. Group IVA phospholipase A2 is associated with the storage of lipids in adipose tissue and liver. Prostaglandins Other Lipid Mediat. 2008 Jun;86(1-4):12–17. doi: 10.1016/j.prostaglandins.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 20.Kakinuma C, Takagaki K, Yatomi T, Nakamura N, Nagata S, Uemura A, et al. Acute toxicity of an anti-Fas antibody in mice. Toxicol Pathol. 1999 Jul-Aug;27(4):412–420. doi: 10.1177/019262339902700404. [DOI] [PubMed] [Google Scholar]
- 21.Kita Y, Ohto T, Uozumi N, Shimizu T. Biochemical properties and pathophysiological roles of cytosolic phospholipase A2s. Biochim Biophys Acta. 2006 Nov;1761(11):1317–1322. doi: 10.1016/j.bbalip.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 22.Kronke M, Adam-Klages S. Role of caspases in TNF-mediated regulation of cPLA(2) FEBS Lett. 2002 Oct 30;531(1):18–22. doi: 10.1016/s0014-5793(02)03407-5. [DOI] [PubMed] [Google Scholar]
- 23.Leithauser F, Dhein J, Mechtersheimer G, Koretz K, Bruderlein S, Henne C, et al. Constitutive and induced expression of APO-1, a new member of the nerve growth factor/tumor necrosis factor receptor superfamily, in normal and neoplastic cells. Lab Invest. 1993 Oct;69(4):415–429. [PubMed] [Google Scholar]
- 24.Li G, Han C, Xu L, Lim K, Isse K, Wu T. Cyclooxygenase-2 prevents fas-induced liver injury through up-regulation of epidermal growth factor receptor. Hepatology. 2009 Sep;50(3):834–843. doi: 10.1002/hep.23052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.MacManus JP, Braceland BM. A connection between the production of prostaglandins during liver regeneration and the DNA synthetic response. Prostaglandins. 1976;11(4):609–620. doi: 10.1016/0090-6980(76)90064-2. [DOI] [PubMed] [Google Scholar]
- 26.Michalopoulos GK. Liver regeneration: molecular mechanisms of growth control. FASEB J. 1990 Feb 1;4(2):176–187. [PubMed] [Google Scholar]
- 27.Michalopoulos GK, Bowen WC, Mule K, Luo J. HGF-, EGF-, and dexamethasone-induced gene expression patterns during formation of tissue in hepatic organoid cultures. Gene Expr. 2003;11(2):55–75. doi: 10.3727/000000003108748964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Michalopoulos GK, Bowen WC, Zajac VF, Beer-Stolz D, Watkins S, Kostrubsky V, et al. Morphogenetic events in mixed cultures of rat hepatocytes and nonparenchymal cells maintained in biological matrices in the presence of hepatocyte growth factor and epidermal growth factor. Hepatology. 1999 Jan;29(1):90–100. doi: 10.1002/hep.510290149. [DOI] [PubMed] [Google Scholar]
- 29.Miller AM, Masrorpour M, Klaus C, Zhang JX. LPS exacerbates endothelin-1 induced activation of cytosolic phospholipase A2 and thromboxane A2 production from Kupffer cells of the prefibrotic rat liver. J Hepatol. 2007 Feb;46(2):276–285. doi: 10.1016/j.jhep.2006.08.026. [DOI] [PubMed] [Google Scholar]
- 30.Miura Y, Fukui N. Prostaglandins as possible triggers for liver regeneration after partial hepatectomy. A review. Cell Mol Biol. 1979;25(3):179–184. [PubMed] [Google Scholar]
- 31.Miyaki M, Sato C, Sakai K, Konishi M, Tanaka K, Muraoka M, et al. Malignant transformation and EGFR activation of immortalized mouse liver epithelial cells caused by HBV enhancer-X from a human hepatocellular carcinoma. Int J Cancer. 2000 Feb 15;85(4):518–522. doi: 10.1002/(sici)1097-0215(20000215)85:4<518::aid-ijc12>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 32.Musallam L, Ethier C, Haddad PS, Bilodeau M. Role of EGF receptor tyrosine kinase activity in antiapoptotic effect of EGF on mouse hepatocytes. Am J Physiol Gastrointest Liver Physiol. 2001 Jun;280(6):G1360–1369. doi: 10.1152/ajpgi.2001.280.6.G1360. [DOI] [PubMed] [Google Scholar]
- 33.Nakanishi M, Rosenberg DW. Roles of cPLA2alpha and arachidonic acid in cancer. Biochim Biophys Acta. 2006 Nov;1761(11):1335–1343. doi: 10.1016/j.bbalip.2006.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Natarajan A, Wagner B, Sibilia M. The EGF receptor is required for efficient liver regeneration. Proc Natl Acad Sci U S A. 2007 Oct 23;104(43):17081–17086. doi: 10.1073/pnas.0704126104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Niederberger M, Gines P, Martin PY, St John J, Woytaszek P, Xu L, et al. Increased renal and vascular cytosolic phospholipase A2 activity in rats with cirrhosis and ascites. Hepatology. 1998;27(1):42–47. doi: 10.1002/hep.510270108. [DOI] [PubMed] [Google Scholar]
- 36.Park SM, Rajapaksha TW, Zhang M, Sattar HA, Fichera A, Ashton-Rickardt PG, et al. CD95 signaling deficient mice with a wild-type hematopoietic system are prone to hepatic neoplasia. Apoptosis. 2008 Jan;13(1):41–51. doi: 10.1007/s10495-007-0149-6. [DOI] [PubMed] [Google Scholar]
- 37.Penzo D, Petronilli V, Angelin A, Cusan C, Colonna R, Scorrano L, et al. Arachidonic acid released by phospholipase A(2) activation triggers Ca(2+)-dependent apoptosis through the mitochondrial pathway. J Biol Chem. 2004 Jun 11;279(24):25219–25225. doi: 10.1074/jbc.M310381200. [DOI] [PubMed] [Google Scholar]
- 38.Refsnes M, Dajani OF, Sandnes D, Thoresen GH, Rottingen JA, Iversen JG, et al. On the mechanisms of the growth-promoting effect of prostaglandins in hepatocytes: the relationship between stimulation of DNA synthesis and signaling mediated by adenylyl cyclase and phosphoinositide-specific phospholipase C. J Cell Physiol. 1995 Sep;164(3):465–473. doi: 10.1002/jcp.1041640304. [DOI] [PubMed] [Google Scholar]
- 39.Refsnes M, Thoresen GH, Dajani OF, Christoffersen T. Stimulation of hepatocyte DNA synthesis by prostaglandin E2 and prostaglandin F2 alpha: additivity with the effect of norepinephrine, and synergism with epidermal growth factor. J Cell Physiol. 1994 Apr;159(1):35–40. doi: 10.1002/jcp.1041590106. [DOI] [PubMed] [Google Scholar]
- 40.Rixon RH, Whitfield JF. An early mitosis-determining event in regenerating rat liver and its possible mediation by prostaglandins or thromboxane. J Cell Physiol. 1982 Nov;113(2):281–288. doi: 10.1002/jcp.1041130216. [DOI] [PubMed] [Google Scholar]
- 41.Rodriguez I, Matsuura K, Khatib K, Reed JC, Nagata S, Vassalli P. A bcl-2 transgene expressed in hepatocytes protects mice from fulminant liver destruction but not from rapid death induced by anti-Fas antibody injection. J Exp Med. 1996 Mar 1;183(3):1031–1036. doi: 10.1084/jem.183.3.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rudnick DA, Perlmutter DH, Muglia LJ. Prostaglandins are required for CREB activation and cellular proliferation during liver regeneration. Proc Natl Acad Sci U S A. 2001;98(15):8885–8890. doi: 10.1073/pnas.151217998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sakaguchi T, Nakamura S, Suzuki S, Oda T, Ichiyama A, Baba S, et al. Participation of platelet-activating factor in the lipopolysaccharide-induced liver injury in partially hepatectomized rats. Hepatology. 1999 Oct;30(4):959–967. doi: 10.1002/hep.510300414. [DOI] [PubMed] [Google Scholar]
- 44.Sapirstein A, Spech RA, Witzgall R, Bonventre JV. Cytosolic phospholipase A2 (PLA2), but not secretory PLA2, potentiates hydrogen peroxide cytotoxicity in kidney epithelial cells. J Biol Chem. 1996;271(35):21505–21513. doi: 10.1074/jbc.271.35.21505. [DOI] [PubMed] [Google Scholar]
- 45.Schaloske RH, Dennis EA. The phospholipase A2 superfamily and its group numbering system. Biochim Biophys Acta. 2006 Nov;1761(11):1246–1259. doi: 10.1016/j.bbalip.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 46.Schiffer E, Housset C, Cacheux W, Wendum D, Desbois-Mouthon C, Rey C, et al. Gefitinib, an EGFR inhibitor, prevents hepatocellular carcinoma development in the rat liver with cirrhosis. Hepatology. 2005 Feb;41(2):307–314. doi: 10.1002/hep.20538. [DOI] [PubMed] [Google Scholar]
- 47.Skouteris GG, McMenamin M. Transforming growth factor-alpha-induced DNA synthesis and c-myc expression in primary rat hepatocyte cultures is modulated by indomethacin. Biochem J. 1992 Feb 1;281(Pt 3):729–733. doi: 10.1042/bj2810729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Skouteris GG, Ord MG, Stocken LA. Regulation of the proliferation of primary rat hepatocytes by eicosanoids. J Cell Physiol. 1988;135(3):516–520. doi: 10.1002/jcp.1041350321. [DOI] [PubMed] [Google Scholar]
- 49.Sommerfeld A, Reinehr R, Haussinger D. Bile acid-induced epidermal growth factor receptor activation in quiescent rat hepatic stellate cells can trigger both proliferation and apoptosis. J Biol Chem. 2009 Aug 14;284(33):22173–22183. doi: 10.1074/jbc.M109.005355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Suda T, Okazaki T, Naito Y, Yokota T, Arai N, Ozaki S, et al. Expression of the Fas ligand in cells of T cell lineage. J Immunol. 1995 Apr 15;154(8):3806–3813. [PubMed] [Google Scholar]
- 51.Tommasini I, Sestili P, Guidarelli A, Cantoni O. Peroxynitrite stimulates the activity of cytosolic phospholipase A2 in U937 cells: the extent of arachidonic acid formation regulates the balance between cell survival or death. Cell Death Differ. 2002 Dec;9(12):1368–1376. doi: 10.1038/sj.cdd.4401123. [DOI] [PubMed] [Google Scholar]
- 52.Tsujii H, Okamoto Y, Kikuchi E, Matsumoto M, Nakano H. Prostaglandin E2 and rat liver regeneration. Gastroenterology. 1993 Aug;105(2):495–499. doi: 10.1016/0016-5085(93)90725-r. [DOI] [PubMed] [Google Scholar]
- 53.Vishwanath BS, Frey FJ, Escher G, Reichen J, Frey BM. Liver cirrhosis induces renal and liver phospholipase A2 activity in rats. J Clin Invest. 1996;98(2):365–371. doi: 10.1172/JCI118801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vogel A, van Den Berg IE, Al-Dhalimy M, Groopman J, Ou CN, Ryabinina O, et al. Chronic liver disease in murine hereditary tyrosinemia type 1 induces resistance to cell death. Hepatology. 2004 Feb;39(2):433–443. doi: 10.1002/hep.20077. [DOI] [PubMed] [Google Scholar]
- 55.Wang X, DeFrances MC, Dai Y, Pediaditakis P, Johnson C, Bell A, et al. A Mechanism of Cell Survival. Sequestration of Fas by the HGF Receptor Met. Mol Cell. 2002 Feb;9(2):411–421. doi: 10.1016/s1097-2765(02)00439-2. [DOI] [PubMed] [Google Scholar]
- 56.Wissing D, Mouritzen H, Egeblad M, Poirier GG, Jaattela M. Involvement of caspase-dependent activation of cytosolic phospholipase A2 in tumor necrosis factor-induced apoptosis. Proc Natl Acad Sci U S A. 1997;94(10):5073–5077. doi: 10.1073/pnas.94.10.5073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wojtaszek PA, Van Putten V, Nemenoff RA. Activation of a novel form of phospholipase A2 during liver regeneration. FEBS Lett. 1995;367(3):228–232. doi: 10.1016/0014-5793(95)00556-o. [DOI] [PubMed] [Google Scholar]
- 58.Wu T. Cyclooxygenase-2 in hepatocellular carcinoma. Cancer Treat Rev. 2006 Feb;32(1):28–44. doi: 10.1016/j.ctrv.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 59.Wu T, Han C, Lunz JG, 3rd, Michalopoulos G, Shelhamer JH, Demetris AJ. Involvement of 85-kd cytosolic phospholipase A(2) and cyclooxygenase-2 in the proliferation of human cholangiocarcinoma cells. Hepatology. 2002 Aug;36(2):363–373. doi: 10.1053/jhep.2002.34743. [DOI] [PubMed] [Google Scholar]
- 60.Yang Y, Nemoto EM, Harvey SA, Subbotin VM, Gandhi CR. Increased hepatic platelet activating factor (PAF) and PAF receptors in carbon tetrachloride induced liver cirrhosis. Gut. 2004 Jun;53(6):877–883. doi: 10.1136/gut.2003.024893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yu J, Ip E, Dela Pena A, Hou JY, Sesha J, Pera N, et al. COX-2 induction in mice with experimental nutritional steatohepatitis: Role as pro-inflammatory mediator. Hepatology. 2006 Apr;43(4):826–836. doi: 10.1002/hep.21108. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.