

Abstract
Actin and myosin are the two main proteins required for cell motility and muscle contraction. The structure of their strongly bound complex - rigor state - is a key for delineating the functional mechanism of actomyosin motor. Current knowledge of that complex is based on models obtained from the docking of known atomic structures of actin and myosin subfragment 1 (S1; the head and neck region of myosin) into low-resolution EM electron density maps, which precludes atomic or side chain level information. Here, we use radiolytic protein footprinting for global mapping of sites across the actin molecules that are impacted directly or allosterically by myosin binding to actin filaments. Fluorescence and electron paramagnetic resonance (EPR) spectroscopies and cysteine actin mutants are used for independent, residue-specific probing of S1 effects on two structural elements of actin. We identify actin residue candidates involved in S1 binding, and provide experimental evidence to discriminate between the regions of hydrophobic and electrostatic interactions. Focusing on the role of the DNase-I binding loop (D-loop) and the W-loop residues of actin in their interactions with S1, we found that the emission properties of acrylodan and the mobility of EPR spin labels attached to cysteine mutants of these residues change strongly and in a residue-specific manner upon S1 binding, consistent with the recently proposed direct contacts of these loops with S1. As documented in this study, the direct and indirect changes on actin induced by myosin are more extensive than known until now and attest to the importance of actin dynamics to actomyosin function.
Keywords: actin, myosin, cryo-EM, modeling, radiation footprinting
Introduction
Actin, a major cytoskeletal protein present in all eukaryotic cells, is involved in many cellular processes, including cell motility, cell adhesion, cell division, exocytosis and endocytosis.1-3 Actin filament (F-actin) is a double strand helical structure assembled from hundreds/thousands of globular actin monomers. Myosin II is a motor protein with the two heavy chains forming a long tail and two globular heads and light chains bridging the head and tail parts of the molecule. Under physiological and low salt conditions myosin forms filaments, with the heads protruding radially outwards. Myosin heads interact with actin to convert the chemical energy of ATP hydrolysis into force or muscle contraction. During the ATPase cycle, the initial weakly bound complex is transformed into a strongly-bound complex, identified as the ‘rigor state’ actomyosin. Thus, mapping specific interactions between actin and myosin and the structural changes on these proteins is critical for understanding the force generation process and actomyosin function.
Successive modelings of the actomyosin rigor complex were pursued4-8 by docking X-ray structures of myosin S1 to the cryo-electron microscopy (EM) density maps of actin filaments saturated with S1. These models provided valuable insights into actomyosin interactions. Biochemical studies, involving mutational9-12 and cross-linking analysis of acto-S1 complexes13, confirmed the proposed involvement of some protein segments in acto-S1 interactions. However, the surface loops of myosin (loops 1-3), which were predicted to interact with actin by biochemical studies, were not identified at the atomic/side-chain level in prior acto-S1 models because the electron density corresponding to these loops was missing in the X-ray structures of S1. In the most recent acto-S1 model14, the surface loops of S1 and the actin D-loop were simulated by molecular dynamics using cryo-EM data as a constraint. This led to a higher resolution map of the acto-S1 interaction surface and the discovery of potentially important contacts between dynamic loop elements of actin and S1. However, the newly proposed interactions await conformation by biochemical approaches.
With this goal in mind, we used radiolytic oxidative protein footprinting and mass spectrometry to probe the global actin surface in the acto-myosin complex with a resolution at the level of single amino acid side chains. In this method, changes in surface accessibility of a protein upon its complex formation with a ligand are monitored by measuring the degree of protection of its reactive residues from oxidation by hydroxyl radicals. This approach has been successfully applied to large proteins and oligomers, including monomer and filament structures of actin, and has identified the binding interfaces for a number of protein complexes15-26, including the Arp2/3-WASp (total mass of 220 kD)26 complex. Actomyosin complex is the largest protein assembly studied so far by this approach; a megadalton filament with each actin monomer of 42 kD binding myosin S1 of 115 kD; i.e., forming a unique sequence of repeating units of 157 kD.
In this study, we analyzed the reactivity of the actin filament residues in the absence and presence of S1 and identified the residues that are likely involved in the rigor interaction with myosin. We provide strong evidence for the predicted hydrophobic and ionic interactions at the acto-S1 interface and show previously unknown allosteric changes on actin due to S1 binding. Focusing on two structural elements of actin proposed to interact with myosin, we show that the emission properties of fluorescent labels and the mobilities of spin probes attached to actin’s D-loop and W-loop (residues 166-170) change in a sequence dependent manner upon S1 binding, consistent with their direct interactions with S1.
Results
Radiolytic protein footprinting identifies site specific changes in actin upon S1 binding to actin filaments
Actin and actin/myosin samples were exposed to a white synchrotron X-ray beam for intervals from 0 to 80 ms. Under these experimental conditions, the reaction of amino acid side chains with hydroxyl radicals occurs at rates 10 to 1000 times faster than hydrogen abstraction from the Cα carbon 18; 27. Therefore, protein backbone cleavage is negligible in our experiments and no structural perturbation is expected. The subsequent tryptic proteolysis of these samples was followed by separation of the peptide mixtures by nano-HPLC, and quantification of oxidized species by mass spectrometry (Thermo’s LTQ-FT, LTQ-OrbiTrap, and LCQ DecaXP). The resulting dose response curves were analyzed to determine the modification rate constants. A total of twenty tryptic peptides of actin covering ~75 % of its sequence were analyzed. Among them, three peptides showed no oxidation throughout. Modification rates for all twenty peptides are listed in Table 1.
Table 1.
Modification rates for actin peptides in absence and presence of S1.
| Peptide | Sequence | Oxidized Residues | Subdomain | Modification rate, k (s-1)
|
Degree of Protection | |
|---|---|---|---|---|---|---|
| F-actin, kA | F-actin+S1, kAS1 | |||||
| 1 – 18 | DEDETTALVCDNGSGLVK | L8, C10, L16, K18 | 1 | 3.0 ± 0.2 | 0.4 ± 1.0 | 7.5 |
| 19-28 | AGFAGDDAPR | F21 | 1 | 0.29± 0.02 | 0 | High |
| 29-37 | AVFPSIVGR | P32 | 1+2 | 0.40 ± 0.12 | 0 | High |
| 29-39 | AVFPSIVGRPR | P32, P38 | 1+2 | 1.1 ± 0.1 | 0 | High |
| 40-50 | HQGVMVGMGQK | H40, M44, M47 | 2 | 1.6 ± 0.21 | 0.6 ± 0.2 | 2.7 |
| 51-61 | DSYVGDEAQSK | - | 2 | 0 | 0 | NA |
| 63-68 | GILTLK | - | 2 | 0 | 0 | NA |
| 69-84 | YPIEHGIITNWDDMEK | Y69, H73, W79, M82 | 2+1 | 6.2 ± 0.4 | 0.38 ± 0.06 | 16.3 |
| 85-95 | IWHHTFYNELR | W86, H87, H88, F90, Y91, R95 | 1 | 1.2 ± 0.1 | 0.04 ± 0.08 | 30 |
| 96-113 | VAPEEHPTLLTEAPLNPK | E99, E100, H101, P102, P112/K113 | 1 | 0.46 ± 0.03 | 0.02 ± 0.03 | 23 |
| 119-147 | MTQIMFETFNVPAMYVAIQAVLSLYASGR | M119, M123, F124, Y143 | 1+3 | 3.5 ± 0.3 | 0.028 ± 0.005 | 125 |
| 148-177a | TTGIVLDSGDGVTHNVPIYEGYALPHAIMR | H161, Y166, Y169, L171, P172, H173, M176 | 3 | 23.6 ± 0.7 | 0.21 ± 0.05 | 114 |
| 184-191 | DLTDYLMK | M190 | 4 | 4.2 ± 1.1 | 0.13 ± 0.03 | 32 |
| 197-206 | GYSFVTTAER | R206 | 4 | 0.19 ± 0.02 | 0.06 ± 0.02 | 3.2 |
| 239-254 | SYELPDGQVITIGNER | Y240, P243, D244, R254 | 4 | 1.7 ± 0.1 | 0 | High |
| 292-312 | DLYANNVMSGGTTMYPGIADR | D292, M299, M305, P307 | 3 | 1.9 ± 0.2 | 0.8 ± 0.1 | 2.4 |
| 316-326 | EITALAPSTMK | P322, M325, K326 | 3 | 0.5 ± 0.1 | 0.0 ± 0.1 | High |
| 329-335 | IIAPPER | - | 3 | 0 | 0 | NA |
| 336-359 | KYSVWIGGSILASLSTFQQMWITK | Y337, L346, L349, F352, M355 | 3+1 | 29 ± 8 | 0 | High |
| 360-372 | QEYDEAGPSIVHR | Y362, P367, H371 | 1 | 1.0 ± 0.1 | 0.5 ± 0.1 | 2.2 |
Since oxidation by hydroxyl radical is a function of a residue’s solvent exposure and its intrinsic reactivity, residues at the interface of a protein/protein complex are expected to be protected and exhibit a decreased rate of modification. Remarkably, all seventeen reactive peptides of actin showed decreased oxidation rates upon myosin binding, with their relative protection from hydroxyl radicals ranging from a two-fold decrease to complete absence of oxidation (Figure 1). Figure 2 depicts the locations of the protected peptides (Figure 2A) and their reactive probes (Figure 2B) superimposed on the structure of an actin dimer from a recent acto-S1 model 14. The most strongly protected sites on actin (Figure 2, in red) include four peptides with complete protection from oxidation and two peptides with >100–fold protection. Notably, four additional peptides showed fluctuations in protections ranging from 2-fold to complete (Figure 2, in orange). Among the remaining actin peptides two showed moderate protection by S1, in the range 15-35-fold (in yellow), and four showed modest protections, in the range of 2-3-fold (in lime).
Figure 1.
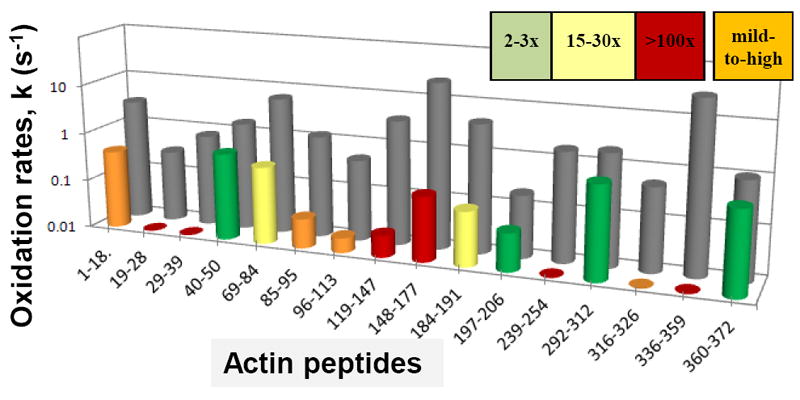
Oxidation rates of F-actin peptides in the presence (front row) and absence (back row) of S1. Oxidation rates are listed in Table 1 and are shown here on a log scale. Color codes refer to different levels of actin peptides protection by S1 from oxidation and are used also in Figures 2 and 3. For the orange code peptides (mild to high protection) the degree of protection listed in Table 1 corresponds to the mean experimental values. The orange bar code reflects the variation observed for these peptides. For F-actin peptide 29-39, the average of oxidation rates of peptides 29-39 and 29-37 (as listed in Table 1) is shown for the F-actin control.
Figure 2.
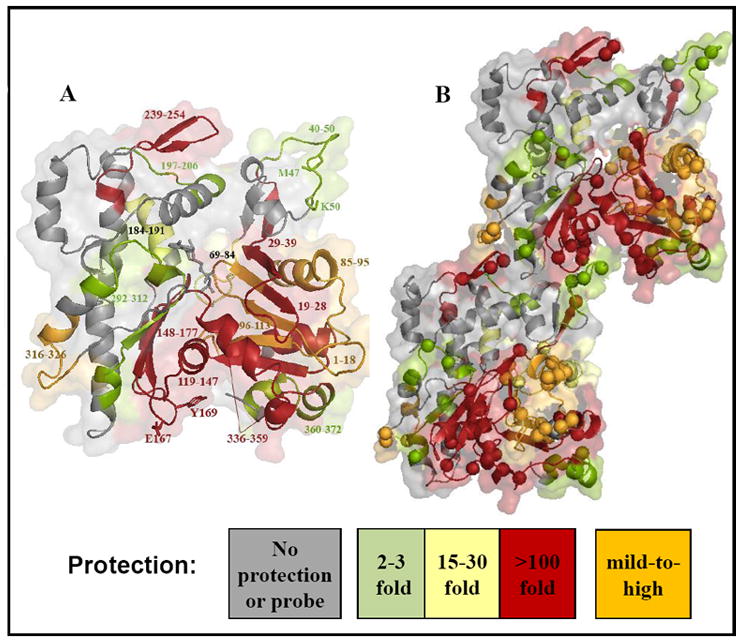
Peptides of actin protected by S1 from oxidation as mapped by radiolytic footprinting. The peptides are shown on the G-actin structure (A) and on the dimer structure (B) adopted from the Lorenz and Holmes acto-S1 model (2010)14. The spheres show amino acids modified on each of the probed peptides.
The oxidized probe sites within each peptide obtained from tandem MS data18; 28 are summarized in Table 1. In our experiments, we were able to detect even trace level modifications using an upgraded beamline with a relatively high flux X-ray, which causes more modifications, and by using highly sensitive mass spectrometers such as LTQ-FT and LTQ-OrbiTrap (see Materials and Methods). For example, we have detected a number of modified glutamic acid residues (Table 1) which usually have relatively low reactivity with hydroxyl radicals27. In total, nearly 60 residues distributed throughout the actin structure were found oxidized, enabling us to evaluate the changes occurring across the entire actin molecule upon S1 binding to the filaments (Figure 2A and B).
Radiolytic footprinting confirms the myosin binding surface and reveals allosteric changes on actin
Previous radiolytic protein footprinting experiments suggest that residues within the interface of a protein/ligand complex experience significant protection from oxidation18; 20-22; 24; 29-31. Thus, highly protected and clustered actin peptides (Figures 2 and 3A, in red and orange), are strong candidates for forming the actomyosin interface. However, significant levels of protection at any particular site can be also observed due to allosteric changes transmitted from a close or distant interface. The fact that all reactive peptides on actin showed protection upon S1 binding indicates that at least some of these peptides cannot simply be masked due to direct S1 binding. However, in the experiments with protein complexes, the presence of more total protein in the sample compared to the free states can also result in up to a 2-3 fold suppression of overall radiolysis32. Protections over this level are then due to formation of an interface or due to allosteric changes of complex formation. We suggest that decreases in oxidation rates at the level of 2-3-fold should be considered only as none to modest protections for this study, meriting independent testing of their significance. This leads us to emphasize the large protections of 10-100 fold (or even more) that are the most significant data.
Figure 3.
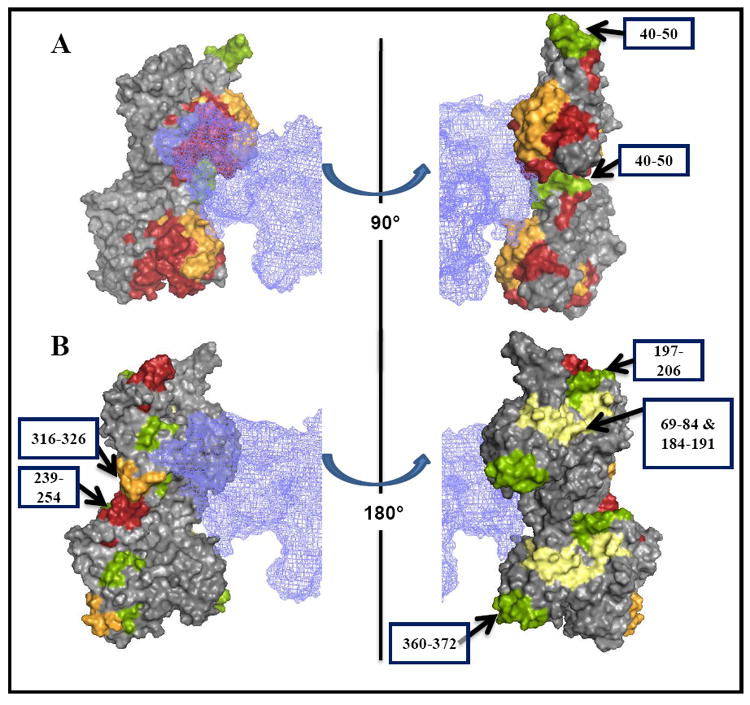
Comparison of radiolytic footprinting data with the acto-S1 model of Lorenz and Holmes (2010) 14 identifies actin segments affected allosterically by S1. S1 protection of actin from oxidation is shown on a surface of actin dimer using the same color code for protection levels as in Figure 1. Myosin S1 is represented by a blue meshwork. Actin peptides involved in direct interactions with S1 are highlighted in (A). The arrows (B) point to the regions of actin that are most likely affected allostericaly by myosin binding.
In light of this, we superimpose (Figure 3) our footprinting results onto the most recent Lorenz and Holmes acto-S1 model to better define the likely interaction sites as opposed to those changes likely due to allostery14 (Figure 3B). As shown in Figures 3A and 3B, actin peptides that are ~100- fold or completely protected (in red) are at or in close proximity to the primary binding site for myosin, except for the 239-254 peptide. The oxidation probes in these peptides were hydrophobic and aromatic residues, including Phe21, Pro32, Pro38, Met119, Met123, Phe124, Tyr143, His161, Tyr166, Tyr169, Leu171, Pro172, His173, Met176, Tyr337, Leu346, Leu349, Phe352, and Met355 (Table 1).
Four actin peptides protected significantly by S1 showed large variations in their oxidation rates (exceeding 100%; Figure 1 and 3; in orange). This can be attributed to a wide distribution of protection levels by S1, from minimal or none to complete protection. Such variations may arise from conformational flexibility of these regions, or perhaps water dynamics/diffusion at the ionic sites environment. The four sites include peptide 1-18 (kA = 3.0 ± 0.2 s-1 and kAS1 = 0.4 ± 1.0 s-1, where kA and kAS1 denote the modification rate constants for actin and actin/myosin S1 respectively) - showing an average 8-fold protection, peptide 85-95 (kA = 1.2 ± 0.1 s-1, kAS1 = 0.04 ± 0.08 s-1) - showing an average 30-fold protection, peptide 96-113 (kA = 0.46 ± 0.03 s-1, kAS1 = 0.02 ± 0.03 s-1) - showing an average 23-fold protection, and peptide 316-326 (kA = 0.5 ± 0.1 s-1, kAS1 = 0.0 ± 0.1 s-1) - showing virtually complete protection. These peptides (Figure 3A, in orange), except for 316-326, flank the clusters of highly protected peptides (Figure 3A, in red), with 1-18 belonging to an upper actin and 85-95 and 96-113 located on a lower actin. Our data is in agreement with previous work that suggests potential electrostatic interactions between these peptides and the myosin loops.6; 8; 14; 33 The probe residues belonging to these peptides are Leu8, Cys10, Leu16, Lys18, Trp86, His87, His88, Phe90, Tyr91, Arg95, Glu99, Glu100, His101, Pro102, Pro112/Lys113, Pro322, Met325, and Lys326 (Table 1).
New vicinal and/or long range allosteric effects of S1 on actin are suggested by the protection of several actin peptides. Among them is the peptide 239-254, which does not overlap with the S1 electron density in the recent14 and previous4; 6; 33 acto-S1 models. Similarly, peptides 69-84, 184-191, 197-206, and 316-326 have significantly reduced oxidation rates in the actomyosin complex. The location of these peptides in cryo-EM based acto-S1 model (Figure 3B) suggests that the observed oxidation effects are of allosteric nature (where they are above 3 fold), confirming extensive changes in actin dynamics by myosin.
Site-directed labeling with fluorescent and EPR spin probes refines the actomyosin interface mapping
In the earlier actomyosin models6; 33, the N-terminal part of the D-loop (residues 40-42) was placed in close proximity to the helix-loop-helix motif of myosin (residues 529-558). However, in the recent acto-S1 model14, it is the C-terminal part of the D-loop (residues 44-50) that is proposed to form hydrogen bonds and electrostatic interactions with S1. Our radiolytic protein footprinting experiments showed only three-fold protection of the D-loop peptide (residues 40-50) by S1. Based on our prior work24 this is not a clear indication of direct binding to the loop, unless there is considerable plasticity of its interaction with S1. Therefore, we employed site-directed labeling method to clarify the difference between our footprinting results and the proposed role of the D-loop in myosin binding in acto-S1 models. For these experiments, single cysteine mutants of yeast actin D-loop residues34; 35 were labeled at a 1:1 mole ratio with a small fluorescent probe acrylodan, and the changes in the emission properties of acrylodan-actin were followed upon S1 binding. Interestingly, S1-induced changes in the emission spectra of acrylodan attached to mutants C45 (V45C/C374S), C46 (G46C/C374S), C47 (M47C/C374S), C48 (G48C/C374A), C49 (Q49C/C374A) and C50 (K50C/C374A) were pronounced, while almost no change was induced in labeled mutants C40 (H40C/C374S), C41 (Q41C/C374S), and C43 (I43C/C374S) (C43 and C47 acrylodan emission spectra are shown as an example in Figure 4A and 4B, respectively). The fluorescence intensity of acrylodan attached to C46 and C47 increased strongly, with the accompanying 12 nm and 21 nm blue shifts (Table 2), respectively, suggesting a significant burial of the label upon S1 binding. In addition, the acrylodan fluorescence intensities and wavelength maxima were changed by S1 for mutants C48, C49, and, C50 (Table 2).
Figure 4.
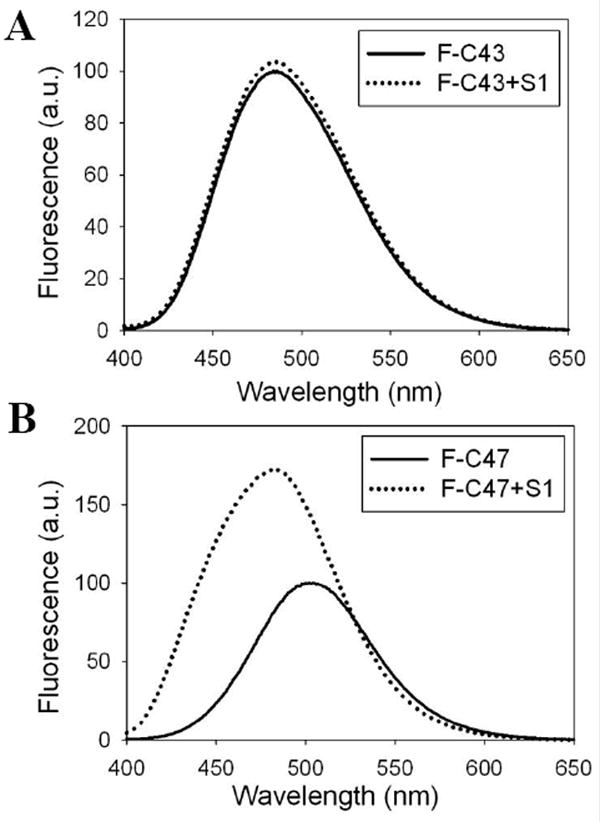
Site-directed fluorescence labeling reveals myosin induced conformational changes in dynamic actin loops. Representative acrylodan emission spectra of yeast F-actin mutants labeled at residue 43 and 47 in the absence (solid lines) and presence (dotted lines) of S1 are given in 4A and 4B, respectively.
Table 2.
Fluorescence properties of acrylodan conjugated D-loop and W-loop cysteine mutants of yeast actin
| Probed residues | λmax emission (nm) | Ratio of Fmax | |
|---|---|---|---|
| D-loop | F-actin | F-actin + S1 | (F-actin+S1)/F-actin |
| C40 | 496 | 495 | 1.0 |
| C41 | 506 | 507 | 1.1 |
| C43 | 485 | 486 | 1.0 |
| C45 | 495 | 502 | 1.0 |
| C46 | 499 | 487 | 1.6 |
| C47 | 504 | 483 | 1.7 |
| C48 | 485 | 507 | 0.9 |
| C49 | 510 | 514 | 0.9 |
| C50 | 512 | 509 | 1.1 |
|
| |||
| W-loop | |||
|
| |||
| C167 | 519 | 517 | 1.20 |
| C169 | 496 | 495 | 1.01 |
To test further the effect of S1 on residues 47-50 on actin, we examined also the mobility of R1 EPR spin labels attached to these residues (Figure 5A and Figure 5B). As seen in Figure 5 (A and B), the EPR spectra reveal that R1 at these positions is relatively mobile and unrestricted in F-actin. However, in the S1 bound filaments the mobility of R1 is greatly reduced, with the EPR spectra showing a population of immobilized spin states that can represent the fraction of spins interacting directly with S1. Taken together, our fluorescence and spin labeling experiments reveal a significant rearrangement of the environment of C-terminal D-loop residues upon S1 binding, supporting the proposed interactions of this site with S1 peptide 543-554. 14
Figure 5.
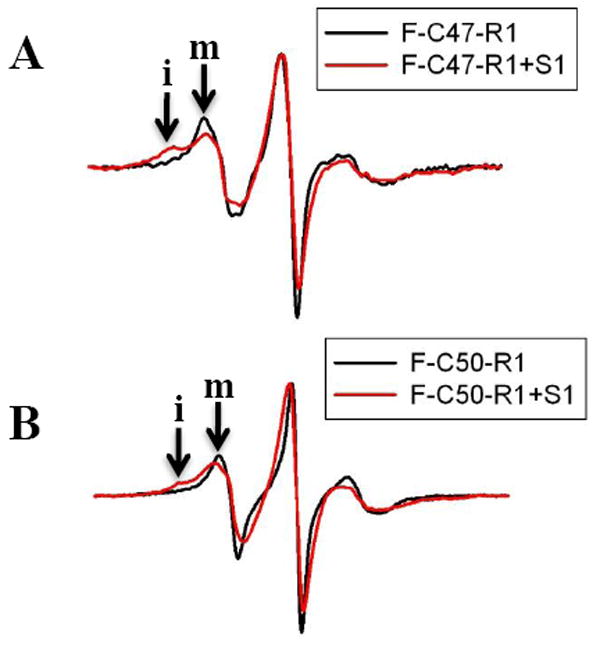
Site-directed spin labels reveal myosin induced conformational changes in actins’ D-loop. EPR spectra of R1 labeled C47 and C50 mutant actin filaments in the absence (black) and presence (red) of S1 are given in 5A and 5B, respectively. Filament spectra in the absence of S1 are normalized to an equal number of spins. In addition, the spectra in the presence of S1 are normalized to the positive EPR central line height of the corresponding F-actin spectrum to emphasize the immobilized spin states observed after S1 binding. The scan width is 100 gauss and “m” and “i: denote the mobile and immobile spin fractions, respectively.
To test the proposed ionic interaction of K50 on actin with E576 on S1, we used the available K50A/D51A yeast actin mutant36 in functional actomyosin assays. While the Vmax (2.8 ± 0.5 s-1) and Km (17 ± 8 μM) were virtually the same for the wt and mutant yeast actin with S1, the strong binding of S1 to the mutant was enhanced ~3-fold (6.0×106 M-1) compared to that to wt actin. In conjunction with this increase, the in vitro motility (sliding) of actin over myosin and the relative force developed in that assay were increased by ~20% for the mutant compared to wt actin (Figure 6). Although these results do not attest to a significant contribution of K50 or D51 on actin to the actomyosin cycle, they show again the sensitivity of actomyosin interactions to any manipulation of D-loop on actin.
Figure 6.
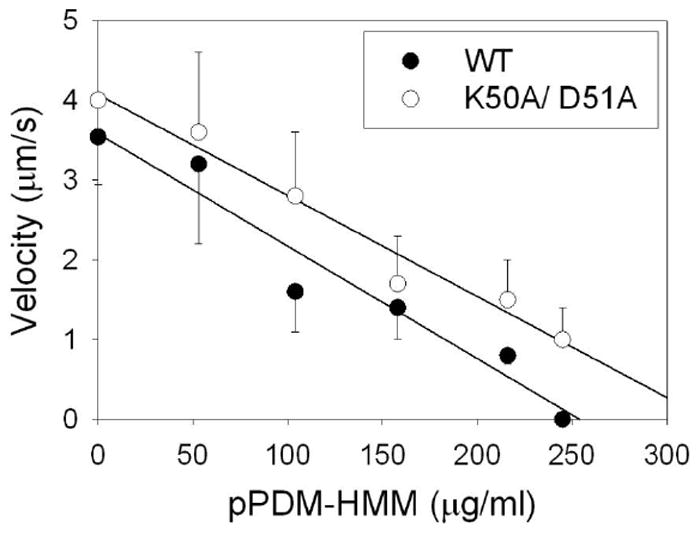
Effect of K50A/D51A mutations on the sliding speed of F-actin over HMM as measured by the pPDM-HMM load. The motilities of actin filaments are measured as the function of pPDM-HMM applied to the assay surface. The sliding of between 100 and 300 filaments is monitored in each case.
The W-loop of actin is an important interaction site for numerous actin binding proteins37, including myosin. It has been recently proposed that Glu167 on the W-loop of actin forms a salt bridge to Lys544 on myosin.14 In our radiolytic footprinting experiments, the tryptic peptide containing the W-loop (148-177 peptide) shows over 100-fold protection by S1, agreeing with the model, and includes Y166 and Y169, which flank G167, as important probe sites. To test the W-loop on actin, we used the previously characterized single cysteine mutants C167 (A167C/C374A) and C169 (F169C/C374A)34 of this loop. We found that the emission of acrylodan attached to C169 was unaffected by S1 binding to F-actin, while a 30% higher fluorescence of the probe at residue 167 was observed upon S1 binding (Table 2). Overall, these results are consistent with the proposed direct interaction of residue 167 with S1.
Discussion
Myosin binding changes the global dynamics of actin
Actin filaments are flexible and dynamic double stranded polymers38, with variations of few degrees in the twist of the long pitch actin helix39. These variations in twist and tilt are believed to create different states of the filament, which may be functionally critical to its interactions with actin binding proteins. Thus, for example, chemical cross-linking of actin’s D-loop on one protomer (residue 41) to the C-terminus (residue 374) of an adjacent protomer in the filament inhibited the in vitro motility of myosin over actin without changing their binding affinity for each other.40 This indicates that the actin filament dynamics and its modulation by myosin, which must be inhibited by the above cross-linking, are essential for force generation by actomyosin. In the present study, the oxidation of all peptides probed on actin is reduced by myosin, suggesting global changes on actin and at least partial restriction of its breathing motions due to S1 binding.
Global and/or transmitted effects of S1 on actin are reflected in the oxidation inhibition of peptides that are outside the previously mapped contact areas of actin and S1 (Table 3). These effects may be related to the general plasticity and couplings of structural elements on actin. For example, there is direct and indirect evidence for the modulation of the actin nucleotide cleft by S1. This evidence includes changes in εATP fluorescence on F-actin induced by S141 and by antibodies to residues 18-29 on actin42 that recognize D24 and D25 (which are involved in S1 binding14). Thus, it is not surprising that the local environment and reactivity of residues surrounding the nucleotide cleft i.e., peptides 69-84 and 184-191, is affected by S1 binding in our radiolysis oxidation experiments. In addition, the recently proposed structural coupling between the actin’s nucleotide cleft and its W-loop 34 might explain the protection of nucleotide cleft peptides from oxidation. Current acto-S1 models (listed in Table 3) suggest a direct interaction between myosin and the W-loop, which may then impact the nucleotide cleft. W-loop-myosin interaction is supported also by our solution results, as discussed below.
Table 3.
Summary of potential contact areas between actin and myosin
| Type of interaction | Actin | aMyosin | |
|---|---|---|---|
| Peptide | Residues probed in this study | Peptides from Cryo-EM models | |
| Ionic | D1-K18 | L8, C10, L16, K18 | Y626-Q647A,B,C,D |
| A19-R28 | F21 | ||
|
| |||
| Hydrophobic and ionic | A29-R39 | P32, P38 | N552-H558A,B |
|
| |||
| Potential hydrogen bonding | G46-K50 | C46, C47, C48, C49, C50 | S549-H558C(partial),D |
|
| |||
| Ionic | I85-R95 | W86, H87, H88, F90, Y91, R95 | K567-H578A,B,C,D |
| V96-K113 | E99, E100, H101, P102, P112/K113 | ||
|
| |||
| Hydrophobic and ionic | M119-R117 | M119, M123, F124, Y143 | P529-K553A,B,C,D& Y403-G416 (for 336-359 peptide)A,B,C,D |
| T148-R177 | H161, Y166, Y169, L171, P172, H173, M176 | ||
| R336-K359 | Y337, L346, L349, F352, M355 | ||
Actin residues are those probed in the present study.
Myosin segments proposed to bind actin in the previous electron microscopy based models of acto-S1 (
Rayment et al., 1993,
Milligan, 1996,
Volkman et al., 2000, and
Lorenz and Holmes, 2010.).
In the same vein, because each S1 binds two actin protomers, it can be expected to change the local environment at the interprotomer interfaces in F-actin. Such changes are most likely behind the effect of S1 on the fluorescence of the pyrene probe attached to C265 on actin43 and – in this work - mild to no protection of peptides 197-206 and 360-372 from oxidation is seen. Ample experimental evidence confirms the effects of S1 on the C-terminal region of actin, including its protection from tryptic cleavage44, the effect on fluorescent probes attached to C37445; 46, and the cross-linking of the C-terminus to the D-loop of an adjacent actin (our unpublished results).
The protection of the 316-326 peptide may be induced through the proposed interaction of D311 and Q314 on actin with R371 on S1.14 Alternatively, the effect of S1 on peptides 316-326 and 239-254 may be attributed to its overall impact on interprotomer interfaces in the filaments. Consistent with this interpretation is the observation that a specific subtilisin cleavage of actin between S234 and S235 i.e., close to the 239-254 peptide and not to the bound S1, reduces by ~20% both the Vmax of acto-S1 ATPase and the in vitro motility of actin over myosin.47 Taken together, the above discussed reactivity changes at several actin sites reveal wide-spread alterations on actin upon myosin binding.
Hydroxyl radical footprinting reveals the ionic contact surface of myosin on actin
We suggest that the peptides 1-18, 19-28, 85-95, and 96-113 of actin engage in ion-pairing interactions with myosin. This hypothesis is supported by prior mutational, cross-linking and cryo-EM studies (Table 3). The charged N-terminal residues of actin (D1, E2, D3, E4, D24, D25) of upper actin are predicted by several EM mappings of the acto-S1 complex4; 6; 14; 33 to pair with the lysine rich surface loop 626-647 on myosin (also called the 50k/20k loop or loop 2). The fact that the N-terminus48 and residues E99 and E10049 of actin can be cross-linked to the 50k/20k loop of myosin supports the EM maps. Similarly, peptides 85-95 and 96-113 have been proposed in prior acto-S1 models4; 6; 14; 33 to form hydrogen bonds and electrostatic interactions with a charged loop 567-578 on myosin (loop 3), presenting the so called secondary actin binding site to myosin in the rigor complex.
Prior studies attempted to assess the importance of some of the above ionic interactions of actin with S1. The substitution of N-terminal acidic actin residues to alanine decreased ~3-fold both the weak (in the presence of MgATP) and strong binding of S111, while the replacement of D24, D25 or E99, E100 decreased only the weak S1 binding12. Yet, the presence of these residues clusters was essential for the in vitro motility of actin over myosin under physiological salt conditions (I = 150 mM). These results led to the conclusion that the main effect of the above charge clusters was on the population of weakly bound actomyosin states, affecting in this way the transition of myosin heads to the force generating step of the cross-bridge cycle 11.
The primary binding site of myosin on actin is identified by hydroxyl radical footprinting
All strongly protected (over 100-fold) actin peptides (29-39, 119-147, 148-177, 239-254, and 336-359), except for peptide 239-254, match the prior predictions of acto-S1 models summarized in Table 3. These models6; 14; 33 predicted that the cleft helices of an upper actin (Tyr143-Thr149, Val339-Gln354) and part of the D-loop of lower actin may interact with the 529-558 ‘helix-loop-helix’ segment of myosin. This segment is thought to be the primary actin binding site on S1 and is aligned with a number of hydrophobic and charged residues on actin. Thus, our footprinting data provides supporting evidence that actin segments 119-147, 148-177, and 336-359 are involved in dominant interactions with myosin.
Moreover, another myosin surface loop, the cardiomyopathy loop (Y403-G416), lies close to actin residues 330-345 in the EM maps.6; 14 A number of charged residues (K328, E334, R335, and K336) in this region appears to interact with oppositely charged residues in the cardiomyopathy loop. This interaction is important for normal actomyosin activity, as revealed by mutations in human β cardiac myosin13, and may contribute to the protection of peptide 336-359 on actin by S1 observed in this study.
Actin D-loop and W-loop interact with myosin in a site-specific manner
The D-loop of actin has been proposed to participate in myosin binding, but a consensus has not been reached regarding the resulting acto-S1 binding interface. Several reports implicated the N-terminal residues in S1 binding4; 6, whereas the more recent study14 identified hydrogen bonding interactions between the C-terminal residues of D-loop and the helix-turn-helix motif of myosin. Unexpectedly, only weak protection of the 40-50 actin peptide by S1 was observed in our oxidation rate measurements. To gain more detailed information on this interface, we have scanned the D-loop residues and their environment via cysteine mutagenesis and fluorescence labeling. The reported changes in fluorescence identified the C-terminal part of the D-loop as the primary region affected by myosin binding. The emission properties of acrylodan were changed much more when the probe was attached to the C-terminal than the N-terminal cysteine mutants (Table 2). Our results explain the low protection (~3-fold) by S1 of the D-loop peptide from oxidation: only one (M47) of three (M47, M44, H40) probe residues are in the myosin binding region identified by cysteine scanning. Moreover, as shown by EPR measurements with spin probes attached to C47 and C50 mutant residues on actin, the C-terminal part of D-loop is dynamic and retains some of its conformational freedom even in the myosin bound state. The EPR spectra reveal populations of immobilized and mobile spin states in the acto-S1 complex (Figure 5). Such conformational plasticity of the D-loop can contribute to its low protection from oxidation. Overall, our results point to the participation of the C-terminal part of D-loop in myosin binding, in direct support of recent EM predictions.14 This conclusion is consistent also with biochemical results showing that subtilisin cleavage of D-loop loop between M47 and G48 inhibits the in vitro motility of cleaved actin filaments over myosin.50; 51
In addition to the D-loop, we have probed also via site-directed acrylodan labeling the involvement of residues 169 and 167 in interactions with myosin. Interestingly, acrylodan attached to cysteine residue 167 (but not 169) was sensitive to myosin binding, agreeing again with the proposed hydrogen bonding interaction between Glu167 and Lys544 on myosin.14
Structural and functional implications
The main approach taken in this study - to map the myosin induced changes on actin via ms time scale oxidative modification of its reactive residues - has the unique advantage of global analysis and simultaneous identification of all regions on actin that are affected by the binding of myosin. The information obtained in this way is free of any possible perturbation or changes that can be introduced by mutational or labeling dependent probing of interaction interfaces. Our results identified a number of actin residues and peptides that form both the ionic and hydrophobic actomyosin interfaces (Figure 2 and 3). Our data provide direct experimental support for the EM/molecular dynamics based model of the acto-S1 interface.14 We present also strong evidence for the involvement of the C-terminal part of actin’s D-loop and W-loop in the interactions with S1. Moreover, in addition to probing the myosin binding surface on actin by footprinting and spectroscopic methods, our data provide the most detailed biochemical mapping of allosteric and/or indirect effects of myosin on actin. These changes are more extensive than known until now and should stimulate interest in the role of actins dynamics and structural transitions in actomyosin function.
Materials and Methods
Sample preparation
Actin from rabbit skeletal muscle was purified by the method of Spudich and Watt52. Myosin subfragment 1 (S1) of rabbit skeletal muscle was prepared by chymotryptic digestion of full-length myosin as described by Weeds and Taylor53. The molar concentrations of actin and myosin S1 were determined from their absorbances at 280 nm (ε1% of 11.0/cm and 7.9/cm respectively) and molecular weights (42 kDa and 115 kDa respectively). These proteins were dialyzed separately against their radiolysis buffers at pH 7.7 (10 mM cacodylate, 50 mM KCl, and 2 mM MgCl2 with additional 0.2 mM ATP for actin) at 4°C overnight. Acto-S1n complex was prepared by mixing equimolar concentrations of actin and myosin subfragment 1 in 1:1 volumetric ratio at 4°C. Immediately thereafter, solutions of actin, acto-S1, and radiolysis buffer were transported at 4-10°C to Brookhaven National Laboratory (Upton, New York), and were subjected to radiolysis the next day. Methionine amide (Met-NH2) was purchased from Bachem Bioscience Inc., and sequencing grade modified trypsin was obtained from Promega (Madison, WI). HPLC grade water and acetonitrile were purchased from Burdick& Jackson (Muskegee, MI). HPLC grade formic acid (99%) was purchased from Acros Organics (New Jersey).
Site-directed labeling and spectroscopic methods
Previously characterized34; 35 single cysteine yeast actin mutants H40C/C374S, Q41C/C374S, I43C/C374S, V45C/C374S, G46C/C374S, M47C/C374S, G48C/C374A, Q49C/C374A, Q50C/C374A, E167C/C374A, F169C/C374Aand the mutant K50A/D51A36 were grown in YPD media and purified using a DNase I affinity column following the previous protocol35. The purified G-actin was kept in buffer A [10 mM HEPES (pH 7.4), 1 mM DTT, 0.2 mM ATP, and 0.2 mM CaCl2] on ice.
Before site-directed labeling, the reducing reagent was removed by applying actin onto a Sephadex G-50 column pre-equilibrated with buffer A. Next, the acrylodan (6-acryloyl-2-dimethylaminonaphthalene, Invitrogen, Eugene, OR) labeling was achieved by reacting DTT free actin with a 1.1-1.2 fold mole excess of label for 2-3 hours, at 24°C. After quenching the labeling reactions with 1mM DTT, actin was converted to Mg-G-actin with 0.05 mM MgCl2 and 0.4 mm EGTA, and then polymerized with 2 mM MgCl2 and equimolar phalloidin. Fluorescence emission spectra of acrylodan labeled actins were recorded between 400-650 nm at 385 nm excitation wavelength in a Photon Technology International (PTI, Lawrenceville, NJ) spectrofluorometer, using FeliX32 Analysis, version 1.1 (PTI).
For site-directed spin labeling, the DTT-free G-actin in buffer A was incubated for an hour at 24°C with a 5-fold molar excess (over actin) of the nitroxide labeling reagent (MTSL [(1-oxy-2,2,5,5-tetramethylpyrrolinyl-3-methyl)methanethiosulfonate])54 which was kindly gifted by Dr. Kálmán Hideg (University of Pécs, Hungary). The unreacted label was removed by passing labeled actin through a Sephadex G-50 column in buffer A. Following this step, spin labeled actin (R1-G-actin) was concentrated to 25-30 μM using Amicon 4 mL concentrators (Millipore, USA). F-actin was prepared from R1-G-actin as described for acrylodan labeled samples and then placed in a glass capillary (VitroCom, Inc., Mountain Lakes, NJ). The X-band EPR spectra were recorded with a Varian 109 spectrometer fitted with a loop gap resonator, using a scan time of 30 s, a field scan of 100 G, a modulation amplitude of 1.0 G at 100 kHz, and an incident microwave power of 2.0 mW.
Synchrotron X-ray radiolysis
Radiolysis experiments of actin, and acto-S1 complex at 15 μM concentration were performed at ambient temperature at X-28C beam-line of the National Synchrotron Light Source, Brookhaven National Laboratory (Upton, New York) at various X-ray exposure times ranging from 0 to 80 ms as described previously.23; 28; 29; 31; 55 10 mM Met-NH2 was added right after exposure to quench secondary oxidation56 and samples were frozen immediately thereafter.
Liquid chromatography - Mass spectrometry
Radiolyzed samples were digested with trypsin29; 31 at an enzyme to protein ratio of 1:20 (w/w) at 37°C overnight. The digestion reaction was terminated by freezing the samples. Just before the mass-spectrometry experiment, the digests were diluted to 200-500 femtomole/μL with 0.1% formic acid. Online RP–HPLC–tandem mass spectrometric analysis of the tryptic digests was performed using LTQ-FT,LTQ-OrbiTrap, and LCQ DecaXP (Thermo Fisher, San Jose, CA) equipped with a nanoelectrospray ionization source and operated with the Xcalibur data acquisition software. LTQ-FT and LTQ-OrbiTrap are coupled to Dionex Ultimate 3000 nano-HPLC, and DecaXP is coupled to Famos-Switchos-Ultimate nano-HPLC, all from LCPackings, Bannockburn, IL. Approximately 1 picomole of protein digest was loaded onto a PepMap C18 capillary trap of 300 μm i.d. × 5 mm (LCPackings, Bannockburn,IL) and desalted (0-5 % acetonitrile, 0.1% formic acid with a loading flow of 20-30μL/min) for 5 min prior to injection onto a PepMap100 C18 column of 75 μm i.d. × 150mm (LCPackings, Bannockburn,IL). Following peptide desalting and injection onto the analytical column, a linear gradient of 2 % per min was carried out at 250-300 nl/min using solvent A (0-5 % acetonitrile/water and 0.1 % formic acid) and solvent B (80-95 % acetonitrile/water and 0.1 % formic acid). Spray voltage and capillary temperature during the gradient run were maintained at 1.8-2.2 kV and 250 °C, respectively. The conventional full scan data-dependent acquisition (DDA) mode was utilized in LTQ-FT and DecaXP over the m/z range 200-2000. A highly accurate survey scan (~2 ppm accuracy) resulted in Fourier transform ion cyclotron resonance (FTICR) detection in the LTQ-FT, followed by parallel MS/MS linear ion-trap analysis of the top five most intense parent ions from the survey scan and three most intense ions from the ‘parent mass list’ provided (actin peptides). DecaXP was used to capture some additional ions that were missed by the LTQ-FT. LTQ-OrbiTrap was used for targeted MS/MS scans. Selected ion monitoring data-dependent acquisition mode (SIM DDA) was utilized with parent ion detected in the OrbiTrap cell (accuracy ~ 2 ppm) over a window of 3.0 Da, followed by the parallel MS/MS linear ion-trap analysis of the most intense ion (found in the preview SIM scan) from the external ‘parent mass list’. The instruments are operated with the automatic gain control (AGC) mode of ion trapping. CID in the linear-ion trap was performed using a 3.0 Da isolation width and 35% normalized collision energy with helium as the target gas.
Modification rate calculation
The ‘fraction unmodified’ of a given peptide from the chromatogram was calculated as the ratio of integrated area of the unmodified peptide to the sum of integrated areas from the modified and unmodified peptides. The dose response curves of ‘fraction unmodified’ vs. ‘X-ray exposure time’ were fitted to fist order decay function with Origin (Origin Labs, Northampton, MA) to determine the modification/oxidation rate constant. The detailed procedures have been described previously. 23; 28; 57; 58
Identification of modification
The sites of oxidation were determined from the MS/MS experiment in the conventional full scan data dependent acquisition (DDA) mode in LTQ-FT and in the SIM DDA mode in the LTQ-OrbiTrap. The data were searched against a data base containing only the rabbit skeletal muscle actin sequence, using Sequest search engine of the BioWorks software (Thermo Electron, Co.). Search input parameter for reported modifications was limited to single modification per peptide to reduce overall false positives. Appropriate filters were set to generate both stringent as well as generous peptide modifications. Final choice of the modifications was based on intelligent analysis of the MS/MS spectra correlating to the predicted fragment ions, retention time of the modified peptide, and score parameters given by Sequest.
Functional Assays
Actin activated S1 ATPase assays and rigor S1 binding measurements with the K50A/D51A and wt yeast actins were carried out as described previously. 12 In vitro motility measurements of actin sliding velocities over myosin in the absence and presence of external load (from pPDM modified HMM; to yield relative force values) were done as reported before.39
Molecular Graphics
Molecular structure representations in the figures were created with Pymol (www.pymol.com).
Highlights.
-
➢
Radiolytic protein footprinting maps myosin rigor binding sites on actin.
-
➢
Residue probing confirms myosin interactions with actin’s W- and D-loops.
-
➢
Footprinting and spectroscopic results agree with recent actomyosin models.
-
➢
Novel mapping of F-actin sites impacted allosterically by myosin binding.
Acknowledgments
This work was supported by the grant from National Institute for Biomedical Imaging and Bioengineering (EB-P41-01979, EB-P30-09998, and EB-R01-09688) to M.R.C., and a grant from USPHS (GM-077190) to E.R. We wish to thank P. Cheung for help with the functional acto-S1 assays and E. Bobkova for carrying out the in vitro sliding and relative force measurement. We also thank Wayne Hubbell for access to their EPR instruments and Christian Altenbach for assisting in the EPR spectra analysis.
Abbreviations
- G-actin
globular actin
- F-actin
filamentous actin
- S1
myosin subfragment-1
- EPR
electron paramagnetic resonance
- D-loop
DNase-I binding loop
- C169
F169C/C374A
- C167
A167C/C374A
- C41
Q41C/C374S
- C43
I43C/C374S
- C45
V45C/C374S
- C46
G46C/C374S
- C47
M47C/C374S
- C48
G48C/C374A
- C49
Q49C/C374A
- C50
K50C/C374A
- WT
wild type
- pPDM
(N,N‘-p-phenylenedimaleimide)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Amann KJ, Pollard TD. Cellular regulation of actin network assembly. Curr Biol. 2000;10:R728–R730. doi: 10.1016/s0960-9822(00)00751-x. [DOI] [PubMed] [Google Scholar]
- 2.Paavilainen VO, Bertling E, Falck S, Lappalainen P. Regulation of cytoskeletal dynamics by actin-monomer-binding proteins. Trends Cell Biol. 2004;14:386–394. doi: 10.1016/j.tcb.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 3.Pollard TD, Blanchoin L, Mullins RD. Molecular mechanisms controlling actin filament dynamics in nonmuscle cells. Annu Rev Biophys Biomol Struct. 2000;29:545–76. doi: 10.1146/annurev.biophys.29.1.545. [DOI] [PubMed] [Google Scholar]
- 4.Rayment I, Holden HM, Whittaker M, Yohn CB, Lorenz M, Holmes KC, Milligan RA. Structure of the actin-myosin complex and its implications for muscle contraction. Science. 1993;261:58–65. doi: 10.1126/science.8316858. [DOI] [PubMed] [Google Scholar]
- 5.Schroder RR, Manstein DJ, Jahn W, Holden H, Rayment I, Holmes KC, Spudich JA. Three-dimensional atomic model of F-actin decorated with Dictyostelium myosin S1. Nature. 1993;364:171–4. doi: 10.1038/364171a0. [DOI] [PubMed] [Google Scholar]
- 6.Volkmann N, Hanein D, Ouyang G, Trybus KM, DeRosier DJ, Lowey S. Evidence for cleft closure in actomyosin upon ADP release. Nat Struct Biol. 2000;7:1147–55. doi: 10.1038/82008. [DOI] [PubMed] [Google Scholar]
- 7.Volkmann N, Ouyang G, Trybus KM, DeRosier DJ, Lowey S, Hanein D. Myosin isoforms show unique conformations in the actin-bound state. Proc Natl Acad Sci U S A. 2003;100:3227–32. doi: 10.1073/pnas.0536510100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Holmes KC, Angert I, Kull FJ, Jahn W, Schroder RR. Electron cryo-microscopy shows how strong binding of myosin to actin releases nucleotide. Nature. 2003;425:423–7. doi: 10.1038/nature02005. [DOI] [PubMed] [Google Scholar]
- 9.Murphy CT, Spudich JA. The sequence of the myosin 50-20K loop affects Myosin’s affinity for actin throughout the actin-myosin ATPase cycle and its maximum ATPase activity. Biochemistry. 1999;38:3785–92. doi: 10.1021/bi9826815. [DOI] [PubMed] [Google Scholar]
- 10.Miller CJ, Doyle TC, Bobkova E, Botstein D, Reisler E. Mutational analysis of the role of hydrophobic residues in the 338-348 helix on actin in actomyosin interactions. Biochemistry. 1996;35:3670–6. doi: 10.1021/bi952645v. [DOI] [PubMed] [Google Scholar]
- 11.Miller CJ, Wong WW, Bobkova E, Rubenstein PA, Reisler E. Mutational analysis of the role of the N terminus of actin in actomyosin interactions. Comparison with other mutant actins and implications for the cross-bridge cycle. Biochemistry. 1996;35:16557–65. doi: 10.1021/bi962388+. [DOI] [PubMed] [Google Scholar]
- 12.Miller CJ, Reisler E. Role of charged amino acid pairs in subdomain-1 of actin in interactions with myosin. Biochemistry. 1995;34:2694–700. doi: 10.1021/bi00008a037. [DOI] [PubMed] [Google Scholar]
- 13.Geisterfer-Lowrance AA, Kass S, Tanigawa G, Vosberg HP, McKenna W, Seidman CE, Seidman JG. A molecular basis for familial hypertrophic cardiomyopathy: a beta cardiac myosin heavy chain gene missense mutation. Cell. 1990;62:999–1006. doi: 10.1016/0092-8674(90)90274-i. [DOI] [PubMed] [Google Scholar]
- 14.Lorenz M, Holmes KC. The actin-myosin interface. Proc Natl Acad Sci U S A. 2010;107:12529–34. doi: 10.1073/pnas.1003604107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hambly DM, Gross ML. Laser flash photolysis of hydrogen peroxide to oxidize protein solvent-accessible residues on the microsecond timescale. J Am Soc Mass Spectrom. 2005;16:2057–2063. doi: 10.1016/j.jasms.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 16.Sharp JS, Becker JM, Hettich RL. Analysis of protein solvent accessible surfaces by photochemical oxidation and mass spectrometry. Anal Chem. 2004;76:672–683. doi: 10.1021/ac0302004. [DOI] [PubMed] [Google Scholar]
- 17.Sharp JS, Guo JT, Uchiki T, Xu Y, Dealwis C, Hettich RL. Photochemical surface mapping of C14S-Sml1p for constrained computational modeling of protein structure. Anal Biochem. 2005;340:201–212. doi: 10.1016/j.ab.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 18.Takamoto K, Chance MR. Radiolytic protein footprinting with mass spectrometry to probe the structure of macromolecular complexes. Annu Rev Biophys Biomol Struct. 2006;35:251–276. doi: 10.1146/annurev.biophys.35.040405.102050. [DOI] [PubMed] [Google Scholar]
- 19.Takamoto K, Kamal JKA, Chance MR. Biochemical implications of a three-dimensional model of monomeric actin bound to magnesium-chelated ATP. Structure. 2006;15:39–51. doi: 10.1016/j.str.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 20.Xu G, Liu R, Zak O, Aisen P, Chance MR. Structural allostery and binding of the transferring receptor complex. Mol Cell Proteomics. 2005;4:1959–1967. doi: 10.1074/mcp.M500095-MCP200. [DOI] [PubMed] [Google Scholar]
- 21.Guan JQ, Chance MR. Structural proteomics of macromolecular assemblies using oxidative footprinting and mass spectrometry. Trends Biochem Sci. 2005;30:583–592. doi: 10.1016/j.tibs.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 22.Guan JQ, Almo SC, Chance MR. Synchrotron Radiolysis and Mass Spectrometry: A new approach to research on the actin cytoskeleton. Acct Chem Res. 2004;37:221–229. doi: 10.1021/ar0302235. [DOI] [PubMed] [Google Scholar]
- 23.Guan JQ, Almo SC, Reisler E, Chance MR. Structural reorganization of proteins revealed by radiolysis and mass spectrometry: G-actin solution structure is divalent cation dependent. Biochemistry. 2003;42:11992–12000. doi: 10.1021/bi034914k. [DOI] [PubMed] [Google Scholar]
- 24.Guan JQ, Takamoto K, Almo SC, Reisler E, Chance MR. Structure and dynamics of the actin filament. Biochemistry. 2005;44:3166–3175. doi: 10.1021/bi048021j. [DOI] [PubMed] [Google Scholar]
- 25.Kiselar JG, Janmey PA, Almo SC, Chance MR. Visualizing the Ca2+-dependent activation of gelsolin by using synchrotron footprinting. Proc Natl Acad Sci U S A. 2003;100:3942–3947. doi: 10.1073/pnas.0736004100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kiselar JG, Mahaffy R, Pollard TD, Almo SC, Chance MR. Visualizing Arp2/3 complex activation mediated by binding of ATP and WASp using structural mass spectrometry. Proc Natl Acad Sci U S A. 2007;104:1552–1557. doi: 10.1073/pnas.0605380104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guan JQ, Chance MR. Structural proteomics of macromolecular assemblies using oxidative footprinting and mass spectrometry. Trends Biochem Sci. 2005;30:583–592. doi: 10.1016/j.tibs.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 28.Maleknia SD, Brenowitz M, Chance MR. Millisecond radiolytic modification of peptides by synchrotron X-rays identified by mass spectrometry. Anal Chem. 1999;71:3965–73. doi: 10.1021/ac990500e. [DOI] [PubMed] [Google Scholar]
- 29.Liu R, Guan JQ, Zak O, Aisen P, Chance MR. Structural reorganization of the transferrin C-lobe and transferrin receptor upon complex formation: the C-lobe binds to the receptor helical domain. Biochemistry. 2003;42:12447–54. doi: 10.1021/bi0352973. [DOI] [PubMed] [Google Scholar]
- 30.Gupta S, Mangel WF, Baniecki ML, McGrath WJ, Takamoto KG, Chance MR. DNA binding provides a molecular strap activating the Adenovirus Proteinase. Mol & Cell Proteomics. 2004 doi: 10.1074/mcp.M400037-MCP200. in press. [DOI] [PubMed] [Google Scholar]
- 31.Liu R, Guan JQ, Zak O, Aisen P, Chance MR. Structural reorganization of Transferrin C-lobe and Transferrin Receptor Upon Complex Formation: C-lobe to the Receptor Helical Domain. Biochemistry. 2003;42:12447–12454. doi: 10.1021/bi0352973. [DOI] [PubMed] [Google Scholar]
- 32.Gupta S, Cheng H, Mollah AK, Jamison E, Morris S, Chance MR, Khrapunov S, Brenowitz M. DNA and protein footprinting analysis of the modulation of DNA binding by the N-terminal domain of the Saccharomyces cerevisiae TATA binding protein. Biochemistry. 2007;46:9886–98. doi: 10.1021/bi7003608. [DOI] [PubMed] [Google Scholar]
- 33.Milligan RA. Protein-protein interactions in the rigor actomyosin complex. Proc Natl Acad Sci U S A. 1996;93:21–6. doi: 10.1073/pnas.93.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kudryashov DS, Grintsevich EE, Rubenstein PA, Reisler E. A nucleotide state-sensing region on actin. J Biol Chem. 2010;285:25591–601. doi: 10.1074/jbc.M110.123869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oztug Durer ZA, Diraviyam K, Sept D, Kudryashov DS, Reisler E. F-actin structure destabilization and DNase I binding loop: fluctuations mutational cross-linking and electron microscopy analysis of loop states and effects on F-actin. J Mol Biol. 2010;395:544–57. doi: 10.1016/j.jmb.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wertman KF, Drubin DG, Botstein D. Systematic mutational analysis of the yeast ACT1 gene. Genetics. 1992;132:337–50. doi: 10.1093/genetics/132.2.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dominguez R. Actin-binding proteins--a unifying hypothesis. Trends Biochem Sci. 2004;29:572–8. doi: 10.1016/j.tibs.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 38.Reisler E, Egelman EH. Actin structure and function: what we still do not understand. J Biol Chem. 2007;282:36133–7. doi: 10.1074/jbc.R700030200. [DOI] [PubMed] [Google Scholar]
- 39.Galkin VE, Orlova A, Lukoyanova N, Wriggers W, Egelman EH. Actin depolymerizing factor stabilizes an existing state of F-actin and can change the tilt of F-actin subunits. J Cell Biol. 2001;153:75–86. doi: 10.1083/jcb.153.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim E, Bobkova E, Miller CJ, Orlova A, Hegyi G, Egelman EH, Muhlrad A, Reisler E. Intrastrand cross-linked actin between Gln-41 and Cys-374. III. Inhibition of motion and force generation with myosin. Biochemistry. 1998;37:17801–9. doi: 10.1021/bi981286b. [DOI] [PubMed] [Google Scholar]
- 41.Root DD, Reisler E. The accessibility of etheno-nucleotides to collisional quenchers and the nucleotide cleft in G- and F-actin. Protein Sci. 1992;1:1014–22. doi: 10.1002/pro.5560010807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Adams SB, Reisler E. Sequence 18-29 on actin: antibody and spectroscopic probing of conformational changes. Biochemistry. 1994;33:14426–33. doi: 10.1021/bi00252a008. [DOI] [PubMed] [Google Scholar]
- 43.Feng L, Kim E, Lee WL, Miller CJ, Kuang B, Reisler E, Rubenstein PA. Fluorescence probing of yeast actin subdomain 3/4 hydrophobic loop 262-274. Actin-actin and actin-myosin interactions in actin filaments. J Biol Chem. 1997;272:16829–37. doi: 10.1074/jbc.272.27.16829. [DOI] [PubMed] [Google Scholar]
- 44.Crosbie RH, Chalovich JM, Reisler E. Interaction of caldesmon and myosin subfragment 1 with the C-terminus of actin. Biochem Biophys Res Commun. 1992;184:239–45. doi: 10.1016/0006-291x(92)91184-r. [DOI] [PubMed] [Google Scholar]
- 45.Crosbie RH, Miller C, Cheung P, Goodnight T, Muhlrad A, Reisler E. Structural connectivity in actin: effect of C-terminal modifications on the properties of actin. Biophys J. 1994;67:1957–64. doi: 10.1016/S0006-3495(94)80678-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Prochniewicz E, Thomas DD. Perturbations of functional interactions with myosin induce long-range allosteric and cooperative structural changes in actin. Biochemistry. 1997;36:12845–53. doi: 10.1021/bi971201r. [DOI] [PubMed] [Google Scholar]
- 47.Vahdat A, Miller C, Phillips M, Muhlrad A, Reisler E. A novel 27/16 kDa form of subtilisin cleaved actin: structural and functional consequences of cleavage between Ser234 and Ser235. FEBS Lett. 1995;365:149–51. doi: 10.1016/0014-5793(95)00446-g. [DOI] [PubMed] [Google Scholar]
- 48.Sutoh K. Mapping of actin-binding sites on the heavy chain of myosin subfragment 1. Biochemistry. 1983;22:1579–85. doi: 10.1021/bi00276a009. [DOI] [PubMed] [Google Scholar]
- 49.Pliszka B, Martin BM, Karczewska E. Ionic interaction of myosin loop 2 with residues located beyond the N-terminal part of actin probed by chemical cross-linking. Biochim Biophys Acta. 2008;1784:285–91. doi: 10.1016/j.bbapap.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 50.Schwyter DH, Kron SJ, Toyoshima YY, Spudich JA, Reisler E. Subtilisin cleavage of actin inhibits in vitro sliding movement of actin filaments over myosin. J Cell Biol. 1990;111:465–70. doi: 10.1083/jcb.111.2.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Borovikov YS, Moraczewska J, Khoroshev MI, Strzelecka-Golaszewska H. Proteolytic cleavage of actin within the DNase-I-binding loop changes the conformation of F-actin and its sensitivity to myosin binding. Biochim Biophys Acta. 2000;1478:138–51. doi: 10.1016/s0167-4838(00)00005-4. [DOI] [PubMed] [Google Scholar]
- 52.Spudich JA, Watt S. The regulation of rabbit skeletal muscle contraction. I. Biochemical studies of the interaction of the tropomyosin-troponin complex with actin and the proteolytic fragments of myosin. J Biol Chem. 1971;246:4866–4871. [PubMed] [Google Scholar]
- 53.Weeds AG, Taylor RS. Separation of subfragment-1 isoenzymes from rabbit skeletal muscle myosin. Nature. 1975;257:54–56. doi: 10.1038/257054a0. [DOI] [PubMed] [Google Scholar]
- 54.Altenbach C, Oh KJ, Trabanino RJ, Hideg K, Hubbell WL. Estimation of inter-residue distances in spin labeled proteins at physiological temperatures: experimental strategies and practical limitations. Biochemistry. 2001;40:15471–82. doi: 10.1021/bi011544w. [DOI] [PubMed] [Google Scholar]
- 55.Kiselar JG, Janmey PA, Almo SC, Chance MR. Structural Analysis of Gelsolin Using Synchrotron Protein Footprinting. Mol Cell Proteomics. 2003;2:1120–1132. doi: 10.1074/mcp.M300068-MCP200. [DOI] [PubMed] [Google Scholar]
- 56.Xu G, Kiselar JG, He Q, Chance MR. Secondary reactions and strategies to improve quantitative protein footprinting. Anal Chem. 2005;77:3029–37. doi: 10.1021/ac048282z. [DOI] [PubMed] [Google Scholar]
- 57.Guan JQ, Vorobiev S, Almo SC, Chance MR. Mapping the G-actin binding surface of cofilin using synchrotron protein footprinting. Biochemistry. 2002;41:5765–75. doi: 10.1021/bi0121104. [DOI] [PubMed] [Google Scholar]
- 58.Kiselar JG, Janmey PA, Almo SC, Chance MR. Structural Analysis of Gelsolin Using Synchrotron Protein Footprinting. Mol Cell Proteomics. 2003;2:1120–1132. doi: 10.1074/mcp.M300068-MCP200. [DOI] [PubMed] [Google Scholar]