

Abstract
Basic research, exploring the hypothesis that 2-(ω-carboxyethyl)pyrrole (CEP) modifications of proteins are generated nonenzymatically in vivo is delivering a bonanza of molecular mechanistic insights into age-related macular degeneration, autism, cancer, and wound healing. CEPs are produced through covalent modification of protein lysyl ε-amino groups by γ-hydroxyalkenal phospholipids that are formed by oxidative cleavage of docosahexaenate-containing phospholipids. Chemical synthesis of CEP-modified proteins and the production of highly specific antibodies that recognize them preceded and facilitated their detection in vivo and enabled exploration of their biological occurrence and activities. This investigational approach –from the chemistry of biomolecules to disease phenotype – is proving to be remarkably productive.
Keywords: Oxidative injury, oxidized phospholipids, biomolecular chemistry, age-related macular degeneration, cancer, wound healing, angiogenesis, inflammation, immune response, innate immunity, carboxyethylpyrrole, carboxypropylpyrrole, Toll-like receptors, rod photoreceptor cells, endothelial cells
INTRODUCTION
We anticipated that a fundamental understanding of the biomolecular chemistry of oxidized (ox) phospholipids (PLs) would provide critical insights into disease processes. Basic research on protein modification by 4-hydroxy-2-nonenal (HNE), a γ-hydroxyalkenal product of phospholipid oxidation, led to the discovery that it forms covalent adducts in vivo that incorporate the ε-amino group of protein lysyl residues in pentylpyrrole modifications (PP-protein in Fig. 1) that accumulate in neurons in the brain of individuals with Alzheimer’s disease and in the blood of individuals with atherosclerosis.1 Analogous reactions of polyunsaturated fatty acyl (PUFA)-derived γ-hydroxyalkenal oxPLs were then shown to produce ω-carboxyalkylpyrrole modifications of proteins after lipolysis of intermediate PL adducts.2 The two most abundant PUFAs in low-density lipoprotein (LDL) are linoleate and arachidonate. For example, oxidation of 1-palmityl-2-linoleyl-sn-glycerophosphocholine (PL-PC) in LDL delivers carboxyheptylpyrrole (CHP)-protein modifications and oxidation of 1-palmityl-2-arachidonyl-sn-glycerophosphocholine (PA-PC) gives carboxypropylpyrrole (CPP)-protein modifications (Fig. 1). Antibodies raised against in vitro CHP and CPP modified proteins were exploited to detect them in oxLDL, and elevated levels of CHPs were detected in blood from individuals with end-stage renal disease or atherosclerosis.2
Figure 1.

Covalent adduction of γ-hydroxyaldehydes with proteins generates alkyl and carboxyalkyl pyrrole modifications.
PART 1: CARBOXYETHYLPYRROLES: FROM HYPOTHESIS TO SYNTHESIS TO DETECTION IN VIVO
The carboxyethylpyrrole hypothesis
In view of these findings, it seemed reasonable to anticipate that oxidation of 1-palmityl-2-docosahexanoyl-sn-glycerophosphocholine (PD-PC) would give rise to carboxyethylpyrrole (CEP) protein modifications (Fig. 1). Although pursuit of CEPs was not directed at understanding a particular disease process, it seemed noteworthy that, although docosahexaenoyl phospholipids are only minor components of lipoproteins and most membranes, they are particularly abundant in certain cells of the brain3 and retina.4, 5 Thus, we anticipated that CEPs might be useful markers for oxidative injury of those tissues.
As will be described in this review, the discoveries of CEPs and their numerous and varied pathological and physiological involvements were not the result of investigating the molecular basis of a particular disease. Rather, they arose from the application of an investigational approach – from basic research on biomolecular chemistry to disease phenotype – that complements the more common opposite paradigm. The resulting studies have now delivered a rich harvest of molecular mechanistic understanding of involvements of lipid oxidation in human diseases including age-related macular degeneration, autism, and cancer, as well as insights into a physiological role of CEPs in wound healing.
Immunological detection of carboxyethylpyrrole protein adducts
Incubation of the γ-hydroxyalkenal HOHA-PC with proteins at 37 °C in pH 7.2 phosphate buffered saline (PBS) generates CEP-modifications of lysyl residues (Fig. 1).6 However, γ-hydroxyalkenals also react with cystine and histidine residues to generate other, i.e., “Michael”, adducts.7 Polyclonal rabbit antibodies specific for CEP-modifications were required to enable their detection in biological samples. Such antigens could be generated by the Paal-Knorr condensation.8 The reaction of 4,7-dioxoheptanoate with proteins produced a relatively low level of CEP modification owing, presumably, to interference by the free carboxylate. Ultimately, we found that condensation of proteins with the 9-floureneylidenemethyl ester of 4,7-dioxoheptanoate, followed by removal of the 9-floureneylidenemethyl group by treatment with 7,11-diazabicyclo[5.4.0]undec-11-ene, delivers higher levels of modification (Fig. 2).9
Figure 2.

Chemical synthesis of CEP-modified proteins.
Despite the close chemical similarity of CEPs and CPPs, antibodies exhibiting extraordinary structural discrimination were obtained.8 Those raised against a CEP-protein modification of keyhole limpet hemocyanin bind with the CEP-modified human serum albumin (HSA) 1000 times more strongly than they bind with CPP-HSA. Those raised against a CPP-protein modification bind with CPP-HSA 80 times more strongly than they bind with CEP-HSA (Fig. 3). Binding of either anti-CEP or anti-CPP antibodies with CHP-HSA is negligible and neither shows any affinity for pentylpyroles.
Figure 3.

Left panel: molecular structures of CEP and CPP derivatives of proteins. Right panel: ELISA comparison of CEP-, CPP-, CHP- and PP-protein modifications with antibodies raised against a CEP-modified protein (DOHA-KLH, left column) or a CPP-modified protein (DOOA-KLH, right column). Data from Table 1 in J Biol Chem (2003) 278, 42027–42035. This research was originally published in The Journal of Biological Chemistry. J Biol Chem. 2003; 278: 42027–42035. © the American Society for Biochemistry and Molecular Biology.
CEPs accumulate in the DHA-rich photoreceptor outer segments of the retina
To test the hypothesis that CEPs are generated in vivo, evidence for their presence was sought in photoreceptor rod outer segment (PhROS) tissue where phospholipids rich in docosahexaenoic acid (DHA) are concentrated (Fig. 4). The detection of light by photoreceptor rod cells is triggered by a photoinduced conformational change in rhodopsin, a protein that is embedded in the lipid bilayer of the photoreceptor PhROS disk membranes. Each rod cell contains a stack of double membrane disks that are organized like a stack of coins surrounded by the PhROS plasma membrane. DHA accounts for more than 80% of the PUFAs in photoreceptor disk membranes. Autoxidation of DHA and consequent formation of CEPs was expected to occur readily because the retina is bathed in a high oxygen tension supplied from the choriocapillaris. Radical initiators of autoxidation can be generated by photosensitization, or as a consequence of the high level of metabolism required to generate the Ca+2 ion gradient that is needed to propagate visual signals. Furthermore, DHA is especially susceptible to free radical-induced oxidation.
Figure 4.

Immunohistochemical analysis of mouse retinal sections with a polyclonal anti-CEP antibody is shown in the left panel and a control tissue section detected with non-immune serum in the right panel.8 Staining indicating CEP is most prominent in the rod photoreceptor outer segments (OS) and retinal pigmented endothelial (RPE) cells. Less intense staining is evident in the inner plexiform layer (IPL), and little if any staining is seen in the outer limiting membrane (OLM), outer nuclear layer (ONL), outer plexiform layer (OPL), inner nuclear layer (INL), or ganglion cell layer (GCL). Based on Fig 1 from Chem Res Toxicol (2006) 19, 262–271 and Fig 5A from J Biol Chem (2003) 278, 42027–42035. This research was originally published in The Journal of Biological Chemistry. Gu, X., Meer, S. G., Miyagi, M., Rayborn, M. E., Hollyfield, J. G., Crabb, J. W. and Salomon, R. G. Carboxyethylpyrrole protein adducts and autoantibodies, biomarkers for age-related macular degeneration. J Biol Chem. 2003; 278: 42027–42035. © the American Society for Biochemistry and Molecular Biology.
The first confirmation of the hypothesis that CEPs are generated in vivo was provided by immunohistochemical staining of mouse retina with rabbit polyclonal anti-CEP antibodies.8 As predicted, CEP immunoreactivity is especially prominent in the PhROSs. In summary, a hypothetical natural product, a CEP-modified protein, was generated through a non-biological chemical synthesis. An immunological tool, created with the aid of this synthetic antigen then provided evidence for the natural occurrence of CEP-modified proteins in vivo.
The paradoxical evolutionary choice of phospholipids rich in the highly oxidizable DHA is apparently dictated by the desirability of the high fluidity engendered by abundant unsaturation. Perhaps, the high fluidity of the disk membranes facilitates rapid lateral diffusion and consequent efficient activation of a cascade of disk membrane-associated enzymes leading to hyperpolarization of the photoreceptor cell plasma membrane. Specifically, many molecules of a G-protein, transducin, must laterally diffuse to be activated by photoactivated rhodopsin that also is mobile. Activated transducin then must laterally diffuse to activate many molecules of a phosphodiesterase that catalyzes the hydrolysis of cyclic GMP, a ligand for plasma membrane Ca+2 channels that close when cGMP levels drop. Ca+2 pumps can then create an action potential by depleting intracellular Ca+2.
CEPs also accumulate in the retinal pigmented endothelium
Prominent CEP immunoreactivity is also apparent in the retinal pigmented endothelial (RPE) cell layer. This is understandable because RPE cells endocytose shed tips of PhROSs that contain old photoreceptor disk membranes (Fig. 4). New disks are assembled at the nuclear end of the stack. In this manner, the entire stack of disks is replaced every 10 days. Thus, the evolutionary strategy of orderly loss of the older disk membranes from the tip of the PhROS is coupled to the continuous assembly of new membranes at the PhROS base. This allows the benefits of a highly fluid DHA-rich membrane to be exploited, in spite of its high susceptibility to oxidative damage, to achieve a valuable capability, vision.
Oxidation of HOHA-PC competes with protein adduction to generate CEPs
Further oxidation competes with protein adduction of 4-hydroxy-7-oxo-5-heptenoyl-phosphatidylcholine (HOHA-PC) and homologous γ-hydroxyalkenal-PCs, i.e., HOOA- and HODA-PC, to produce carboxyalkylpyrroles (CAPs in Fig. 5). A previous study established that HOOA- and HODA-PC, as well as the more oxidized derivatives referred to collectively as oxPCCD36 (Fig. 5), are high affinity ligands for the scavenger receptor CD36 that mediates endocytosis of oxidized low-density lipoprotein (oxLDL) by macrophage cells.10–12
Figure 5.

Oxidation competes with covalent adduction of γ-hydroxyalkenals, i.e., HOHA-PC and homologs, with proteins to deliver carboxyalkylpyrroles (CAPs). The γ-hydroxyalkenals and their more oxidized derivatives, collectively called oxPCCD36, are also ligands for CD36.
HOHA-PC and more oxidized derivatives promote endocytosis of oxidatively damaged photoreceptor rod outer segments by RPE cells
The biological significance of γ-hydroxyalkenal phospholipids is not limited to their role as precursors of CEPs and homologous carboxyalkylpyrroles. Rather, the γ-hydroxyalkenals and their more oxidized derivatives, collectively called oxPCCD36 (Fig. 5), also promote accumulation of CEPs in RPE cells. Plasma LDL and oxLDL are delivered to the basal side of the RPE from the choriocapillaris (Fig. 4). RPE cells express both LDL receptors and CD36 scavenger receptors13 that mediate endocytosis of LDL and oxLDL respectively.14 It seemed reasonable to presume that endocytosis of oxidatively damaged PhROSs by RPE cells can also be promoted by oxPCCD36. Each RPE cell interacts with the tips of about 50 PhROSs, and 10% of each outer segment is shed daily and phagocytosed by the RPE. Clearance by the RPE of shed rod PhROS tips is critical to the maintenance and normal function of the retina. Using a panel of analytically pure oxidatively truncated phospholipids, available through chemical syntheses6, 15, 16, oxPCCD36 were shown to serve as endogenous ligands on PhROSs for uptake by RPE via CD36.17 LC-MS/MS comparison, with authentic analytically pure samples, of retinal lipids recovered from dark-adapted rats following physiological light exposure demonstrated in vivo formation of oxPCCD36. Uptake studies using RPE cells isolated from wild-type versus CD36 null mice suggest that CD36 plays a role in engulfment of PhROSs via oxPCCD36. Thus, light exposure apparently promotes “oxidative tagging” of photoreceptor outer segments with structurally defined oxidatively truncated choline glycerophospholipids that serve as a physiological signal for CD36-mediated phagocytosis under oxidant stress conditions (Fig. 6). It should be noted that, to be recognized by RPE cells, oxPCCD36 must be located in the PhROS plasma membrane. Apparently, oxidative damage in photoreceptor cells is not limited to the DHA-rich disk membranes. This may be because lipid hydroperoxides generated during oxidation of the disk lipids can release hydroxyl radicals (•OH) that diffuse to and promote oxidation of the PhROS plasma membrane, especially at the tip that interacts with the apical process of RPE cells.
Figure 6.

Endocytosis of shed packets of photoreceptor rod outer segment (PhROS) tips by an RPE cell is promoted by binding of oxidized lipids, especially oxPCCD36 that protrude from the surface of oxidatively damaged photoreceptor outer segment plasma membranes, with CD36 scavenger receptors on RPE cell plasma membranes. Illustration by David Schumick, BS, CMI. Reprinted with the permission of the Cleveland Clinic Center for Medical Art & Photography © 2011. All Rights Reserved.
CEP-modified protein levels are elevated in AMD retina and blood
Having secured evidence for the production of CEPs in vivo in tissues rich in DHA, the most pressing question was their role, if any, in human disease. Age-related macular degeneration (AMD) is the most common cause of legal blindness in the elderly population in developed countries18, with about 35% of people 75 years or older having some degree of AMD.19 A recent clinical trial20 showed that the progression of AMD could be slowed in some individuals by high daily doses of antioxidant vitamins and zinc. This and other observations21 suggested that oxidative stress contributes to the pathogenesis of AMD resulting in the accumulation of CEP. Therefore, CEP levels in retina tissue from individuals with AMD and from controls with no eye disease were assessed. Separation of protein extracts from RPE/Bruch’s membrane/choroid tissues by SDS-PAGE followed by Western blot revealed massive accumulations of CEP-modified protein in retinas from individuals with AMD but not in those from age- and sex-matched individuals with no eye disease (Fig. 7). Elevated CEP immunoreactivity was also detectable in blood from by enzyme-linked immunosorbent assay (ELISA) in 916 AMD and 488 control donors. Mean CEP adduct levels were found to be elevated in AMD plasma by ~60%.8, 22
Figure 7.

Western analysis of proteins from AMD and normal retina. Human Bruch’s membrane/RPE/choroid from the macular region of AMD and normal donor eyes was subjected to SDS-PAGE, electroblotted to poly(vinylidene difluoride), and probed with the rabbit polyclonal anti-CEP antibody. CEP-modified human serum albumin (CEP-HSA) was used as a positive control. The age and sex of the donor eyes are listed at the top of each lane. More immunoreactivity is evident in the AMD samples than in the normals. This research was originally published in PNAS. Crabb, J. W., Miyagi, M., Gu, X., Shadrach, K., West, K. A., Sakaguchi, H., Kamei, M., Hasan, A., Yan, L., Rayborn, M. E., Salomon, R. G. and Hollyfield, J. G. (2002) Drusen proteome analysis: an approach to the etiology of age-related macular degeneration. Proc Natl Acad Sci U S A 99, 14682–14687. © The National Academy of Sciences of the United States of America.
CEP-modified proteins are present in drusen that are a risk factor for AMD
Immunocytochemical analysis demonstrated CEP immunoreactivity in extracellular deposits, called drusen and Bruch’s membrane.23 The RPE forms an integral part of the blood-retinal barrier and is responsible for vectorial transport of nutrients to the photoreceptor cells and waste products to the blood. Presumably, CEPs accumulate in the RPE cells because oxidative damage impairs their ability to process and degrade waste products from the photoreceptors. Increased deposition of CEP-modified proteins in the choriocapillaris interface with the circulatory system and in drusen, behind the neural retina on Bruch’s membrane (Fig. 4) may be a consequence of such impairment. An abundance of drusen in the macula, the small portion of the central retina responsible for high-acuity vision, is considered to be a significant risk factor for the development of AMD.23 Perhaps the normal protective mechanisms against oxidative stress are compromised in AMD.
CEP-modified proteins are associated with neurofilaments in autistic brain
Besides their extraordinary abundance in photoreceptor disc membranes, docosahexaenoyl phospholipids are also exceptionally plentiful in certain cells of the brain.3 Oxidative stress is implicated in neuronal damage in many neurodegenerative diseases24, and the developing brain appears at increased risk for oxidative damage because of the immaturity of antioxidant defenses.25 Therefore, we anticipated that CEP-modified proteins might accumulate in autistic brain where oxidative injury might contribute to aberrant neuronal development.
Although direct evidence was sorely lacking for oxidative injury in autistic brain, there was ample evidence of oxidative injury in autistic peripheral tissues.26, 27 Under normal conditions, reactive oxygen species (ROS) are cleared from cells by the action of superoxide dismutase (SOD), catalase, and glutathione peroxidase (GPx). SOD removes superoxide anion by converting it into hydrogen peroxide (H2O2). Catalase promotes the dismutation of H2O2 to water and O2 and GPx promotes the reduction of H2O2 and hydroperoxides to water and alcohols, respectively. Erythrocytes from autistic individuals contain significant elevations in ZnCuSOD28 and severely depressed levels of GPx (-44.4%) compared with controls.29, 30 As a result, the H2O2 formed by the action of SOD would not be efficiently removed owing to diminished levels of GPx. The imbalance is exacerbated by the low plasma levels of reduced glutathione (GSH) in ASD patients (4.1 ± 0.5 μmol/L)31, 32 compared with healthy controls (7.6 ± 1.4 μmol/L), p < 0.001, because GSH is a cosubstrate needed for the GPx-mediated reduction of H2O2. In the presence of redox active Cu or Fe, SOD can promote oxidative injury owing to Cu-catalyzed Haber-Weiss or Fe-catalyzed Fenton reactions of H2O2 to generate •OH, a potent ROS.
Immunohistochemical studies with anti-CEP antibodies provided the first direct evidence of increased oxidative stress in the autistic brain.33 Significant CEP-related immunoreactivity in the white matter, often extending well into the grey matter of axons, was found in every case of autism examined with white matter being most intensely labeled. White matter consists primarily of axons serving to connect different sections of the brain. In fact, anatomical studies revealed abnormal alterations in white matter in autistic brain. Depending on specific regions, the white matter abnormalities in autism include smaller white matter volumes in some regions such as corpus callosum29, 30 but also dysregulation and larger white matter volumes in other regions such as cerebellum.34–38 Prominent staining by anti-CEP antibody was observed in all cases of autism examined (n = 5), but not any of the control specimens as shown in representative cortical staining patterns from 3 of the autism cases aged 9, 7, and 5 years (Fig. 8D, E, F) and 3 control cases aged 5, 6 and 11 years (Fig. 8A, B, C). Often, cell bodies were also labeled (Fig. 8D). A striking thread-like localization of CEP appears to be a hallmark of the autistic brain. Immunoprecipitation with anti-CEP followed by SDS-PAGE and Western blot with SMI32, an antibody against neurofilament heavy chain, shows an intense immunoreactive band at approximately 200 kDa, confirming that neurofilament is a major protein target for CEP-modification in autistic brain.
Figure 8.
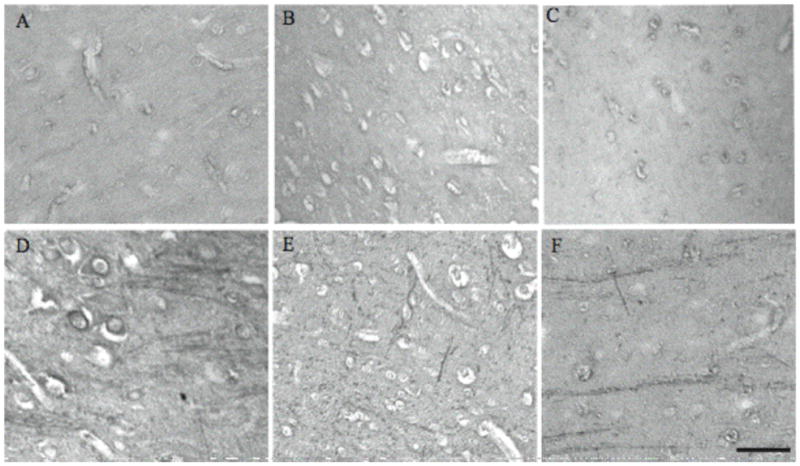
CEP-modified protein is detected in the brains of all autism cases while no specific labeling is seen in controls. Representative cases of autism aged 9, 7, and 5 years (D, E, F) and controls aged 5, 6, and 11 years (A, B, C) are shown. In some autism cases, CEP is also present in neuronal cell bodies (D). Scale bar = 50 μm. This research was originally published in Am J Biochem Biotech. Evans, T. A., Siedlak, S. L., Lu, L., Fu, X., Wang, Z., McGinnis, W. R., Fakhoury, E., Castellani, R. J., Hazen, S. L., Walsh, W. J., Lewis, A. T., Salomon, R. G., Smith, M. A., Perry, G. and Zhu, X. (2008) The Autistic Phenotype Exhibits a Remarkably Localized Modification of Brain Protein by Products of Free Radical-Induced Lipid Oxidation. Am J Biotech Biochem 4, 61–72. © Science Publications.
Notably, no significant increase in levels of CEP was detectable by ELISA of total tissue protein extracts.33 Thus, the dramatic local accumulation of CEPs observed immunohistochemically represents a subtle, albeit potentially important oxidative modification in the autistic brain. This situation is reminiscent of a localized dramatic elevation of oxidative damage (assessed by the reaction of free carbonyls with 2,4-dinitrophenylhydrazine) in the cell bodies of neurons of Alzheimer’s disease brain that contrasts with barely detectable differences between levels of this oxidative modification in total tissue protein extracts from Alzheimer’s versus age-matched control brain.39
CEPs are abundant in melanoma and accumulate in ageing vasculature and healing wounds
As will be detailed in part 2 of this review, the accumulation of CEPs in AMD retina led to the hypothesis and subsequent demonstration that CEPs induce the angiogenesis into the retina that is associated with advanced stages of AMD (vide infra). We next examined the association of CEPs with other vasculature including that associated with tumor growth –because it is promoted by angiogenesis, and it seemed reasonable to expect that stimulation of angiogenesis by CEP would be general. CEP levels were found to increase with the extent of tissue inflammation and vascularization, as well as with ageing of vasculature.40 Thus, CEP is present in melanoma, showing excessive vascularization and inflammation (assessed by CD31 and CD68 staining, respectively), at levels elevated six fold over normal skin (Fig. 9a). CEP is also present in mouse arteriolar smooth muscle cells, and levels increase with ageing (Fig. 9b). CEPs are transiently present during wound healing in mouse skin, reaching a maximum 3 days after injury, in parallel with intense wound vascularization (assessed by CD31), before returning to original levels when the wound has healed (Fig. 9c).
Figure 9.

Immunostaining of CEP (green), of CD31 endothelial cells (red) or CD61 macrophage cells (red) in (a) human skin and melanoma, (b) mouse arteriolar, and (c) wound tissues. This research was originally published in Nature. West, X. Z., Malinin, N. L., Merkulova, A. A., Tischenko, M., Kerr, B. A., Borden, E. C., Podrez, E. A., Salomon, R. G. and Byzova, T. V. (2010) Oxidative stress induces angiogenesis by activating TLR2 with novel endogenous ligands. Nature 467, 972–976. © Nature Publishing Group.
PART 2: BIOLOGICAL ACTIVITIES OF CARBOXYETHYLPYRROLES
A. Age-related macular degeneration
A1. Autoantibodies against CEP-modified “self proteins” are abundant in AMD plasma
RPE cells are located immediately adjacent to the blood-retinal barrier. By carrying CEP-rich oxidatively damaged PhROSs into RPE cells, HOHA-PC and other OxPCCD36 induce the transport of these antigens into perilously close proximity to the blood-retina barrier. Leakage of that barrier could deliver CEP modified-self proteins into the choroidal vasculature. Alternatively, HOHA-PC could slip through the barrier and then generate CEP-modified proteins in the blood. Our results show that serum albumin is the most abundant CEP-modified protein present in blood.22 It seemed plausible that CEP-modified proteins might trigger an immune response resulting in an immune-mediated attack on and destruction of RPE and photoreceptor cells that are abundantly decorated with CEP modifications. A pilot clinical study confirmed the predicted association of CEP autoimmunity with the AMD phenotype. Plasma from the AMD donors exhibited a 2-fold higher average CEP autoantibody titer (3.4 ± 3.1 S.D., n = 19)) than plasma from age-matched normal controls (1.5 ± 0.4 S.D., n = 19), and the difference was statistically significant (p = 0.02). Notably, a large fraction (63%) of the AMD donors exhibited both elevated CEP immunoreactivity and high anti-CEP autoantibody titer, i.e., above the mean for non-AMD controls, compared with only 5% of age-matched controls.8
A2. CEP autoantibody levels parallel those of retinal pathology in a mouse model of dry AMD
The finding of elevated levels of CEP autoantibodies in AMD plasma was congruent with previous suggestions that immune-mediated events may be associated with the pathogenesis of AMD.41–43 As noted above, CEP-modified proteins are more abundant in AMD than in normal Bruch’s membrane and were found associated with drusen proteins.23 Proteins associated with inflammation and immune-mediated processes, e.g., complement components C3, C5, and C9, Ig kappa, and Ig lambda, are prevalent among drusen-associated constituents.23, 41, 42, 44 Mutations/polymorphisms in genes coding for complement pathway proteins and its regulators are present in approximately 50% of AMD patients.45–50 Furthermore, dendritic cells, potent antigen-presenting cells, are intimately associated with drusen development, and complement activation is a key pathway that is active both within drusen and along the RPE-choroid interface.
To test the hypothesis that CEPs might be the inflammatory signal generated in the outer retina that directs the immune system to attack this tissue in AMD, mice were immunized with CEP-modified mouse serum albumin (CEP-MSA) and Freunds’ adjuvant in an attempt to raise the level of sensitivity to endogenously generated CEP.51, 52 Our hypothesis was that immunized mice would generate a stronger immune response to CEP adducts, making the outer retina more vulnerable to immune-mediated damage. Titers of antibody were six to eight times higher in the CEP-MSA-immunized mice compared to naive and control mice immunized with MSA or Freund’s adjuvant alone. The outer retina pathology included vesiculation and swelling of individual or multiple adjacent cells, cell lysis, and the presence of monocytes (Fig 10b–g, i) in the interphotoreceptor matrix, some of which were identified as macrophages. Although macrophages were found near some RPE lesions, it is unclear whether they initiate the pathology observed or are only a response to it, as many lesions occurred in the absence of these cells. Macrophage movement into this compartment may occur due to the release of cytokines from lysed cells. Indeed, melanin-containing macrophages were observed, suggesting that there is debris removal after RPE lysis (Fig. 10c). In some regions the RPE was missing (Fig. 10j). Notably, the severity of the pathology observed was closely correlated with the antibody titer.
Figure 10.
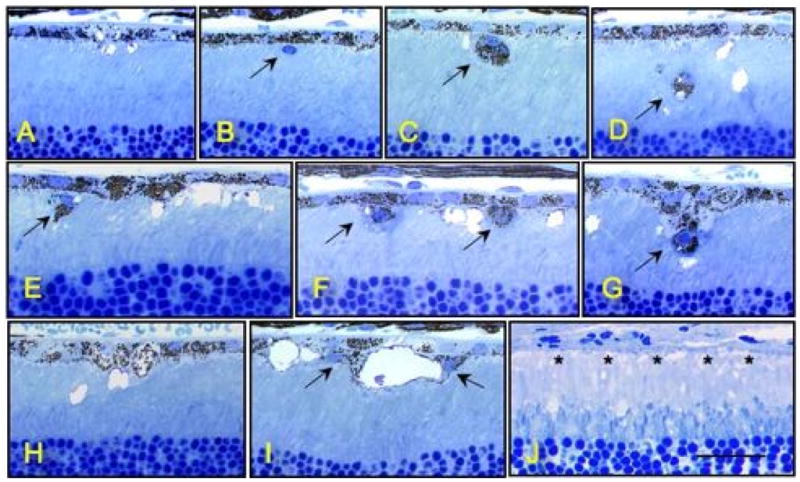
Several inflammatory cells immediately adjacent to the RPE or in the interphotoreceptor matrix (large arrows). Large empty vacuoles are also evident (C–I), some of which appear to be intracellular (I). In (J) the RPE has degenerated (asterisks). Bar in lower right of (J) represents 25 mm.
A3. CEP-MSA-immunized mice exhibit many characteristics of “dry” AMD
The most prevalent form of AMD, known as “dry” AMD is characterized by geographic atrophy. The CEP-MSA-induced mouse model also exhibited another characteristic of “dry” AMD, a drusen-like sub-RPE deposition that accumulated with ageing. Thus, 12–14 months after a single CEP-MSA immunization, an approximately 3–5 μm thick “dry” AMD-like sub-RPE basal laminar deposit accumulated that was not observed in the control eyes or in mice given multiple immunizations during 2–3 months. Thus, an immune response to a CEP-adducted self-protein is sufficient to cause localized AMD-like lesions in mice without the need for genetic, dietary, or lighting stress that had been shown previously to result in similar sub-RPE deposits.53–56 In addition, immunohistochemical analysis revealed the deposition below the RPE of C3d, a degradation product of C3b, a key complement protein. CEP-MSA immunized mice showed ten times more immunoflourescence indicative of C3d in Bruch’s membrane than did control mice (Fig 11). Immunized mice also showed an AMD-like decreased a- and b-wave amplitudes in response to light.52
Figure 11.
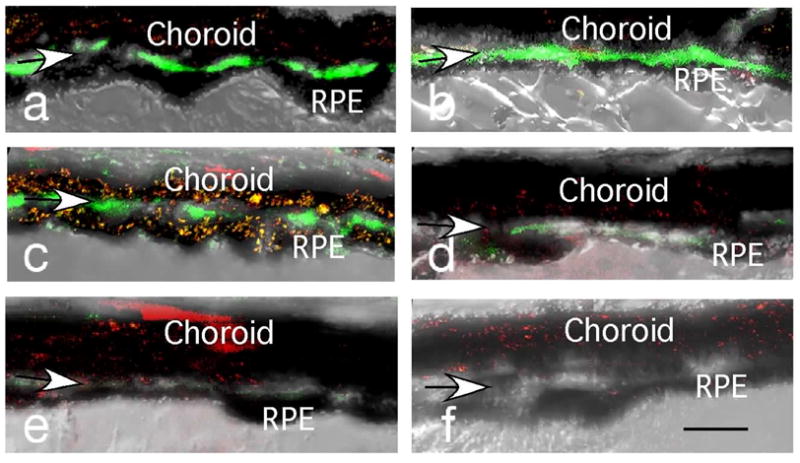
Immunocytochemistry of the outer retina of mice immunized with CEP-MSA (a–c); mice immunized with MSA (d–e); and an age-matched naïve mouse probed with C3d antibody (green) and propidium iodide (red). (f). Arrows indicate Bruch’s membrane. Bar length represents 10 μm. With kind permission from Springer Science+Business Media: Mol. Neurobiol., A hapten generated from an oxidation fragment of docosahexaenoic acid is sufficient to initiate age-related macular degeneration, 41, 2010, 290–298, Hollyfield, J. G., Perez, V. L. and Salomon, R. G., Fig. 3.
A4. CEPs promote choroidal neovascularization into the neural retina
Choroidal neovascularization (CNV, also called “wet” AMD) is characterized by new blood vessels that break through Bruch’s membrane. When these new blood vessels hemorrhage, a blood clot accumulates between the RPE and foveal photoreceptors causing immediate blindness 57, 58. In view of the massive accumulation of CEPs in photoreceptor outer segments, RPE cells, and drusen on Bruch’s membrane, it seemed plausible that they may contribute to the pathogenesis of “wet” AMD (Fig. 12). No evidence for CNV was observed in the mouse model of “dry” AMD described above. Presumably this is because damage of the blood-retina barrier is needed to initiate CNV. To determine whether the increased levels of CEP-HSA in Bruch’s membrane of individuals with AMD might contribute to the development of CNV, the effect of CEP-MSA was examined in a different mouse model.59 Mice with laser-induced rupture of Bruch’s membrane were given a subretinal injection of PBS, MSA, CEP-MSA, or vascular endothelial growth factor (VEGF). CEP-MSA significantly augmented the CNV area (Fig. 13c) when compared with injections of PBS (Fig. 13a) or unmodified MSA (Fig. 13b) and was similar to that obtained with injections of VEGF (Fig. 13d) that has been known to contribute to CNV in AMD. Subretinal injections of CEP-MSA, MSA, or VEGF in the absence of laser injury did not induce any CNV. Treating primary RPE cells or ARPE19 cells with CEP-HSA did not induce VEGF secretion.59 Thus, CEP induces angiogenesis through a unique pathway not involving VEGF.
Figure 12.

Choroidal neovascularization in “wet” AMD. Illustration by David Schumick, BS, CMI. Reprinted with the permission of the Cleveland Clinic Center for Medical Art & Photography © 2011. All Rights Reserved.
Figure 13.
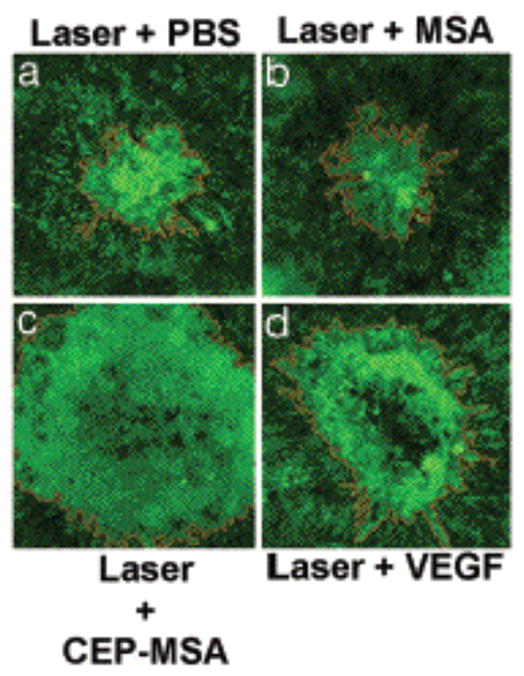
Exacerbation of CNV lesions (light green) by CEP-MSA in a mouse model of CNV. Representative photographs of mouse choroids at 14 days after laser photocoagulation and subretinal injections of (a) PBS, (b) MSA (1 μg), (c) CEP-MSA (1 μg), and (d) VEGF (20 ng). This research was originally published in PNAS. Ebrahem, Q., Renganathan, K., Sears, J., Vasanji, A., Gu, X., Lu, L., Salomon, R. G., Crabb, J. W. and Anand-Apte, B. (2006) Carboxyethylpyrrole oxidative protein modifications stimulate neovascularization: Implications for age-related macular degeneration. Proc Natl Acad Sci U S A 103, 13480–13484. © The National Academy of Sciences of the United States of America.
B. Corneal vascularization
CEPs promote corneal vascularization
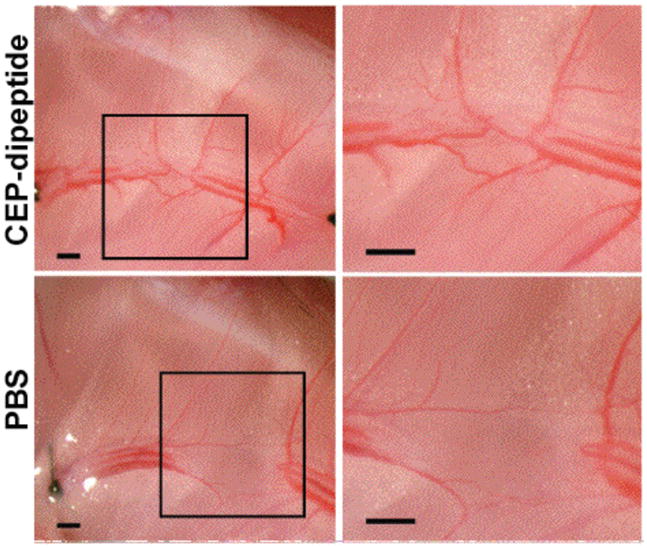
Corneal vascularization is a risk factor for transplant rejection because it compromises the immune privilege of this normally avascular tissue. A dramatic demonstration of the angiogenicity of CEPs was achieved in a corneal micropocket assay.59 Hydron/sucralfate pellets containing test compounds were implanted 1 mm from the limbus of rat cornea. CEP-HSA stimulated limbal blood vessel growth towards the pellet (Fig 14A) while unmodified HSA (Fig. 14B) or dipeptide (Fig. 14C) did not. To further test the specificity of the angiogenic response, the assay was repeated with pellets containing premixed antibody and CEP-HSA or CEP-dipeptide. Control nonimmune mouse antibodies did not show inhibition of CEP-HSA mediated corneal neovascularization (Fig 14D) while monoclonal anti-CEP antibody almost completely neutralized the formation of new blood vessels from CEP-HSA implants (Fig. 14E). Modification of proteins with γ-hydroxyalkenal phospholipids, e.g., HOHA-PC, promiscuously generates a complex mixture of biologically active CEP modifications in vivo. To assess the importance of the protein moiety on activity, the angiogenicity of a CEP-dipeptide (CEP-modified acetyl-Gly-Lys-O-methyl ester) and the unmodified dipeptide were also assessed. Although the dipeptide is inactive, the CEP-dipeptide is strongly angiogenic (Fig 14F).
Figure 14.

CEP-HSA as well as CEP-dipeptide (CEP adduct of Ac-Gly-Lys-OH) induces angiogenesis in a rat corneal micropocket assay. Shown are representative photographs of mouse corneas at 7 days after implantation of pellets containing (a) HSA, (b) HSA, (c) dipeptide (d) CEP-HSA + nonimmune IgM, (e) CEP-HSA + anti-CEP IgM, (f) CEP-dipeptide.
C. Cancer
CEPs promote tumor angiogenesis and growth
The abundance of CEPs in melanoma, the importance of angiogenesis for tumor growth, and the angiogenicity of CEPs suggested that they might promote tumor growth. VEGF-induced angiogenesis promotes tumor growth, and anti-VEGF mAb or VEGF receptor inhibitors, e.g., AAL-993, are clinically useful to block tumor growth. The ability of anti-CEP mAb to block the angiogenic action of CEPs suggested that they similarly might have medicinal utility for inhibiting tumor growth. Tumor size and vascular area (as indicated by anti-CD31 staining) generated ten days after subcutaneous implantation of murine melanoma cells into mice were compared (Fig. 15) to those produced with simultaneous intraperitoneal injection of AAL-993, anti-CEP mAb or isotype control IgG2a.40 Inhibition of endogenous CEP or VEGF decreased tumor size and vascularization. The effects were additive, i.e., the combination of anti-CEP mAb with a VEGF receptor inhibitor caused a 50% decrease (p<0.001) in vascularization over the level achieved with the VEGF inhibitor alone.40
Figure 15.
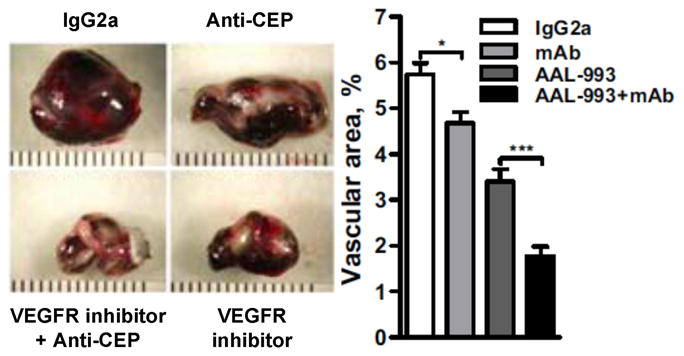
The effects of AAL-993, anti-CEP mAb, or isotype control IgG2a on tumor size (left panel) and their vascularization (right panel). This research was originally published in Nature. West, X. Z., Malinin, N. L., Merkulova, A. A., Tischenko, M., Kerr, B. A., Borden, E. C., Podrez, E. A., Salomon, R. G. and Byzova, T. V. (2010) Oxidative stress induces angiogenesis by activating TLR2 with novel endogenous ligands. Nature 467, 972–976. © Nature Publishing Group.
D. The cell biology of CEP- and CPP-induced angiogenesis
D1. CEPs and CPPs stimulate endothelial cell adhesion and migration in vitro
Integrin-dependent adhesion and migration of endothelial cells (ECs) are major cellular events mediating new vessel formation.60 To further characterize the angiogenic activities of carboxyalkyl pyrroles, cell adhesion induced by CEP-MSA, CEP-dipeptide, CPP-HSA, and for comparison VEGF, was examined using an established assay with human umbilical vein ECs.61 Low concentrations of CEPs and CPPs, e.g., 0.5 μg/ml of CEP dipeptide, were sufficient to produce a 50% increase in adhesion over control.40 A Boyden chamber assay demonstrated induction of EC migration that was 100% over control. Preincubation with the tripeptide RGD inhibited adhesion confirming integrin dependence. Induction of EC migration during angiogenesis requires activation of Rac1, a small G protein, by conversion into the GTP-bound form. Treatment with CEP resulted in generation of a substantial subpopulation of Rac1 in complex with GTP.40
D2. CEPs induce angiogenesis by activating TLR2
As previously found for VEGF receptor-mediated angiogenesis, CEPs induce endothelial cell proliferation, migration, and tube formation (Fig. 16).40 Although signaling through the VEGF receptor (R) resulting in angio-genesis also involves activation of Rac1, in contrast to VEGF, stimulation of ECs with CEP did not result in phosphorylation of the VEGFR. Furthermore, CEP-induced angiogenic activities in vitro (tube formation, proliferation, or migration) and in vivo were not decreased by the VEGFR inhibitor AAL-993 at a concentration sufficient to block VEGF effects.40 Notably, signaling through the pattern recognition scavenger receptor (SR) CD36 or SR-B1, for which the phospho-lipid precursors HOHA-PC and HOOA-PC of CEPs and CPPs (see Fig. 1) are high affinity ligands, also involves Rac1 activation.62, 63 However, the involvement of CD36 or SR-B1 in the angiogenic activities of CEPs was ruled out because the responses of ECs from both CD36 and SR-B1 knockout mice were not diminished compared with those from wild type mice.
Figure 16.

CEP-induced angiogenesis is triggered by binding to TLR2, possibly as a heterodimer with TLR1, resulting in activation, migration, proliferation, and tube formation. Illustration by David Schumick, BS, CMI. Reprinted with the permission of the Cleveland Clinic Center for Medical Art & Photography © 2011. All Rights Reserved.
The pattern recognition Toll-like receptors TLR2 and TLR4 also signal through Rac1 activation64–66, are expressed on the endothelium, and TLR2 was recently implicated in angiogenesis.67 Antibodies against TLR2, but not TLR4, inhibited CEP-induced, but not VEGF-induced EC tube formation and migration.40 In contrast, ECs from TLR2 knockout mice did not respond to treatment with CEP in several in vitro assays, whereas VEGF-triggered responses were not affected by the lack of TLR2. TLR2 often functions in concert with TLR1 or TLR6 through heterodimerization. The possible involvement of TLR2/TLR1, but not TLR2/TLR6 heterodimers, in CEP recognition is suggested by the ability of anti-TLR1, but not anti-TLR6, to diminish the CEP-induced angiogenic EC tube formation response (Fig. 16). Activation of TLR2 signaling by CEP was shown to be mediated by MyD88 and to result in Rac1 and NF-κB activation40 that lead to cytokine secretion, the inflammatory response, and angiogenesis. In contrast, VEGF-induced angiogenesis does not require MyD88.
E. Wound healing
CEP-induced angiogenesis promotes wound healing
As noted above, CEP is transiently present during wound healing, reaching a maximum 3 days after injury before returning to original levels when the wound has healed. Since high levels of CEP coincide with intense wound vascularization and CEP is angiogenic, it appeared that CEP promotes wound healing and administration of CEP-dipeptide might have therapeutic utility. This hypothesis was tested by unilateral femoral artery ligation and excision, resulting in hind limb ischemia in wild type (TLR2+/+) mice followed by treatment with CEP-dipeptide in PBS every other day after ischemia or the same amount of PBS as control.40 After 4 weeks of wound healing with injection of CEP-dipeptide, the excision area was reconnected and dendritic growth resulted in more vascular branches (Fig. 17). In contrast, no obvious reconnection or new vascular branches were noted with injection of PBS. A significant increase of hind limb blood flow, measured by a Doppler near-infrared laser, was confirmed in the CEP-dipeptide treated but not the PBS treated mice. In contrast, CEP-dipeptide was ineffective in restoring blood flow in transgenic mice lacking the Toll-like receptor (TLR2−/−).
Figure 17.
Wound healing 4 weeks after surgery at the site of mouse artery ligation and excision of the artery and vein between the caudally branching deep femoral artery and the tibial artery branching. The tissues were treated with CEP-dipeptide or vehicle (PBS, as control) by intramuscular injections distal to the ligation site, with injections every 48 h starting at the day of operation. On the right-magnified view of the artery excision area indicated on the photograph on the left; scale bars are 500 μm. This research was originally published in West, X. Z., Malinin, N. L., Merkulova, A. A., Tischenko, M., Kerr, B. A., Borden, E. C., Podrez, E. A., Salomon, R. G. and Byzova, T. V. (2010) Oxidative stress induces angiogenesis by activating TLR2 with novel endogenous ligands. Nature 467, 972–976. © Nature Publishing Group.
CONCLUSIONS
It is becoming abundantly clear that lipid oxidation contributes to the pathogenesis of many diseases, and may have important roles in physiological processes as well. An especially effective approach to unraveling these processes on a molecular level exploits: (1) the prediction of products of lipid oxidation and their reactions with biomolecules; (2) chemical synthesis of the putative oxidized lipids and their adducts with biomolecules, and their exploitation to guide and enable the development of analytical methods for their detection in vivo; (3) exploitation of these insights and tools to clinically establish biomedical relevance; and (4) cell biological and animal studies to unravel the biological mechanisms involved.
Studies evolving from the carboxyethylpyrrole hypothesis have now established that these lipid-derived oxidative protein modifications occur widely in vivo; in the retina of individuals with AMD, in the brain of autistic children, in human melanoma, in ageing vasculature, and in healing wounds. In a murine model, a single immunization with CEP-MSA suffices to induce autoantibody production, dry AMD-like retinal atrophy, decreased a- and b-wave amplitudes in response to light and deposition below the RPE of a degradation product of C3b, a key complement protein. CEP-MSA also promotes a wet AMD-like CNV, i.e., angiogenesis from the choriocpillaris into the retina. CEP is generated in tumors and wounds where it promotes tumor growth and wound healing by fostering angiogenesis.
FUTURE PERSPECTIVES
Although both VEGF and CEP both promote the angiogenesis associated with wet AMD and tumor growth, they do so through orthogonal signaling pathways, i.e., through VEGF and Toll-like receptors, respectively. Therapeutic modalities targeted against the VEGF pathway, i.e., anti-VEGF antibodies or VEGF receptor inhibitors, have proven clinically beneficial for the treatment of wet AMD and cancer. Similar approaches targeted at the CEP pathway, i.e., anti-CEP antibodies or Toll-like receptor inhibitors, seem likely to have clinical utility. Because blocking one of these pathways may lead to compensation by the other, combination therapies may be most effective for inhibiting pathological angiogenesis as well as for achieving optimal control of the precarious balance between the pathological and physiological roles of these signaling pathways since angiogenesis can either promote host defense and tissue repair or exacerbate organ dysfunction resulting in disease. Furthermore, CEP derivatives may have clinical utility for promoting wound healing through TLR2-mediated angiogenesis.
Besides the obvious therapeutic potential of inhibiting CEP-induced angiogenesis for preventing tumor growth or the choroidal neovascularization of “wet” AMD, anti-CEP antibodies and TLR2 inhibitors may have utility for treatment of other oxidation-driven pathologies such as atherosclerosis, in which arterial thickening can depend on its microvasculature. Notably, TLR2 knockout mice are protected from atherosclerosis68 and expression of TLR2 is markedly enhanced in human atherosclerotic plaques.69 It is tempting to speculate that TLR2-mediated activation of innate immunity promotes the deposition of C3d in AMD retinas. TLR2-mediated activation of adaptive immunity70 by CEP may also contribute to the pathogenesis of “dry” AMD.
Two threads of our chemistry hypothesis-driven research on CEPs converge on neuronal pathology. First, as noted at the outset, our basic research on protein modification by HNE led to the discovery that it forms covalent adducts in vivo that incorporate the ε-amino group of protein lysyl residues in pentylpyrrole modifications (PP-protein in Fig. 1), and we found that PPs accumulate in neurons in the brain of individuals with Alzheimer’s disease.1 Because of their structural similarities and mechanisms of formation (see Fig. 1), the co-production of CEPs with PPs is highly likely. Furthermore, we detected CEPs in brain from autistic individuals.33 In the second place, TLR2 expression is increased in cerebral cortical neurons in response to ischemia/reperfusion injury71 that coincidentally involves oxidative damage of lipids and proteins. The amount of brain damage and neurological deficits caused by a stroke are significantly less in mice deficient in TLR2 compared with wild-type control mice. Furthermore, TLR2 is expressed on microglial cells72, and activation of microglial cell TLR2 by a clinically relevant bacterium, Group B Streptococcus, causes the generation of NO that induces neuronal death in neonatal meningitis.73 TLR2 also mediates signaling in microglial response to putative “endogenous ligand(s)” generated consequent to axonal injury.74 In an in vitro model in hippocampal slices, ischemia upregulates the expression of TLR2 that then promotes neuronal cell death by fostering the excessive generation of the pro-inflammatory cytokine interleukin-1β.75 Since CEPs are also TLR2 ligands, it seems reasonable to anticipate that CEPs, generated under conditions of oxidative stress, contribute to brain injury, e.g., through activation of microglial TLR2.
Acknowledgments
Funding Sources
We are grateful to the National Institutes of Health for Grants GM021249 and EY016813 in support of research in the laboratory of RGS presented in this review. Research in the laboratory of JGH is supported by National Institutes of Health Grant R01-EY014240; BRTT from the State of Ohio (05-29); Foundation Fighting Blindness, Research to Prevent Blindness, Macular Vision Research Foundation, Wolf Family Foundation, and Llura and Gordon Gund Foundation.
ABBREVIATIONS
- AMD
age-related macular degeneration
- CAPs
carboxyalkylpyrroles
- CEP
carboxyethylpyrrole
- CEP-HSA
CEP-modified human serum albumin
- CEP-MSA
CEP-modified mouse serum albumin
- CHP
carboxyheptylpyrrole
- CNV
choroidal neovascularization
- CPP
carboxypropylpyrrole
- DHA
docosahexaenoic acid
- ECs
endothelial cells
- ELISA
enzyme-linked immunosorbent assay
- GPx
glutathione peroxidase
- HNE
4-hydroxy-2-nonenal
- HOHA-PC
4-hydroxy-7-oxo-5-heptenoyl-phosphatidylcholine
- HSA
human serum albumin
- LDL
low-density lipoprotein
- ox
oxidized
- PA-PC
1-palmityl-2-arachidonyl-sn-glycerophosphocholine
- PBS
phosphate buffered saline
- PD-PC
1-palmityl-2-docosahexanoyl-sn-glycerophosphocholine
- PLs
phospholipids
- PL-PC
1-palmityl-2-linoleyl-sn-glycerophosphocholine
- PhROS
photoreceptor rod outer segment
- PUFA
polyunsaturated fatty acyl
- ROS
reactive oxygen species
- RPE
retinal pigmented endothelial
- SOD
superoxide dismutase
- TLR
Toll-like receptor
- VEGF
vascular endothelial growth factor
Footnotes
DISCLOSURE
The authors declare competing financial interests. Anti-CEP antibodies and antigens and their use for clinical assessment of CEP and CEP autoantibody levels, and the mouse model for age-related macular degeneration described in this review are protected for commercialization by the Cleveland Clinic. The inventors include R.G.S. and J.G.H.
References
- 1.Sayre LM, Sha W, Xu G, Kaur K, Nadkarni D, Subbanagounder G, Salomon RG. Immunochemical evidence supporting 2-pentylpyrrole formation on proteins exposed to 4-hydroxy-2-nonenal. Chem Res Toxicol. 1996;9:1194–1201. doi: 10.1021/tx960094j. [DOI] [PubMed] [Google Scholar]
- 2.Kaur K, Salomon RG, O’Neil J, Hoff HF. (Carboxyalkyl)pyrroles in human plasma and oxidized low-density lipoproteins. Chem Res Toxicol. 1997;10:1387–1396. doi: 10.1021/tx970112c. [DOI] [PubMed] [Google Scholar]
- 3.Skinner ER, Watt C, Besson JA, Best PV. Differences in the fatty acid composition of the grey and white matter of different regions of the brains of patients with Alzheimer’s disease and control subjects. Brain. 1993;116(Pt 3):717–725. doi: 10.1093/brain/116.3.717. [DOI] [PubMed] [Google Scholar]
- 4.Alvarez RA, Aguirre GD, Acland GM, Anderson RE. Docosapentaenoic acid is converted to docosahexaenoic acid in the retinas of normal and prcd-affected miniature poodle dogs. Invest Ophthalmol Vis Sci. 1994;35:402–408. [PubMed] [Google Scholar]
- 5.Wang N, Anderson RE. Enrichment of polyunsaturated fatty acids from rat retinal pigment epithelium to rod outer segments. Curr Eye Res. 1992;11:783–791. doi: 10.3109/02713689209000751. [DOI] [PubMed] [Google Scholar]
- 6.Gu X, Sun M, Gugiu B, Hazen S, Crabb JW, Salomon RG. Oxidatively truncated docosahexaenoate phospholipids: total synthesis, generation, and peptide adduction chemistry. J Org Chem. 2003;68:3749–3761. doi: 10.1021/jo026721t. [DOI] [PubMed] [Google Scholar]
- 7.Hoff HF, O’Neil J, Wu Z, Hoppe G, Salomon RL. Phospholipid hydroxyalkenals: biological and chemical properties of specific oxidized lipids present in atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 2003;23:275–282. doi: 10.1161/01.atv.0000051407.42536.73. [DOI] [PubMed] [Google Scholar]
- 8.Gu X, Meer SG, Miyagi M, Rayborn ME, Hollyfield JG, Crabb JW, Salomon RG. Carboxyethylpyrrole protein adducts and autoantibodies, biomarkers for age-related macular degeneration. J Biol Chem. 2003;278:42027–42035. doi: 10.1074/jbc.M305460200. [DOI] [PubMed] [Google Scholar]
- 9.Lu L, Gu X, Hong L, Laird J, Jaffe K, Choi J, Crabb J, Salomon RG. Synthesis and structural characterization of carboxyethylpyrrole-modified proteins: mediators of age-related macular degeneration. Bioorg Med Chem. 2009;17:7548–7561. doi: 10.1016/j.bmc.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Salomon RG, Gu X. Critical insights into cardiovascular disease from basic research on the oxidation of phospholipids: the γ-hydroxyalkenal phospholipid hypothesis. Chem res toxicol. 2011 doi: 10.1021/tx200207z. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Podrez EA, Poliakov E, Shen Z, Zhang R, Deng Y, Sun M, Finton PJ, Shan L, Febbraio M, Hajjar DP, Silverstein RL, Hoff HF, Salomon RG, Hazen SL. A novel family of atherogenic oxidized phospholipids promotes macrophage foam cell formation via the scavenger receptor CD36 and is enriched in atherosclerotic lesions. J Biol Chem. 2002;277:38517–38523. doi: 10.1074/jbc.M205924200. [DOI] [PubMed] [Google Scholar]
- 12.Podrez EA, Poliakov E, Shen Z, Zhang R, Deng Y, Sun M, Finton PJ, Shan L, Gugiu B, Fox PL, Hoff HF, Salomon RG, Hazen SL. Identification of a novel family of oxidized phospholipids that serve as ligands for the macrophage scavenger receptor CD36. J Biol Chem. 2002;277:38503–38516. doi: 10.1074/jbc.M203318200. [DOI] [PubMed] [Google Scholar]
- 13.Hayes KC, Lindsey S, Stephan ZF, Brecker D. Retinal pigment epithelium possesses both LDL and scavenger receptor activity. Invest Ophthalmol Vis Sci. 1989;30:225–232. [PubMed] [Google Scholar]
- 14.Gordiyenko N, Campos M, Lee JW, Fariss RN, Sztein J, Rodriguez IR. RPE cells internalize low-density lipoprotein (LDL) and oxidized LDL (oxLDL) in large quantities in vitro and in vivo. Invest Ophthalmol Vis Sci. 2004;45:2822–2829. doi: 10.1167/iovs.04-0074. [DOI] [PubMed] [Google Scholar]
- 15.Choi J, Laird JM, Salomon RG. An efficient synthesis of γ-hydroxy-α,β-unsaturated aldehydic esters of 2-lysophosphatidylcholine. Bioorg Med Chem. 2011;19:580–7. doi: 10.1016/j.bmc.2010.10.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deng Y, Salomon RG. Total synthesis of γ-hydroxy-α,β-unsaturated aldehydic esters of cholesterol and 2-lysophosphatidylcholine. J Org Chem. 1998;63:7789–7794. [Google Scholar]
- 17.Sun M, Finnemann SC, Febbraio M, Shan L, Annangudi SP, Podrez EA, Hoppe G, Darrow R, Organisciak DT, Salomon RG, Silverstein RL, Hazen SL. Light-induced oxidation of photoreceptor outer segment phospholipids generates ligands for CD36-mediated phagocytosis by retinal pigment epithelium: a potential mechanism for modulating outer segment phagocytosis under oxidant stress conditions. J Biol Chem. 2006;281:4222–4230. doi: 10.1074/jbc.M509769200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Evans JR. Risk factors for age-related macular degeneration. Prog Retin Eye Res. 2001;20:227–253. doi: 10.1016/s1350-9462(00)00023-9. [DOI] [PubMed] [Google Scholar]
- 19.Klein R, Klein BE, Linton KL. Prevalence of age-related maculopathy. The Beaver Dam Eye Study. Ophthalmology. 1992;99:933–943. doi: 10.1016/s0161-6420(92)31871-8. [DOI] [PubMed] [Google Scholar]
- 20.AREADS_Research_Group. A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, beta carotene, and zinc for age-related macular degeneration and vision loss: AREDS report no. 8. Arch Ophthalmol. 2001;119:1417–1436. doi: 10.1001/archopht.119.10.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beatty S, Koh H, Phil M, Henson D, Boulton M. The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv Ophthalmol. 2000;45:115–34. doi: 10.1016/s0039-6257(00)00140-5. [DOI] [PubMed] [Google Scholar]
- 22.Gu J, Paeur GJ, Yue X, Narendra U, Sturgill GM, Bena J, Gu X, Peachey NS, Salomon RG, Hagstrom SA, Crabb JW, Clinical G Proteomic Study G. Assessing susceptibility to age-related macular degeneration with proteomic and genomic biomarkers. Mol Cell Proteomics. 2009;8:1338–1349. doi: 10.1074/mcp.M800453-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crabb JW, Miyagi M, Gu X, Shadrach K, West KA, Sakaguchi H, Kamei M, Hasan A, Yan L, Rayborn ME, Salomon RG, Hollyfield JG. Drusen proteome analysis: an approach to the etiology of age-related macular degeneration. Proc Natl Acad Sci U S A. 2002;99:14682–14687. doi: 10.1073/pnas.222551899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Back SA, Luo NL, Mallinson RA, O’Malley JP, Wallen LD, Frei B, Morrow JD, Petito CK, Roberts CT, Jr, Murdoch GH, Montine TJ. Selective vulnerability of preterm white matter to oxidative damage defined by F2-isoprostanes. Ann Neurol. 2005;58:108–120. doi: 10.1002/ana.20530. [DOI] [PubMed] [Google Scholar]
- 25.Meagher EA, FitzGerald GA. Indices of lipid peroxidation in vivo: strengths and limitations. Free Radic Biol Med. 2000;28:1745–1750. doi: 10.1016/s0891-5849(00)00232-x. [DOI] [PubMed] [Google Scholar]
- 26.Chauhan A, Chauhan V. Oxidative stress in autism. Pathophysiology. 2006;13:171–181. doi: 10.1016/j.pathophys.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 27.Keller F, Persico AM. The neurobiological context of autism. Mol Neurobiol. 2003;28:1–22. doi: 10.1385/MN:28:1:1. [DOI] [PubMed] [Google Scholar]
- 28.Zoroglu SS, Armutcu F, Ozen S, Gurel A, Sivasli E, Yetkin O, Meram I. Increased oxidative stress and altered activities of erythrocyte free radical scavenging enzymes in autism. Eur Arch Psychiatry Clin Neurosci. 2004;254:143–147. doi: 10.1007/s00406-004-0456-7. [DOI] [PubMed] [Google Scholar]
- 29.Golse B, Debray-Ritzen P, Durosay P, Puget K, Michelson AM. Alterations in two enzymes: superoxide dismutase and glutathion peroxidase in developmental infantile psychosis (infantile autism) (author’s transl) Rev Neurol (Paris) 1978;134:699–705. [PubMed] [Google Scholar]
- 30.Yorbik O, Sayal A, Akay C, Akbiyik DI, Sohmen T. Investigation of antioxidant enzymes in children with autistic disorder. Prostaglandins Leukot Essent Fatty Acids. 2002;67:341–343. doi: 10.1054/plef.2002.0439. [DOI] [PubMed] [Google Scholar]
- 31.James SJ, Cutler P, Melnyk S, Jernigan S, Janak L, Gaylor DW, Neubrander JA. Metabolic biomarkers of increased oxidative stress and impaired methylation capacity in children with autism. Am J Clin Nutr. 2004;80:1611–1617. doi: 10.1093/ajcn/80.6.1611. [DOI] [PubMed] [Google Scholar]
- 32.James SJ, Melnyk S, Jernigan S, Cleves MA, Halsted CH, Wong DH, Cutler P, Bock K, Boris M, Bradstreet JJ, Baker SM, Gaylor DW. Metabolic endophenotype and related genotypes are associated with oxidative stress in children with autism. Am J Med Genet B Neuropsychiatr Genet. 2006;141B:947–956. doi: 10.1002/ajmg.b.30366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Evans TA, Siedlak SL, Lu L, Fu X, Wang Z, McGinnis WR, Fakhoury E, Castellani RJ, Hazen SL, Walsh WJ, Lewis AT, Salomon RG, Smith MA, Perry G, Zhu X. The Autistic Phenotype Exhibits a Remarkably Localized Modification of Brain Protein by Products of Free Radical-Induced Lipid Oxidation. Am J Biotech Biochem. 2008;4:61–72. [Google Scholar]
- 34.Castelli F, Frith C, Happe F, Frith U. Autism, Asperger syndrome and brain mechanisms for the attribution of mental states to animated shapes. Brain. 2002;125:1839–1849. doi: 10.1093/brain/awf189. [DOI] [PubMed] [Google Scholar]
- 35.Just MA, Cherkassky VL, Keller TA, Kana RK, Minshew NJ. Functional and anatomical cortical underconnectivity in autism: evidence from an FMRI study of an executive function task and corpus callosum morphometry. Cereb Cortex. 2007;17:951–961. doi: 10.1093/cercor/bhl006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Just MA, Cherkassky VL, Keller TA, Minshew NJ. Cortical activation and synchronization during sentence comprehension in high-functioning autism: evidence of underconnectivity. Brain. 2004;127:1811–1821. doi: 10.1093/brain/awh199. [DOI] [PubMed] [Google Scholar]
- 37.Koshino H, Carpenter PA, Minshew NJ, Cherkassky VL, Keller TA, Just MA. Functional connectivity in an fMRI working memory task in high-functioning autism. Neuroimage. 2005;24:810–821. doi: 10.1016/j.neuroimage.2004.09.028. [DOI] [PubMed] [Google Scholar]
- 38.Mottron L, Burack JA, Iarocci G, Belleville S, Enns JT. Locally oriented perception with intact global processing among adolescents with high-functioning autism: evidence from multiple paradigms. J Child Psychol Psychiatry. 2003;44:904–913. doi: 10.1111/1469-7610.00174. [DOI] [PubMed] [Google Scholar]
- 39.Smith MA, Sayre LM, Anderson VE, Harris PL, Beal MF, Kowall N, Perry G. Cytochemical demonstration of oxidative damage in Alzheimer disease by immunochemical enhancement of the carbonyl reaction with 2,4-dinitrophenylhydrazine. J Histochem Cytochem. 1998;46:731–735. doi: 10.1177/002215549804600605. [DOI] [PubMed] [Google Scholar]
- 40.West XZ, Malinin NL, Merkulova AA, Tischenko M, Kerr BA, Borden EC, Podrez EA, Salomon RG, Byzova TV. Oxidative stress induces angiogenesis by activating TLR2 with novel endogenous ligands. Nature. 2010;467:972–976. doi: 10.1038/nature09421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hageman GS, Luthert PJ, Victor Chong NH, Johnson LV, Anderson DH, Mullins RF. An integrated hypothesis that considers drusen as biomarkers of immune-mediated processes at the RPE-Bruch’s membrane interface in aging and age-related macular degeneration. Prog Retin Eye Res. 2001;20:705–732. doi: 10.1016/s1350-9462(01)00010-6. [DOI] [PubMed] [Google Scholar]
- 42.Johnson LV, Ozaki S, Staples MK, Erickson PA, Anderson DH. A potential role for immune complex pathogenesis in drusen formation. Exp Eye Res. 2000;70:441–449. doi: 10.1006/exer.1999.0798. [DOI] [PubMed] [Google Scholar]
- 43.Penfold PL, Madigan MC, Gillies MC, Provis JM. Immunological and aetiological aspects of macular degeneration. Prog Retin Eye Res. 2001;20:385–414. doi: 10.1016/s1350-9462(00)00025-2. [DOI] [PubMed] [Google Scholar]
- 44.Mullins RF, Russell SR, Anderson DH, Hageman GS. Drusen associated with aging and age-related macular degeneration contain proteins common to extracellular deposits associated with atherosclerosis, elastosis, amyloidosis, and dense deposit disease. Faseb J. 2000;14:835–846. [PubMed] [Google Scholar]
- 45.Edwards AO, Ritter R, 3rd, Abel KJ, Manning A, Panhuysen C, Farrer LA. Complement factor H polymorphism and age-related macular degeneration. Science. 2005;308:421–424. doi: 10.1126/science.1110189. [DOI] [PubMed] [Google Scholar]
- 46.Gold B, Merriam JE, Zernant J, Hancox LS, Taiber AJ, Gehrs K, Cramer K, Neel J, Bergeron J, Barile GR, Smith RT, Hageman GS, Dean M, Allikmets R. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat Genet. 2006;38:458–462. doi: 10.1038/ng1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hageman GS, Anderson DH, Johnson LV, Hancox LS, Taiber AJ, Hardisty LI, Hageman JL, Stockman HA, Borchardt JD, Gehrs KM, Smith RJ, Silvestri G, Russell SR, Klaver CC, Barbazetto I, Chang S, Yannuzzi LA, Barile GR, Merriam JC, Smith RT, Olsh AK, Bergeron J, Zernant J, Merriam JE, Gold B, Dean M, Allikmets R. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci U S A. 2005;102:7227–7232. doi: 10.1073/pnas.0501536102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Haines JL, Hauser MA, Schmidt S, Scott WK, Olson LM, Gallins P, Spencer KL, Kwan SY, Noureddine M, Gilbert JR, Schnetz-Boutaud N, Agarwal A, Postel EA, Pericak-Vance MA. Complement factor H variant increases the risk of age-related macular degeneration. Science. 2005;308:419–421. doi: 10.1126/science.1110359. [DOI] [PubMed] [Google Scholar]
- 49.Klein RJ, Zeiss C, Chew EY, Tsai JY, Sackler RS, Haynes C, Henning AK, SanGiovanni JP, Mane SM, Mayne ST, Bracken MB, Ferris FL, Ott J, Barnstable C, Hoh J. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308:385–389. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yates JR, Sepp T, Matharu BK, Khan JC, Thurlby DA, Shahid H, Clayton DG, Hayward C, Morgan J, Wright AF, Armbrecht AM, Dhillon B, Deary IJ, Redmond E, Bird AC, Moore AT. Complement C3 variant and the risk of age-related macular degeneration. N Engl J Med. 2007;357:553–561. doi: 10.1056/NEJMoa072618. [DOI] [PubMed] [Google Scholar]
- 51.Hollyfield JG, Bonilha VL, Rayborn ME, Yang X, Shadrach KG, Lu L, Ufret RL, Salomon RG, Perez VL. Oxidative damage-induced inflammation initiates age-related macular degeneration. Nat Med. 2008;14:194–198. doi: 10.1038/nm1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hollyfield JG, Perez VL, Salomon RG. A hapten generated from an oxidation fragment of docosahexaenoic acid is sufficient to initiate age-related macular degeneration. Mol Neurobiol. 2010;41:290–298. doi: 10.1007/s12035-010-8110-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ambati J, Anand A, Fernandez S, Sakurai E, Lynn BC, Kuziel WA, Rollins BJ, Ambati BK. An animal model of age-related macular degeneration in senescent Ccl-2- or Ccr-2-deficient mice. Nat Med. 2003;9:1390–1397. doi: 10.1038/nm950. [DOI] [PubMed] [Google Scholar]
- 54.Cousins SW, Espinosa-Heidmann DG, Alexandridou A, Sall J, Dubovy S, Csaky K. The role of aging, high fat diet and blue light exposure in an experimental mouse model for basal laminar deposit formation. Exp Eye Res. 2002;75:543–553. doi: 10.1006/exer.2002.2047. [DOI] [PubMed] [Google Scholar]
- 55.Gottsch JD, Bynoe LA, Harlan JB, Rencs EV, Green WR. Light-induced deposits in Bruch’s membrane of protoporphyric mice. Arch Ophthalmol. 1993;111:126–129. doi: 10.1001/archopht.1993.01090010130039. [DOI] [PubMed] [Google Scholar]
- 56.Malek G, Johnson LV, Mace BE, Saloupis P, Schmechel DE, Rickman DW, Toth CA, Sullivan PM, Bowes Rickman C. Apolipoprotein E allele-dependent pathogenesis: a model for age-related retinal degeneration. Proc Natl Acad Sci U S A. 2005;102:11900–11905. doi: 10.1073/pnas.0503015102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bressler SB, Maguire MG, Bressler NM, Fine SL. Relationship of drusen and abnormalities of the retinal pigment epithelium to the prognosis of neovascular macular degeneration. The Macular Photocoagulation Study Group. Arch Ophthalmol. 1990;108:1442–1447. doi: 10.1001/archopht.1990.01070120090035. [DOI] [PubMed] [Google Scholar]
- 58.Sarks SH, Van Driel D, Maxwell L, Killingsworth M. Softening of drusen and subretinal neovascularization. Trans Ophthalmol Soc U K. 1980;100:414–422. [PubMed] [Google Scholar]
- 59.Ebrahem Q, Renganathan K, Sears J, Vasanji A, Gu X, Lu L, Salomon RG, Crabb JW, Anand-Apte B. Carboxyethylpyrrole oxidative protein modifications stimulate neovascularization: Implications for age-related macular degeneration. Proc Natl Acad Sci U S A. 2006;103:13480–13484. doi: 10.1073/pnas.0601552103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Avraamides CJ, Garmy-Susini B, Varner JA. Integrins in angiogenesis and lymphangiogenesis. Nat Rev Cancer. 2008;8:604–617. doi: 10.1038/nrc2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Somanath PR, Byzova TV. 14-3-3beta-Rac1-p21 activated kinase signaling regulates Akt1-mediated cytoskeletal organization, lamellipodia formation and fibronectin matrix assembly. J Cell Physiol. 2009;218:394–404. doi: 10.1002/jcp.21612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Osada Y, Sunatani T, Kim IS, Nakanishi Y, Shiratsuchi A. Signalling pathway involving GULP, MAPK and Rac1 for SR-BI-induced phagocytosis of apoptotic cells. J Biochem. 2009;145:387–394. doi: 10.1093/jb/mvn176. [DOI] [PubMed] [Google Scholar]
- 63.Wilkinson B, Koenigsknecht-Talboo J, Grommes C, Lee CY, Landreth G. Fibrillar beta-amyloid-stimulated intracellular signaling cascades require Vav for induction of respiratory burst and phagocytosis in monocytes and microglia. J Biol Chem. 2006;281:20842–20850. doi: 10.1074/jbc.M600627200. [DOI] [PubMed] [Google Scholar]
- 64.Arbibe L, Mira JP, Teusch N, Kline L, Guha M, Mackman N, Godowski PJ, Ulevitch RJ, Knaus UG. Toll-like receptor 2-mediated NF-kappa B activation requires a Rac1-dependent pathway. Nat Immunol. 2000;1:533–540. doi: 10.1038/82797. [DOI] [PubMed] [Google Scholar]
- 65.Eitel J, Krull M, Hocke AC, N’Guessan PD, Zahlten J, Schmeck B, Slevogt H, Hippenstiel S, Suttorp N, Opitz B. Beta-PIX and Rac1 GTPase mediate trafficking and negative regulation of NOD2. J Immunol. 2008;181:2664–2671. doi: 10.4049/jimmunol.181.4.2664. [DOI] [PubMed] [Google Scholar]
- 66.Equils O, Madak Z, Liu C, Michelsen KS, Bulut Y, Lu D. Rac1 and Toll-IL-1 receptor domain-containing adapter protein mediate Toll-like receptor 4 induction of HIV-long terminal repeat. J Immunol. 2004;172:7642–7646. doi: 10.4049/jimmunol.172.12.7642. [DOI] [PubMed] [Google Scholar]
- 67.Grote K, Schuett H, Salguero G, Grothusen C, Jagielska J, Drexler H, Muhlradt PF, Schieffer B. Toll-like receptor 2/6 stimulation promotes angiogenesis via GM-CSF as a potential strategy for immune defense and tissue regeneration. Blood. 115:2543–2552. doi: 10.1182/blood-2009-05-224402. [DOI] [PubMed] [Google Scholar]
- 68.Mullick AE, Tobias PS, Curtiss LK. Modulation of atherosclerosis in mice by Toll-like receptor 2. J Clin Invest. 2005;115:3149–3156. doi: 10.1172/JCI25482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Edfeldt K, Swedenborg J, Hansson GK, Yan ZQ. Expression of toll-like receptors in human atherosclerotic lesions: a possible pathway for plaque activation. Circulation. 2002;105:1158–1161. [PubMed] [Google Scholar]
- 70.Schjetne KW, Thompson KM, Nilsen N, Flo TH, Fleckenstein B, Iversen JG, Espevik T, Bogen B. Cutting edge: link between innate and adaptive immunity: Toll-like receptor 2 internalizes antigen for presentation to CD4+ T cells and could be an efficient vaccine target. J Immunol. 2003;171:32–36. doi: 10.4049/jimmunol.171.1.32. [DOI] [PubMed] [Google Scholar]
- 71.Tang SC, Arumugam TV, Xu X, Cheng A, Mughal MR, Jo DG, Lathia JD, Siler DA, Chigurupati S, Ouyang X, Magnus T, Camandola S, Mattson MP. Pivotal role for neuronal Toll-like receptors in ischemic brain injury and functional deficits. Proc Natl Acad Sci U S A. 2007;104:13798–13803. doi: 10.1073/pnas.0702553104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jack CS, Arbour N, Manusow J, Montgrain V, Blain M, McCrea E, Shapiro A, Antel JP. TLR signaling tailors innate immune responses in human microglia and astrocytes. J Immunol. 2005;175:4320–4330. doi: 10.4049/jimmunol.175.7.4320. [DOI] [PubMed] [Google Scholar]
- 73.Lehnardt S, Henneke P, Lien E, Kasper DL, Volpe JJ, Bechmann I, Nitsch R, Weber JR, Golenbock DT, Vartanian T. A mechanism for neurodegeneration induced by group B streptococci through activation of the TLR2/MyD88 pathway in microglia. J Immunol. 2006;177:583–592. doi: 10.4049/jimmunol.177.1.583. [DOI] [PubMed] [Google Scholar]
- 74.Babcock AA, Wirenfeldt M, Holm T, Nielsen HH, Dissing-Olesen L, Toft-Hansen H, Millward JM, Landmann R, Rivest S, Finsen B, Owens T. Toll-like receptor 2 signaling in response to brain injury: an innate bridge to neuroinflammation. J Neurosci. 2006;26:12826–12837. doi: 10.1523/JNEUROSCI.4937-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yao H, Felfly H, Wang J, Zhou D, Haddad GG. DIDS protects against neuronal injury by blocking Toll-like receptor 2 activated-mechanisms. J Neurochem. 2009;108:835–846. doi: 10.1111/j.1471-4159.2008.05838.x. [DOI] [PMC free article] [PubMed] [Google Scholar]